

Нефтехимия, 2019, T. 59, № 3, стр. 315-324
Перспективные процессы производства биотоплив типа “drop in” и продуктов нефтехимии из возобновляемого сырья (обзор)
Т. А. Паланкоев 1, *, К. И. Дементьев 1, С. Н. Хаджиев 1
1 Институт нефтехимического синтеза им. А.В. Топчиева РАН
Москва, Россия
* E-mail: tpalankoev@ips.ac.ru
Поступила в редакцию 09.11.2018
После доработки 01.12.2018
Принята к публикации 10.12.2018
Аннотация
Рассмотрены основные виды возобновляемых источников углерода, пригодных для производства биотоплив типа “drop in” – углеводородных топлив с минимальным содержанием кислорода, полностью совместимых с традиционными топливами. Обсуждены методы переработки различных видов сырья, указаны их основные преимущества и недостатки. Показано, что получение “drop in” биотоплив из растительных масел и отходов производства биодизеля возможно в рамках существующих процессов без значительных капитальных затрат. Переработка продуктов пиролиза лигноцеллюлозной биомассы сопряжена с рядом технических и технологических проблем и в настоящее время находится в стадии активной научно-технической проработки.
ВВЕДЕНИЕ
Мировая экономика традиционно базируется на ископаемом сырье – уголь, нефть и газ обеспечивают более двух третей мирового производства энергии и на 100% закрывают потребности человечества в углеводородах (УВ) – ценном химическом сырье [1]. Трудно представить себе сегодня мир без транспорта с двигателями внутреннего сгорания, без полимерных материалов, лаков, лекарств и других важнейших продуктов. Соответственно, развитие мировой экономики приводит к увеличению объемов добычи ископаемого сырья.
С 2005 по 2012 гг. мировые разведанные запасы нефти увеличились с 187.5 до 230.3 млрд т [2]. Однако, начиная с 2012 г., объем прироста запасов крайне мал; более того, в 2015 г. было отмечено снижение общемировых запасов нефти на 300 млн т по сравнению с 2014 г. В России ситуация с запасами нефти не менее тревожна – падение разведанных запасов началось в 2007 г. и продолжается; текущий уровень составляет 14.0 млрд т, что на 500 млн т меньше, чем в 2007 г. Кроме того, большая часть вновь открытых запасов представлена сравнительно тяжелыми видами нефтей, сосредоточенными в малых и средних месторождениях. Большие нефтяные бассейны легкой нефти открываются все реже; существующие месторождения постепенно исчерпываются [3]. Подобные тенденции актуальны и для нашей страны. Так, например, добыча на крупнейшем в России Самотлорском месторождении пережила свой пик в 1980-х, сейчас же добыча снижается, причем добываемая нефть крайне обводнена [4].
Другой проблемой, связанной с широким использованием ископаемых топлив, является выделение большого количества техногенного диоксида углерода в атмосферу. Ежегодно в мире его выбрасывается более 33 млрд т (и эта цифра ежегодно увеличивается), что составляет около 1.8% от общего количества этого газа в атмосфере. Концентрация CO2 в атмосфере повысилась с 277 ppm (1750 г.) до 395 ppm (2013 г.), причем подавляющая часть этого роста приходится на период индустриальной активности человечества [5]. По всей видимости, биота Земли не справляется с утилизацией такого количества техногенного углекислого газа. Дальнейшее развитие экономики, основанной только на ископаемом сырье, продолжение и усиление антропогенного воздействия на окружающую среду грозит развитием непредсказуемых (возможно, катастрофических!) последствий для человечества.
Единственной реальной заменой ископаемых УВ представляется возобновляемое сырье – биомасса растений. Биомасса по элементному составу содержит углерод, водород и кислород; соответственно, ее можно использовать как для производства электроэнергии, так и (потенциально) для производства топлив и химических продуктов. Основными преимуществами использования биомассы в качестве источника энергии и химического сырья являются:
– чрезвычайно большой объем доступных ресурсов; так, ежегодный объем только лесотехнических отходов и только в России составляет не менее 1 млрд м3 [6];
– быстрое воспроизводство биосырья и возможность экстенсивного расширения его производства – ежегодный общемировой прирост биомассы составляет около 220 млрд т; используется едва лишь десятая часть [7];
– отсутствие влияния на углеродный баланс в биосфере при сжигании топлив, полученных из биомассы. Генерация биомассы осуществляется в результате поглощения и усваивания атмосферного диоксида углерода. Соответственно, при сжигании топлива в атмосферу попадет лишь уловленный из нее газ, который вновь может быть переведен в тот же объем биомассы. В настоящее время биомасса уже используется для производства ряда компонентов моторных топлив на ее основе: в промышленности освоены технологии получения компонентов автобензина из возобновляемого сырья (биоэтанола, биобутанола) и биодизеля. Их суммарное мировое производство в настоящее время составляет более 84 млн т/год (в нефтяном эквиваленте) [2].
Однако традиционные биотоплива обладают рядом существенных недостатков:
– низкая теплота сгорания, по сравнению с УВ, что приводит к повышенному расходу топлива;
– необходимость перенастройки двигателя;
– заметная коррозионная активность, необходимость применения специальных конструкционных материалов для двигателей и топливных систем;
– плохая совместимость с топливами на основе ископаемого сырья (биоэтанол, биобутанол);
– неудовлетворительные низкотемпературные свойства (биодизель).
Все перечисленные недостатки биотоплив обусловлены наличием в их составе кислорода. В связи с этим большую популярность набирает так называемая концепция “drop in”: производство из биомассы топлив с минимальным содержанием кислорода, полностью совместимых с нефтяными фракциями. В качестве сырья для получения таких топлив могут служить различные продукты, полученные из биомассы: растительные масла, биоспирты и жидкие продукты пиролиза.
Растительные масла. Они являются весьма привлекательным сырьем для производства биотоплив [8–10]. Наиболее простым и нашедшим промышленное применение способом является переэтерификация рапсового или соевого масла метанолом в присутствии кислотных или основных катализаторов с получением смеси метиловых эфиров жирных кислот – компонента дизельного топлива [11]. Мировой объем производства биодизеля в 2016 г. составил свыше 31 млн т [12]; лидирующими производителями являются США и Бразилия [13, 14].
Процесс производства биодизеля является достаточно грязным: в качестве сырья используется метанол, в качестве катализатора – щелочи, а в процессе реакции переэтерификации образуется большое количество водных отходов. Альтернативным способом переработки растительных масел в биотопливо является гидродеоксигенация. Сообщается, что при использовании катализаторов на основе интерметаллических комплексов Pt–Sn, нанесенных на оксид алюминия, возможно полное подавление реакций декарбоксилирования и декарбонилирования с селективностью по УВ вплоть до количественной [15]. Получаемый концентрат УВ нормального строения может быть далее путем гидроизомеризации переработан в высококачественное бессернистое реактивное и дизельное топливо [16]. Использование кислотных носителей на основе цеолита ZSM-5 позволяет получать концентрат ароматических УВ с селективностью до 70% из растительных масел в одну стадию [17].
Каталитический крекинг также является перспективным процессом с точки зрения производства моторных топлив из растительных масел [18–20]. Так, в работе [13] исследован каталитический крекинг соевого масла на коммерческом ZSM-5 (Si/Al = 20) в стационарном слое при 450°С. Показана возможность исчерпывающей конверсии масла в УВ бензинового и дизельного ряда, а также УВ С3–С4. Выход легких олефинов в зависимости от условий крекинга при этом может составлять до 50%, жидкий продукт представляет собой концентрат ароматических УВ. Крекинг пальмового масла и отработанных пищевых масел изучен в работе [19]. Выход УВ в подобном процессе существенно зависит от кислотности катализатора и его селективности по размеру реагирующих молекул. Наибольшим потенциалом в катализе такого процесса обладают цеолиты (ZSM-5 и Y) и мезопористые материалы (MCM-41, SBA-15). Тем не менее, в процессе крекинга растительных масел наблюдается активное образование оксидов углерода, что свидетельствует об интенсивном протекании реакций декарбоксилирования и декарбонилирования. Основные показатели крекинга различных видов масел на цеолитных катализаторах приведены в табл. 1.
Таблица 1.
Типичный состав основных продуктов каталитического крекинга растительных масел [21]. Условия: Т = 450°С, W = 2.5 ч–1
| Сырье | Пальмовое масло | Отработанное пальмовое масло | Косточковое пальмовое масло | Смесь жирных кислот в пальмовом масле | ||||
|---|---|---|---|---|---|---|---|---|
| Катали-затор | ZSM-5 | композит ZSM-5/ MCM-41 | MCM-41 | композит ZSM-5/ MCM-41 | MCM-41 | SBA-15 | Al-SBA | Al-MCM-41 |
| Выход продуктов, мас. % | ||||||||
| Газ | 8.2 | 11.4 | 19.4 | 13.4 | 8.9 | 9.5 | 16.2 | 6.7 |
| Фр. НК-200 | 77.9 | 64.2 | 56.9 | 57.1 | 73.8 | 41 | 57.8 | 59.3 |
| Кокс | 1.7 | 5.4 | 10.9 | 10.7 | 10.8 | 7.2 | 6.3 | 9.8 |
Растительное масло может быть введено в процесс крекинга как в чистом виде, так и в смеси с традиционным сырьем каталитического крекинга. Показано, что введение 10% смеси хлопкового и подсолнечного масел в вакуумный дистиллят приводит к увеличению выхода бензиновой фракции на 4% [22]. При этом бензиновая фракция обогащается олефиновыми, ароматическими и изопарафиновыми УВ, что приводит, в свою очередь к увеличению октанового числа бензина на 2.5 п. В качестве компонента сырья крекинга могут быть использованы также непищевые и отработанные масла [23–26]. С химической точки зрения компоненты растительных масел – триглицериды жирных кислот – подвергаются декарбоксилированию с образованием углеводородных карбокатионов, претерпевающих в дальнейшем различные превращения: изомеризацию, расщепление, циклизацию и т.д.
Благодаря хорошей смешиваемости растительных масел с нефтяными фракциями, а также их высокой конверсии в условиях крекинга, переработка масел в процессе каталитического крекинга может быть организована на основе существующих нефтеперерабатывающих установок с минимальными модификациями.
Превращение оксигенатов – отходов биопроизводства. В свете набирающей популярность технологии производства биотоплив актуальной становится проблема переработки побочных продуктов таких процессов. Так, при производстве биодизеля переэтерификацией растительных масел, побочным продуктом является глицерин в виде водного раствора, дополнительно содержащего метанол, остатки масел и жирных кислот. Производство биобутанола в процессе ферментативного превращения биомассы приводит к образованию смеси кислородсодержащих побочных продуктов, основным из которых является ацетон. Сегодня утилизация подобных отходов осуществляется сжиганием в метане с выработкой электроэнергии [27]. Этот способ характеризуется низкой эффективностью, поскольку теплота сгорания глицерина относительно невелика.
С целью разработки более выгодного пути переработки вышеупомянутых отходов за последние несколько лет проведено значительное количество работ по кислотно-каталитической конверсии кислородсодержащих соединений в полезное сырье для нефтехимии (непредельные УВ) и компоненты топлив. Показано, что ацетон претерпевает на цеолитных катализаторах превращения при температурах 450–500°С с образованием олефинов С2–С4 и ароматических УВ (толуола, ксилола) с высокой конверсией (95–98%) [28, 29]. При этом существенное влияние на выход и селективность превращения оказывают параметры цеолитного катализатора: структура и размер пор, соотношение Si/Al. В зависимости от этих характеристик, процесс каталитического крекинга будет преимущественно протекать либо с образованием ароматических УВ и парафинов, либо с образованием преимущественно легких олефинов.
Технический глицерин также является перспективным сырьем производства сырья для нефтехимии, в силу своей низкой стоимости и возобновляемости [30]. В частности, сообщается о превращении водного раствора глицерина на цеолите типа ZSM-5 при температурах порядка 600°С с образованием газообразных и жидких УВ, а также оксидов углерода [31]. Селективность по целевым продуктам может регулироваться введением различных металлов в состав катализатора, однако селективность по олефинам даже для лучших образцов не превышает 20%. Известны также работы по превращению глицерина на цеолитных катализаторах в акролеин, ацетальдегид, формальдегид, ацетол [32–34]. Оптимизация состава катализатора и условий проведения реакции позволяет достичь селективности по акролеину и ацетальдегиду в интервале 25–30%.
Более перспективным представляется проведение конверсии кислородсодержащих отходов совместно с нефтяным сырьем для повышения селективности превращения по УВ. Показано [35], что при введении в вакуумный дистиллят до 10 мас. % кислородсодержащей добавки (ацетон, глицерин, метанол) последняя полностью превращается в газообразные продукты, богатые С3–С4-олефинами, причем селективность по пропилену достигает 60%. Кислород целиком утилизируется в виде воды. Изменение концентрации добавки приводит возрастанию выхода газа (вплоть до 5 мас. % в случае ацетона), выход жидких продуктов при этом практически не изменяется. Повышение содержания глицерина в сырье до 50% приводит к снижению конверсии сырья на 25–30% за счет уменьшения селективности по бензиновой фракции и газу [36].
Переработка продукта пиролиза биомассы. Наиболее широко распространенный возобновляемый ресурс – лигноцеллюлозная биомасса. Одним из основных способов первичной переработки биомассы (например, древесных опилок и прочих отходов лесного хозяйства) является пиролиз в отсутствие воздуха при атмосферном давлении, температурах в диапазоне 450–550°С и коротком времени пребывания в реакционной зоне. Данный эффективный способ переработки биомассы с высоким выходом жидких продуктов (до 70–80%) рассматривается также как первый этап технологии превращения биомассы в моторные топлива и сырье для нефтехимии [37]. Получаемый в результате пиролиза биомассы жидкий продукт (биомасло, бионефть, в отечественной литературе – жижка пиролиза) представляет собой многокомпонентную смесь соединений различных классов, образовавшихся при деполимеризации и разложении целлюлозы, гемицеллюлозы и лигнина: кислот, спиртов, альдегидов, эфиров, кетонов, фенолов, гваяколов, сахаров и пр. Элементный состав и некоторые свойства биомасла приведены в табл. 2.
Таблица 2.
Типичные свойства масла пиролиза древесины [37]
| Параметр | Значение |
|---|---|
| Содержание воды, мас. % | 15–30 |
| pH | 2.5–4.0 |
| Вязкость при 50°С (сП) | 40–100 |
| Элементный состав, мас. % | |
| C | 54–58 |
| H | 5.5–7.0 |
| O | 35–40 |
| N | 0–0.2 |
| Остаток перегонки, мас. % | До 50 |
Высокая кислотность обуславливает значительную коррозионную активность бионефти; для ее транспортировки и переработки необходимо использовать специальные виды материалов. Другая проблема бионефти – высокое содержание кислорода, который распределен по более, чем 300 компонентам. Наличие в составе бионефти термолабильных соединений – альдегидов, кетонов, сахаров и ангидросахаров – приводит к высокой степени термической нестабильности этого вида биосырья.
Поскольку бионефть представляет собой многокомпонентную смесь молекул различной реакционной способности с различными функциональными группами, одним из подходов к ее переработке является разделение на фракции с последующим превращением отдельных компонентов. При этом процесс разгонки неприменим из-за резкого ускорения полимеризации компонентов биосырья даже при небольшом нагревании, поэтому альтернативным методом может быть экстракция. Так, с помощью экстракции водой можно выделить нерастворимые в воде олигомеры лигнина, а растворенные в воде альдегиды и кетоны могут быть отделены от сахаров с помощью эфира [38].
Исследована возможность [39] выделения некоторых групп компонентов водорастворимой части бионефти путем экстрагирования органическими растворителями: гексаном, петролейным эфиром, хлороформом, этилацетатом. Сообщается об эффективном разделении при применении двухстадийной экстракции хлороформом и этилацетатом. Хлороформом удается эффективно экстрагировать фураны, фенолы и кетоны, а этилацетатом – органические кислоты. В остатке преимущественно остается левоглюкозан и сахара в водном растворе. Выделенные соединения могут быть использованы в качестве сырья для каталитического облагораживания фуранов и производных фенола в УВ, ферментирования левоглюкозана в спирты и диолы, а также производства водорода из органических кислот. Экстракция водонерастворимой части бионефти дихлорметаном позволяет выделить фенол и его производные, а также компоненты пиролитического лигнина [40], которые могут быть использованы для последующего синтеза фенольных смол.
На сегодняшний день существует ряд схем последовательной экстракции бионефти рядом растворителей (вода, диэтиловый эфир, дихлорметан и пр.), использующихся для рутинного анализа ее группового состава [41]. Тем не менее, с технологической точки зрения экстрактивное разделение бионефти с получением каких-либо ценных компонентов ограничивается стоимостью растворителей и нерешенной проблемой стабильности получаемых фракций.
Помимо выделения отдельных компонентов для последующей переработки, бионефть может быть облагорожена путем этерификации низкомолекулярными спиртами [42–46]. При этом удается достичь значительного снижения количества кислотных групп за счет превращения кислот в соответствующие эфиры, что, в целом, положительно сказывается на свойствах сырья: отмечается снижение вязкости, кислотности, увеличение химической стабильности [47–49]. Этерификация характеризуется низкой скоростью реакции (реакционная смесь может выдерживаться в течение 2–3 месяцев), поэтому ряд работ направлен на изучение каталитической этерификации с использованием кислотных и основных катализаторов [50], а также цеолитов и алюмосиликатов [51, 52]. Благодаря дешевизне реагентов и катализаторов и простоте процесса этот подход может стать приемлемым способом стабилизации биосырья.
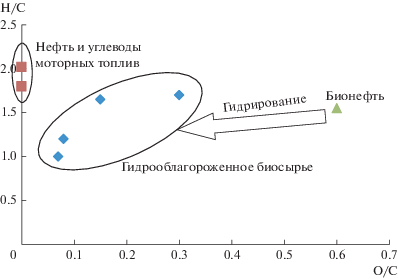
Помимо высокой кислотности и низкой термической стабильности, эффективному вовлечению бионефти в переработку с получением УВ препятствует также низкое отношение Н/С по сравнению с традиционной нефтью (рисунок). В связи с этим большое количество работ посвящено гидрооблагораживанию бионефти, при этом целью большинства исследований является достижение следующих условий: максимально полное удаление кислорода и увеличение отношения Н/С при максимально мягких условиях и низком расходе водорода.
По аналогии с нефтепереработкой, катализаторами такого процесса могут служить традиционные катализаторы гидрооблагораживания нефтяных продуктов на основе Co, Mo и Ni на носителе. Изучению этого процесса на примере модельных соединений – в основном, фенола и гваякола – посвящено достаточно много работ [55–62]. Так, например, изучена гидроконверсия гваякола на сульфиде молибдена и биметаллическом сульфированном молибдене, модифицированном кобальтом, при температуре 300°С и давлении водорода 40 атм. При превращении на MoS2 без носителя выход продуктов, не содержащих кислород (циклогексен, бензол, циклогексан), возрастает с конверсией и достигает 55 мол. %; основным кислородсодержащим продуктом при этом является фенол (выход – до 30 мол. %). Промотирование катализатора кобальтом способствует повышению активности катализатора и увеличению интенсивности реакций отщепления метокси-группы с образованием фенола. Однако к увеличению выхода продуктов, не содержащих кислород, такая модификация не приводит [56]. Традиционные сульфидные катализаторы достаточно быстро дезактивируются в отсутствие сульфирующего агента из-за окисления активной фазы [58]. Помимо этого, в условиях конверсии бионефти нестабилен и сам носитель, так как исходные кислородсодержащие соединения и образующаяся вода взаимодействуют с оксидом алюминия [57].
В связи с этим активно изучаются альтернативные носители для катализаторов в условиях гидроконверсии модельных соединений бионефти. При использовании углеродных нанотрубок в качестве носителей для никель-кобальтовых катализаторов при температуре 200°С и давлении водорода не менее 50 атм, конверсия гваякола достигает 93%, а селективность по основному продукту – циклогексанолу – порядка 85% [63]. В другой работе [64] испытана серия катализаторов на основе никеля на различных подложках (δ-Al2O3, SiO2, SiO2–ZrO2, CeO2–ZrO2) в процессе гидроконверсии гваякола при температуре 320°С и давлении водорода 170 атм. В терминах конверсии гваякола и степени удаления кислорода наилучшие результаты получены на катализаторах на основе никеля на SiO2 и ZrO2–SiO2–La2O3 в качестве носителя. Так, максимальная конверсия исходного кислородсодержащего соединения достигла 97.5%, а степень удаления кислорода – 97.3%. Основными продуктами указанных превращений являлись алифатические УВ С5–С7, а также бензол. Наибольший выход кокса получен на катализаторе, содержащем только никель на оксиде кремния. С другой стороны, наименьшее содержание кокса получено для аналогичного катализатора, но модифицированного медью, что и иллюстрирует ее влияние. Наиболее перспективным в процессе конверсии гваякола, с точки зрения степени удаления кислорода и выхода кокса, признан катализатор на основе модифицированного медью никеля на матрице ZrO2–SiO2–La2O3. Основной продукт превращения гваякола на этом катализаторе – циклогексан, выход которого достиг 58.9 мол. %, а также бензол (9.8 мол. %), диметилциклопропан (9.3 мол. %), циклопентан (5.4 мол. %).
Другое направление исследований – подбор катализаторов, не нуждающихся в сульфировании для создания каталитически активных центров. В качестве таких катализаторов могут служит частично восстановленные оксиды [65–68], а также карбиды, нитриды и фосфиды переходных металлов [68–71]. Достаточно высокой активностью обладают благородные металлы – Pd, Pt, Ru и др. В частности, показано, что палладиевые и платиновые катализаторы эффективны в катализе реакций декарбоксилирования и декарбонилирования [69]. Использование благородных металлов позволяет проводить облагораживание бионефти в среде водорода при сравнительно мягких условиях; так, в присутствии Pd гидрирование фенола и его производных в соответствующие ароматические или насыщенные УВ осуществляется при температуре 250–350°С и давлении водорода 15 атм [72, 73].
Перспективным методом деоксигенации бионефти является также каталитический крекинг. Перспективность этого метода обусловлена гибкостью процесса, возможностью превращения различных видов сырья путем модификации цеолитных катализаторов крекинга и подбора оптимальных условий.
Каталитический крекинг непосредственно бионефти достаточно затруднен [74]. Наблюдается высокий выход оксидов углерода, причем выход СО превышает выход СО2 (50–60 мас. % против 30–40), т.е. цеолитные катализаторы способствуют преимущественному протеканию реакций декарбонилирования. Вместе с тем, имеет место проблема активного коксообразования. Превращение бионефти в процессе каталитического крекинга с учетом ее элементного состава можно представить в виде эмпирического уравнения [75]:
CH1.4O0.4 → 0.9“CH1.2” + 0.1CO2 + 0.2H2O
где “CH1.2” – углеводородный продукт.
С учетом полного удаления кислорода, по стехиометрии приведенного уравнения реакции, теоретический выход углеводородного продукта не превышает 42 мас. %. Причиной такого ограничения является низкое отношение Н/С, которое также влияет и на качество получаемого продукта. Так, эффективное отношение Н/С для получаемых из бионефти УВ, рассчитываемое по формуле
Так, в работе [77] показана возможность превращения модельных соединений – уксусной кислоты, гидроксиацетона, фенола – при введении их в обычное сырье крекинга при концентрации до 10 мас. %. При этом возрастает выход бензиновой фракции на 2.3–3.7 мас. % (в случае фенола), и выход С3–С4-углеводородов на 3.3 мас. % (в случае уксусной кислоты). Однако какого-либо синергетического эффекта авторами обнаружено не было. При превращении добавок на катализаторах, содержащих цеолиты Y и ZSM-5, обнаружено некоторое снижение выхода пропилена и бутиленов, то есть в присутствии кислородсодержащих соединений нивелируется интенсификация образования легких олефинов при введении ZSM-5. Авторы связывают это с селективной дезактивацией ZSM-5 при превращении кислородсодержащих соединений.
По данным работ [36, 77], при крекинге смесей нефтяное сырье–кислородсодержащая добавка (10 мас. %) не наблюдается существенных изменений в продуктах каталитического крекинга по сравнению с нефтяным сырьем; тем не менее, с увеличением доли добавки до 30 мас. % наблюдается изменение выхода газа и кокса, причем изменения носят неаддитивный характер. Авторами обсуждаются пути превращения кислородсодержащих соединений в условиях каталитического крекинга. Реакции, в результате которых образуются газообразные продукты, разделяются на три типа: реакции дегидрирования, реакции, генерирующие водород, и реакции, поглощающие водород. Управление балансом между этими тремя путями необходимо, например, для получения ценных легких олефинов из биосырья посредством каталитического крекинга.
Также исследовано влияние на протекание крекинга УВ более трудно превращаемого соединения – гваякола [78]. Смесь гваякол–нефтяное сырье подвергали крекингу в присутствии катализаторов на основе цеолитов типа Y и ZSM-5. Было показано, что гваякол оказывает негативное влияние на процесс крекинга УВ, что вызвано дезактивацией катализатора. Этот эффект особо выражен при использовании добавки ZSM-5. Также можно отметить увеличение выхода бензиновой фракции, что логично ожидать, так как образующиеся из гваякола фенол, бензол, а также сам непревращенный гваякол входят в интервалы кипения этой фракции. О схожем эффекте сообщается при превращении фенола в качестве добавки.
Подобное исследование проведено авторами работы [79], посвященной крекингу уксусной кислоты, гваякола, фурфурола, ацетола совместно с УВ – н-гептаном, циклогексаном. С помощью широкого набора физико-химических методов анализа исследовано превращение указанных соединений в присутствии цеолита ZSM-5. Детально изучен механизм дезактивации катализатора: во-первых, на поверхности формируется прекурсор, состоящий из большой алифатической цепи и небольшой ароматической части. Далее, по мере покрытия активных центров катализатора тип кокса сменяется с термического на каталитический и постепенно закупоривает каналы молекулярных сит. Этот эффект ускоряет дезактивацию катализатора.
Исследование возможности вовлечения продуктов пиролиза биомассы в условиях, приближенных к промышленным, позволяет избежать ряда проблем, в частности, связанных с подачей сырья. Так, авторами работы [80] отмечается, что на лабораторных установках подразумевается обычно совместная подача двух видов сырья, которые плохо смешиваются друг с другом, что может быть причиной наблюдения отрицательных эффектов от введения биосырья в процесс каталитического крекинга (в частности, повышенного коксообразования). В связи с этим, в указанной работе исследована возможность совместной переработки продукта пиролиза опилок и стандартного вакуумного дистиллята (смеси с 5 и 10 мас. % биосырья) в присутствии коммерческого равновесного катализатора крекинга на демонстрационной установке с лифт-реактором при 540°С. В полученном авторами распределении продуктов отмечается общая для таких работ закономерность: значительно снижается выход сухого (кроме СО и СО2, выход которых возрастает по сравнению с нефтяным сырьем) и жирного газов, увеличивается доля легких олефинов, выходы жидких продуктов отличаются слабо. Что касается выхода кокса, то при введении 5 мас. % биосырья его выход снижается (с 6.3–7.2 до 5.6–6.3 мас. % в зависимости от конверсии); введение 10 мас. % биосырья не влияет существенным образом на выход кокса. Авторы предполагают, что при 5% добавки преобладает эффект разбавления биосырья, что подавляет его коксообразование, тогда как при 10% этот эффект нивелируется. Анализ изотопного состава углерода показал, что примерно 30 мас. % углерода из биосырья переходит в состав жидких продуктов крекинга, что сопоставимо с оценочной эффективностью процесса гидрооблагораживания, согласно которой 30–50 мас. % углерода попадает в состав облагороженных продуктов. Кислород, вводимый в процесс в составе бионефти, почти полностью удаляется в основном в виде воды и, в меньшей степени, – СО и СО2.
Аналогичный эффект, проявляющийся в уменьшении выхода кокса при превращении смеси нефтяного сырья и масла пиролиза (до 20 мас. % в смеси), сообщается авторами работы [81]. Испытания проводили в установке, имитирующей лифт-реактор, с движущимся псевдоожиженным равновесным катализатором крекинга (15% цеолита HY на матрице), при 500°С. Снижение выхода кокса может быть вызвано эффектом присутствующей в бионефти водой, а также взаимодействием кислородсодержащих соединений и УВ в реакциях переноса водорода. На активное протекание этих реакций указывает также сниженный по сравнению с аддитивным расчетом выход СО и СО2. Изучение образовавшегося кокса на поверхности дезактивированного катализатора показало, что кокс, образовавшийся при крекинге нефтяного сырья, имеет ароматическую природу и обладает большими и более упорядоченными кластерами, нежели кокс, образовавшийся при превращении бионефти, который отличается меньшей ароматичностью и наличием кислородсодержащих групп.
Введение в сырье крекинга бионефти существенным образом влияет не только на выход жидких продуктов, но и на их групповой состав. Жидкий продукт обогащается ароматическими и изопарафиновыми УВ, наблюдается также снижение содержания олефинов (табл. 3). Октановое число бензина при этом сохраняется, что дает возможность использовать его в качестве основы для производства высокооктановых бензинов.
Таблица 3.
Групповой состав бензина крекинга, полученного из сырья с добавкой бионефти [82].
| Сырье | ВД | ВД + 3% бионефти |
|---|---|---|
| Парафины | 3.2 | 3.2 |
| Изопараифны | 24.3 | 24.5 |
| Ароматика | 49.2 | 50.5 |
| Нафтены | 9.3 | 9.3 |
| Олефины | 14.0 | 12.6 |
| ОЧИМ/ОЧММ | 92.5/81.6 | 92.1/81.5 |
Кислородсодержащие соединения в жидком продукте крекинга фиксируются в количестве менее 0.2% и представлены в основном фенолами. Существует прямая зависимость между содержанием бионефти в сырье крекинга и содержанием фенолов в бензиновой фракции (табл. 4). Практически все производные фенолов концентрируются в тяжелой части бензина крекинга (Тн.к.> 80°C); будучи эффективными антидетонаторами, фенолы способствуют увеличению октанового числа бензина на 1.0–1.1 пунктов как по моторному, так и по исследовательскому методу [83].
Таблица 4.
| Добавка бионефти в сырье | – | 5% | 10% | 20% |
|---|---|---|---|---|
| Фенол | 362 | 844 | 1681 | 3423 |
| п-Метоксифенол | 36 | 0 | 0 | 0 |
| 4-Метилфенол | 562 | 724 | 1338 | 3266 |
| 2-Метилфенол | 526 | 652 | 1087 | 2329 |
| 3,4-Диметилфенол | 55 | 122 | 213 | 300 |
| 3,5-Диметилфенол | 90 | 413 | 597 | 424 |
| 2,3/2,6-Диметилфенол | 235 | 127 | 1175 | 929 |
| 2,4/2,5-Диметилфенол | 399 | 439 | 566 | 1550 |
| Всего фенолов: | 2265 | 3321 | 6657 | 12 221 |
Каталитический крекинг совместно с УВ нефти можно рассматривать как один из перспективных подходов к переработке бионефти с получением ценных продуктов в одну стадию. В целом, на сегодняшний день показана возможность вовлечения до 10 мас. % кислородсодержащих соединений в качестве добавок к нефтяному сырью. Однако при этом с повышением доли кислородсодержащих добавок в смесевом сырье или при введении трудно превращаемых соединений (гваякола, фенола и пр.), наблюдается негативное влияние: происходит снижение конверсии сырья из-за повышенного коксообразования, а также дезактивация катализатора с увеличением содержания ароматических соединений в продуктах. Для устранения негативного влияния добавки биосырья и увеличения эффективности его переработки необходимо увеличение отношения Н/С в сырье. Не меньшей проблемой является термическая нестабильность бионефти, обуславливающая невозможность ее использования в качестве компонента сырья на промышленных установках крекинга.
Эффективная переработка бионефти в моторные топлива и сырье для нефтехимии – нетривиальная задачя, которая вряд ли может быть решена с использованием одностадийной технологии. Перспективным подходом может являться, вероятно, комбинирование различных процессов. Обзор таких работ выполнен в работе [85]. Так, в работе [54] изучена комбинация процессов гидрирования и каталитического крекинга для превращения бионефти. Согласно полученным результатам, для данного подхода достаточно мягкого гидрирования на Ru/C-катализаторе с неполным удалением кислорода для успешного вовлечения полученного полупродукта в каталитический крекинг в качестве добавки до 20 мас. % к нефтяному сырью. Также исследуется и возможность вовлечения в крекинг продукта каталитического пиролиза биомассы, получаемого в присутствии цеолитных катализаторов. В работе [86] показано, что до 10 мас. % добавки продукта каталитического пиролиза (19.5 мас. % кислорода) подвергается крекингу без значительных изменений в селективности или каких-либо технологических трудностей. При этом продукт каталитического пиролиза обладает хорошей смешиваемостью с нефтяной фракцией при 60°С. Совместный с нефтяным сырьем каталитический крекинг гидрооблагороженной бионефти (22 мас. % кислорода), исходной бионефти (36.5 мас. % кислорода) и продукта каталитического пиролиза биомассы (22 мас. % кислорода) исследован в работе [87]. Введение до 20 мас. % добавки биосырья не привело к обнаружению оксигенатов в жидких продуктах крекинга, причем бензин крекинга обогащается ароматическими и изопарафиновыми УВ. Наибольший выход жидких продуктов получен при крекинге сырья с добавкой гидрооблагороженной бионефти, для нее же наблюдался наименьший выход кокса, что было связано с меньшим содержанием сахаров и более высоким (Н/С)эфф. Тем не менее, количество работ, посвященных совместной переработке продукта каталитического пиролиза, на данный момент невелико и требуются дополнительные исследования для оценки преимуществ этого подхода.
ЗАКЛЮЧЕНИЕ
Исследование возможности использования возобновляемого сырья для производства “drop in” топлив вызывает большой интерес в силу его невысокой стоимости, а также в свете возрастающей сложности извлечения ископаемых УВ и экологических факторов. На сегодняшний день практически готовы к внедрению технологии синтеза “drop in” биотоплив из растительных масел различного происхождения и отходов производства биодизеля. Технология переработки продуктов пиролиза лигноцеллюлозной биомассы находится в стадии научно-технической проработки вследствие наличия ряда нерешенных проблем: высокой термолабильности продукта пиролиза, высокого содержания кислорода и низкого соотношения (Н/С)эфф. Наиболее перспективным выглядит комплексный подход, состоящий из частичной гидродеоксигенации продукта пиролиза в мягких условиях с последующим каталитическим крекингом деоксигенированного продукта. Степень вовлечения возобновляемого сырья в нефтепереработку в рамках данного подхода может составлять до 20%; помимо биотоплива возможно производство нефтехимического сырья (легкие олефины, ароматические УВ).
Список литературы
2016 Key World Energy Statistics. http://www.iea.org/publications/freepublications/24.
BP Statistical Review of World Energy 2018. https:// www.bp.com/content/dam/bp/en/corporate/excel/ energy-economics/statistical-review/bp-stats-review-2018-all-data.xlsx.
Hook M., Hirsch R., Aleklett K. // Energy Policy. 2009. V. 37. P. 2262.
Technically recoverable shale oil and shale gas resources: an assessment of 137 shale formations in 41 countries outside the United States // U.S. Energy Information Administration, 2013.
Le Quéré C., Moriarty R., Andrew R. M. et al. // Earth Syst. Sci. Data. V. 7. P.47.
Перспективы развития ВИЭ в России. Программа Европейского проекта TACIS для Российской Федерации. М.: Атмограф, 2009. 455 с.
Панцхава Е.С., Беренгартен М.Г., Ванштейн С.И. / Биогазовые технологии. Проблемы экологии, энергетики, сельскохозяйственного производства. М.: МГУИЭ, ЗАО Центр “ЭКОРОС”, 2008. 217 с.
Rico J., Sauer I. // Renewable and Sustainable Energy Reviews. 2015. V. 45. P. 513.
Baskar G., Aiswarya R. // Renewable and Sustainable Energy Reviews. 2016. V. 57. P. 496.
Mardhiah H., Ong H., Masjuki H., Lim S., Lee H. // Renewable and Sustainable Energy Reviews. 2017. V. 67. P. 1225.
Issariyakul T., Dalai A.K. // Renewable and Sustainable Energy Reviews. 2014. V. 31. P. 446.
EIA International Energy Statistics. https://www. eia.gov/beta/international.
Emori E.Y., Hirashima F.H., Zandonai C.H., Ortiz-Bravo C.A., Fernandez-Machado N.R., Olsen-Scaliante M.H. // Catalysis Today. 2017. V. 279. P. 168.
Brazil Biofuels Annual 2018 – USDA GAIN reports. https://gain.fas.usda.gov/Recent%20GAIN%20Publications/Biofuels%20Annual_Sao%20Paulo%20ATO_ Brazil_8-10-2018.pdf.
Цодиков М.В., Чистяков А.В., Губанов М.А., Жарова П.А., Шаповалов С.С., Пасынский А.А., Кривенцов В.В., Моисеев И.И. // Известия Академии наук. Серия хим. 2015. № 9. С. 2062.
Galadima A., Muraza O. // J. of Industrial and Engineering Chemistry. 2015. V. 29. P. 12.
Цодиков М.В., Чистяков А.В., Губанов М.А., Мурзин В.Ю., Букина З.М., Колесниченко Н.В., Хаджиев С.Н. // Нефтехимия. 2013. Т. 53. № 1. С. 50.
Buzetzki E., Sidorova K., Cvengrošová Z., Kaszonyi A., Cvengroš J. // Fuel Processing Technology. 2011. V. 92. P. 1623.
Ong Y.K., Bhatia S. // Energy. 2010. V. 35. P. 111.
Pyl S.P., Schietekat C.M., Reyniers M.-F., Abhari R., Marin G.B., Van Geem K.M. // Chemical Engineering J. 2011. V. 176–177. P. 178.
Хаджиев С.Н., Герзелиев И.М., Дементьев К.И. // Нефтехимия. 2013. Т. 53. № 6. С. 403.
Abbasov V., Mammadova T., Aliyeva N., Abbasov M., Movsumov N, Joshi A., Lvov Y., Abdullayev E. // Fuel. 2016. V. 181. P. 55.
Suurs A.A.R., Hekkert M.P. // Energy. 2009. V. 34. P. 669.
Tamunaidu P., Bhatia S. // Bioresource Technology. 2007. V. 98. № 18. P. 3593.
Ooi Y.S., Bhatia S. // Microporous and Mesoporous Materials. 2007. V. 102. № 1–3. P. 310.
Ooi Y.S., Zakaria R., Mohamed A.R., Bhatia S. // Biomass and Bioenergy. 2004. V. 27. № 5. P. 477.
O’Driscoll C. // Biofuels Bioproducts and Biorefining. 2007. V. 1. № 1. P. 6.
Viswanadham N., Saxena S. // Fuel. 2013. V. 105. P. 490.
Tago T., Konno H., Ikeda S., Yamazaki S., Ninomiya W., Nakasaka Y., Masuda T. // Catalysis Today. 2011. V. 164. P. 158.
Zakaria Z.Y., Saidina Amin N.A., Linnekoski J. // Biomass and Bioenergy. 2013. V. 55. P. 370.
Zakaria Z.Y., Linnekoski J., Saidina Amin N.A. // Chem. Engineering J. 2012. V. 207–208. P. 803.
Corma A., Huber G.W., Sauvanaud L., O’Connor P. // J. of Catalysis. 2008. V. 257. P. 163.
Blass S.D., Hermann R.J. Persson N.E., Bhan A., Schmidt L.D. // Applied Catalysis A: General. 2014. V. 475. P. 10.
Pathak K., Reddy K.M., Bakhshi N.N., Dalai A.K. // Applied Catalysis A: General. 2010. V. 372. P. 224.
Хаджиев С.Н., Герзелиев И.М., Дементьев К.И. // Нефтехимия. 2014. Т. 54. С. 3.
Corma A., Huber G.W., Sauvanaud L., O’Connor P. // J. of Catalysis. 2007. V. 247. P. 307.
Qi Z., Jie C., Tiejun W., Ying X. // Energy Conversion and Management. 2007. V. 48. P. 87.
Oasmaa A., Kuoppala E., Gust S., Solantausta Y. // Energy and Fuels. 2003. V. 17. P. 1.
Ren S., Ye X.P., Borole A. // J. of Analytical and Applied Pyrolysis. 2017. V. 123. P. 30.
Wang S., Wang Y., Cai Q., Wang X., Jin H., Luo Z. // Separation and Purification Technology. 2014. V. 122. P. 248.
Oasmaa A., Kuoppala E., Solantausta Y. // Energy and Fuels. 2003. V. 17. P. 433.
Oasmaa A., Kuoppala E., Selin J.F., Gust S., Solantausta Y. // Energy and Fuels. 2004. V. 18. № 5. P. 1578.
Mahfud F., Melian-Cabrera I., Manurung R., Heeres H. // Process Safety and Environmental Protection. 2007. V. 85. № 5. P. 466.
Xu J., Jiang J., Sun Y., Lu Y. // Biomass and Bioenergy. 2008. V. 32. № 11. P. 1056.
Tang Y., Yu W., Mo L., Lou H., Zheng X. // Energy and Fuels. 2008. V. 22. № 5. P. 3484.
Xiong W.M., Zhu M.Z., Deng L., Fu Y., Guo Q.X. // Energy and Fuels. 2009. V. 23. P. 2278.
Zhang Q., Chang J., Wang T., Xu Y. // Energy and Fuels. 2006. V. 20. P. 2717.
Wang J.-J., Chang J., Fan J. // Energy and Fuels. 2010. V. 24. P. 3251.
Hu X., Gunawan R., Mourant D., Hasan M., Wu L., Song Y., Lievens C., Li C.-Z. // Fuel Processing Technology. 2017. V. 155. P. 2.
Zhang Q., Chang J., Wang T.J. // Energy and Fuels. 2006. V. 20. P. 2717.
Peng J., Chen P., Lou H., Zheng X. // Energy and Fuels. 2008. V. 22. P. 3489.
Peng J., Chen P., Lou H., Zheng X. // Bioresource Technology. 2009. V. 100. P. 3415.
Santos R.G., Bordado J.C., Mateus M.M. // J. of Cleaner Production. 2016. V. 137. P. 195.
Mercader F.M., Groeneveld M.J, Kersten S.R.A., Way N.W.J, Schaverien C.J., Hogendoorn J.A. // Applied Catalysis B: Environmental. 2010. V. 96. P. 57.
Li X., Chen G., Liu C., Ma W., Yan B., Zhang J. // Renewable and Sustainable Energy Reviews. 2017. V. 71. P. 296.
Bui N.V., Laurenti D., Afanasiev P., Geantet C. // Applied Catalysis B: Environmental. 2011. V. 101. P. 239.
Bu Q., Lei H., Zacher H.A., Wang L., Ren S., Liang J., Wei Y., Liu Y., Tang J., Zhang Q., Ruan R. // Bioresource Technology. 2012. V. 124. P. 470.
Echeandia S., Arias L.P., Barrio I.V., Pavelec B., Fierro J.L.G. // Applied Catalysis B: Environmental. 2010. V. 101. P. 1.
Senol I.O., Viljava R.T., Krause A.O. // Applied Catalysis A: General. 2007. V. 326. P. 236.
Senol I.O., Viljava R.T., Krause A.O. // Catalysis Today. 2005. V. 106. P. 186.
Senol I.O., Viljava R.T., Krause A.O. // Catalysis Today. 2005. V. 100. P. 331.
Ferrari M., Bosmans S., Maggi R., Delmon B., Grange P. // Catalysis Today. 2001. V. 65. P. 257.
Zhou M., Wang Y., Wang Y., Xiao G. // J. of Energy Chemistry. 2015. V. 24. P. 425.
Bykova V.M., Ermakov Y.D., Kaichev V.V., Bulavchenko A.O., Saraev A.A., Lebedev M.Yu., Yakovlev V.A. // Applied Catalysis B: Environmental. 2012. V. 113–114. P. 296.
Bunch Y.A., Ozkan U.S. // J. of Catalysis. 2002. V. 206. P. 177.
Prasomsri T., Nimmanwudipong T., Roman-Leshkov Y. // Energy Environ. Sci. 2013. V. 6. P. 1732.
Zhang X., Tang J., Zhang Q., Liu Q., Li Y., Chen L., Wang Ch., Ma L. // Catalysis Today. 2019. V. 319. P. 41.
Whiffen M V., Smith K.J. // Energy and Fuels. 2010. V. 24. P. 4728.
Li X., Luo X., Jin Y., Li J., Zhang H., Zhang A., Xie J. // Renewable and Sustainable Energy Reviews. 2018. V. 82. P. 3762.
Zhao H.Y., Li D., Bui P., Oyama S.T. // Applied Catalysis A: General. 2011. V. 391. P. 305.
Boullosa-Eiras S., Lodeng R., Bergem H., Stöcker M., Hannevold L., Blekkan E.A. // Catalysis Today. 2014. V. 223. P. 44.
Wang X.Y., Leng J., Wang S.M., Asiri A.M., Marwani H.M., Qin H.-L. // Tetrahedron Letters. 2017. V. 58. P. 2340.
Echeandia S., Pawelec B., Barrio I.V. // Fuel. 2014. V. 117. P. 1061.
Mante O., Agblevor F., McClung R. // Biomass Conversion and Biorefinery. 2011. V. 1. P. 189.
Mortensena P.M., Grunwaldt J.D., Jensen P.A., Knudsen K.G., Jensen A.D. // Applied Catalysis A: General. 2011. V. 407. P. 1.
Huber G.W., Corma A. // Angew. Chem. Int. Ed. 2007. V. 46. P. 7184.
Graca I., Ribeiro F., Cerqueira H.S., Lam Y.L., Almeida M.B.B. // Applied Catalysis B: Environmental. 2009. V. 90. P. 556.
Graca I., Lopes J.M., Ribeiro M.F., Ribeiro F.R., Cerqueira H.S., Almeida M.B.B. // Applied Catalysis B: Environmental. 2011. V. 101. P. 613.
Chen G., Zhang R., Ma W., Liu B., Li X., Yan B., Cheng Z., Wang T. // Science of the Total Environment. 2018. V. 631–632. P. 1611.
Pinho A.R., Almeida M.B., Mendes F.L., Casavechia L.C., Talmadge M.S., Kinchin C.M., Chum H.L. // Fuel. 2017. V. 188. P. 462.
Ibarra A., Veloso A., Bilbao J., Arandes J.M., Castano P. // Applied Catalysis B: Environmental. 2016. V. 182. P. 336.
Bryden K., Weatherbee G., Habib T.E. // Grace Davison Catalagram. 2013. №113. P. 3.
de Rezende Pinho A., de Almeida M.B.B., Mendes F.L., Ximenes V.L., Casavechia L.C. // Fuel Processing Technology. 2015. V. 131. P. 159.
de Rezende Pinho A., de Almeida M.B.B., Mendes F.L., Casavechia L.C., Talmadge M.S., Kinchin C.M., Chum H.L. // Fuel. 2017. V. 188. P. 462.
Stefanidis S.D., Kalogiannis K., Lappas A. // WIREs Energy and Environment. 2017. V. 7. P. e281.
Wang C., Li M., Fang Y. // Industrial & Engineering Chemistry Research. 2016. V. 55. № 12. P. 3525.
Lindfords C., Paasikallio V., Kuoppala E., Solantausta Y. // Energy and Fuels. 2015. V. 29. № 6. P. 3707.
Дополнительные материалы отсутствуют.