

Нефтехимия, 2019, T. 59, № 4, стр. 460-465
Особенности дезактивации силикоалюмофосфата SAPO-34 в процессе превращения метанола в низшие олефины в реакторах различного типа
В. С. Павлов 1, *, С. В. Коннов 1, П. А. Коц 2, И. И. Иванова 1, 2
1 Институт нефтехимического синтеза им. А.В. Топчиева РАН
Москва, Россия
2 Московский государственный университете имени М.В. Ломоносова, Химический факультет
Москва, Россия
* E-mail: pavlov@ips.ac.ru
Поступила в редакцию 10.03.2019
После доработки 13.03.2019
Принята к публикации 14.03.2019
Аннотация
В работе проведен сравнительный анализ дезактивации катализатора SAPO-34 в конверсии метанола в низшие олефины в проточных реакторах с неподвижным слоем катализатора, в реакторах с кипящим слоем и в сларри-реакторе с катализатором, диспергированным в среде полидиметилсилоксана (ПДМС). Установлено, что скорость дезактивации катализатора существенным образом зависит от типа реактора. Равномерное контактирование метанола и катализатора в кипящем слое позволяет увеличить стабильность работы во времени в 1.5 раза по сравнению с неподвижным слоем катализатора. Проведение же реакции в сларри-реакторе в среде ПДМС приводит к значительному ускорению дезактивации, обусловленное высокой растворимостью олефинов и метанола в ПДМС, приводящее к увеличению времени контакта катализатора с реагентами и продуктами реакции. Предложен метод измерения растворимости газов в жидких полимерах при высоких температурах с использованием масс-спектрометрического детектирования выходящих газов.
В настоящее время основным источником получения легких олефинов (этилена и пропилена) является крекинг нефтяных фракций. Однако увеличивающийся спрос на легкие олефины стимулирует исследователей к разработке альтернативных методов синтеза легких олефинов, в частности, получению этилена и пропилена из продуктов переработки природного газа. Данный процесс превращения метанола в низшие олефины впервые был предложен фирмой Mobil в 1977 г. [1] с использованием цеолитного катализатора со структурой MFI. В настоящее время данный процесс внедрен рядом крупных компаний [2, 3] (UOP, ExxonMobil, Lurgi, NorskHydro, HaldorTopsøe, DCIP). Ключевым недостатком наиболее селективного катализатора данного процесса на основе силикофлюмофасфата со структурой CHA (SAPO-34) является его стремительная дезактивация, что приводит к необходимости конструирования реакторов сложных как с инженерной точки зрения, так и с точки зрения масштабирования процесса [3]. Согласно существующим на данный момент представлениям, дезактивация катализатора на основе SAPO-34 связана с преобразованием находящихся в порах катализатора полиметилзамещенных бензолов, являющихся активными интермедиатами процесса, в более тяжелые ароматические углеводороды (УВ) – нафталины, пирены и антрацены, блокирующие пористую структуру катализатора [4].
Аппаратурное оформление процесса конверсии метанола в УВ существенным образом зависит от структуры катализатора. Так, промышленная реализация процесса конверсии метанола в УВ на цеолите структуры MFI, как правило, проводится в реакторе с неподвижным слоем катализатора (РНСК) [5], в то время как для промышленной реализации процесса на SAPO-34 преимущественно используется реактор с непрерывной регенерацией и кипящим слоем катализатора, конструкция которого исходно была разработана для каталитического крекинга нефтяных фракций [3]. В таком реакторе частицы находятся в псевдоожиженном слое и, в зависимости от масштаба реактора, перемещаются в кипящем или турбулентном режиме. По мере закоксовывания катализатора частицы в таком реакторе оседают и отправляются на регенерацию. Важно отметить, что закономерности каталитического действия и дезактивации катализатора SAPO-34, как правило, изучают в РНСК, что вызывает вопросы о применимости этих результатов для используемого в промышленности реактора с псевдоожиженным слоем катализатора (РПСК). В работе [6] проводили сравнение каталитических характеристик SAPO-34 в РПСК и в РНСК. Было установлено, что, несмотря на небольшое уменьшение выхода метана в РПСК, свидетельствующее о снижении интенсивности гидридного переноса по сравнению с РНСК, в обоих реакторах катализатор демонстрирует одинаковое время стабильной работы. Была охарактеризована общая кинетика накопления кокса, близкая в обоих типах реакторов, однако детального анализа природы коксовых отложений проведено не было.
Альтернативой РНСК и РПСК может служить сларри-технология, характеризуемая низкими капитальными затратами на сооружение установок, превосходным тепло- и массопереносом, хорошим контролем температуры и высокой производительностью каталитического процесса. В сларри-реакторе (СЛР) тонкодисперсный гетерогенный катализатор диспергирован в инертной жидкости, и реакция реализуется в трехфазной проточной системе (газ−жидкость−твердый катализатор). Ультрадисперсные суспендированные каталитические системы по своим свойствам близки к гомогенным катализаторам, что дает возможность вести гетерогенно-каталитические реакции в более мягких условиях с высокими показателями (производительностью, селективностью).
Цель настоящей работы – детальное сравнение дезактивации силикоалюмофосфатного катализатора со структурой CHA (SAPO-34) в ходе превращения метанола в низшие олефины в реакторах различного типа: СЛР, РНСК и РПСК.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Силикоалюмофосфат SAPO-34 был получен методом гидротермального синтеза в автоклавах, вращающихся со скоростью 5 об./мин, из реакционной смеси мольного состава Al2O3–2P2O5–0.6SiO2–4(C2H5)4NOH–70H2O в течение 25 ч при температуре 190°С в соответствии с методикой [10]. В качестве исходных реагентов использовали изопропоксид алюминия, силиказоль (40 мас. %), фосфорную кислоту (85 мас. %) и гидроксид тетраэтиламмония (35%-ный водный раствор). После кристаллизации твердый продукт отделяли от жидкой фазы центрифугированием, отмывали, высушивали при 70°C в течение 12 ч и прокаливали при 600°C в течение 6 ч в токе сухого воздуха.
Фазовый анализ силикоалюмофосфата проводили с использованием рентгенофазового анализа на дифрактометре D2 PHASER фирмы BRUKER с медным анодом (λСuKα1 = 1.5418 Å). Съемку дифрактограмм осуществляли при вращении образца в горизонтальной плоскости в интервале углов 2θ 5ο–50ο с шагом 0.05ο, ширина щелей на выходе из трубки 0.6 мм и перед детектором 1.15 мм, время регистрации в точке 3 с.
Исследование пористой структуры образца проводили методом низкотемпературной адсорбции азота на установке ASAP-2010 фирмы Micromeritics. Предварительно образец вакуумировали при температуре 350°С до 0.4 Па. Адсорбцию N2 проводили при 77 К.
Кислотные свойства изучали методом термопрограммированной десорбции аммиака на универсальном сорбционном газовом анализаторе УСГА-101 фирмы Унисит. Навеску образца (~0.1 г в виде фракции 0.25–0.5 мм) помещали в кварцевый реактор, нагревали в токе гелия до температуры 250°С со скоростью нагрева 10°С/мин и прокаливали при этой температуре в течение 1 ч в токе гелия, после чего охлаждали до 60°С. Насыщение аммиаком проводили в токе осушенной смеси NH3/N2 (1 : 9) в течение 15 мин. Удаление физически адсорбированного аммиака проводили при 100°С в токе сухого гелия в течение 1 ч. После этого образец охлаждали в токе осушенного гелия (скорость подачи 30 мл/мин) до 60°С и проводили линейный подъем температуры в реакторе со скоростью 8°С/мин до 800°С.
Элементный анализ выполняли на рентгенофлуоресцентном спектрометре ARL PERFORM’X фирмы TermoScientific с родиевой трубкой и ускоряющим напряжением 2.5 кВ.
Каталитические свойства изучали в реакторах трех типов. Каталитический эксперимент в РНСК проводили на фракции катализатора размером 0.5–1 мм при массовой скорости подачи сырья 2 ч–1 и температуре 400°С. Разбавление метанола азотом составляло метанол/N2 = 1/5 мольн.
Эксперимент в РПСК осуществляли в кварцевом реакторе в аналогичных условиях на гранулах катализатора размером 0.1–0.25 мм и подаче смеси азота и метанола снизу. Такое соотношение размера гранул катализатора и подачи газа обеспечивало равномерное перемешивание частиц катализатора и препятствовало их уносу из реактора.
Для исследований в системе СЛР использовали реактор барботажного типа. В качестве среды для диспергирования катализатора и проведения реакциив СЛР был выбран полидиметилсилоксан (марка Syltherm-1000), химически инертное вещество, устойчивое к воздействию высоких температур. Для приготовления однородной каталитической суспензии навеску катализатора в виде порошка вначале диспергировали в н-гексане, растворимом в ПДМС, но имеющем меньшую вязкость. Затем получившуюся суспензию смешивали с ПДМС и удаляли гексан перед проведением каталитического эксперимента в течение 30 мин при 90°С. Для диспергирования 1 г катализатора использовали около 15 мл гексана и 50 мл ПДМС. Эксперименты проводили в токе азота при температуре 400°С. Пары метанола, разбавленные азотом в соотношении метанол/N2 = = 1/5 моль, барботировали в каталитическую суспензию сквозь пористый фильтр Шота. Массовая скорость подачи метанола составляла 2 ч–1. Отбор проб производили каждые 35–40 мин.
Продукты реакции анализировали с использованием метода ГЖХ на хроматографе “Кристалл 5000” фирмы “Хроматэк Аналитик”, снабженном ПИД и капиллярной колонкой с PoraBondQ. Содержание водорода и оксидов углерода определяли на хроматографе “Кристалл 5000”, снабженном детектором по теплопроводности и набивной колонкой, содержащей фазу PorapakQ.
Конверсию рассчитывали как мольную долю атомов углерода в продуктах реакции, рассматривая диметиловый эфир и метанол в качестве реагентов. Индекс гидридного сдвига был рассчитан как мольная доля алканов среди продуктов реакции в соответствии с формулой, предложенной в работе [11].
Анализ коксовых отложений на образцах после проведения каталитических экспериментов проводили термогравиметрическим методом на приборе “TASDTQ600”. Температурно-программируемое окисление проводили в токе воздуха (100 мл/мин) в интервале температур 25–800°С при скорости нагрева 10°С/мин. Образцы, дезактивированные в СЛР, вначале отмывали от ПД-МС гексаном, а затем высушивали и анализировали методом дифференциального термического анализа.
Растворимость метанола и пропилена в ПД-МС измеряли с помощью масс-спектрометра MS7-200 с программой анализа RGA 200. Потоки гелия (газ-разбавитель, 20 мл/мин), аргона (для измерения “мертвого” объема, 5 мл/мин) и исследуемого вещества пропускали через ПДМС объемом 50 мл. Перед началом подачи растворяемого вещества и аргона ПДМС нагревали до заданной температуры и выдерживался в течение 30 мин в токе гелия. После начала подачи смеси исследовали зависимость концентрации аргона и растворяемого вещества от времени на выходе с реактора. Растворимость измеряли путем интегрирования ионного тока, соответствующегоконцентрации растворяемоговещества с корректировкой на мертвый объем. Концентрацию в газовой фазе рассчитывали, исходя из уравнения состояния идеального газа и условий эксперимента.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
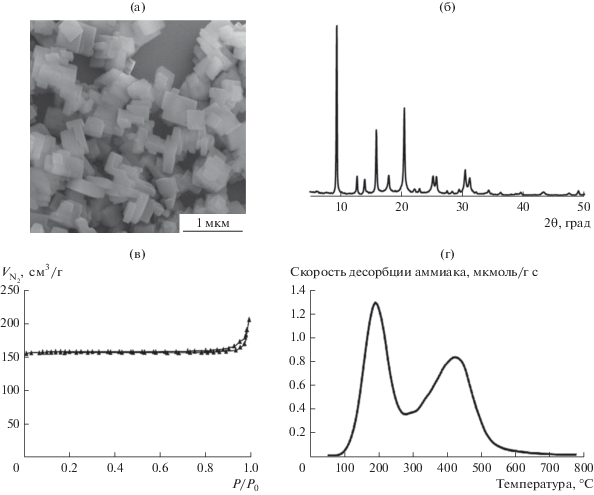
По данным элементного анализа, сканирующей электронной микроскопии (1а) а также порошковой дифракции (1б), полученный образец имеет структуру CHA, характеризуется мольным соотношением (Al + P)/Si = 8 и размером кристаллов 300–400 нм.
Кубическая морфология кристаллов типична для силикоалюмофосфата SAPO-34. Изотерма низкотемпературной адсорбции азота для SAPO-34 (рис 1в) соответствует классической изотерме для микропористых образцов. Петля гистерезиса на уровне 0.9–1 P/P0 отвечает заполнению межкристаллического пространства между наноразмерными кристаллами силикоалюмофосфата. Объем микропор составляет 0.24 см3/г, что соответствует образцам SAPO-34 с высокой степенью кристалличности. Спектр ТПД аммиака для образца приведен на рис. 1г. Общее количество кислотных центров составляет 1830 мкмоль/г.
Рис. 1.
Физико-химические свойства свежепрокаленного образца SAPO-34: (а) микрофотография СЭМ; (б) дифрактограмма; (в) изотерма низкотемпературной адсорбции азота; (г) профиль ТПД аммиака.
На рис. 2 представлены результаты каталитических экспериментов по превращению метанола в олефины на катализаторе SAPO-34, проведенных в реакторах различного типа. Из рис. 2а видно, что конверсия метанола падает с различной скоростью: в РПСК наблюдается наиболее медленная дезактивация, несколько быстрее катализатор дезактивируется в РНСК и, наконец, наиболее стремительная дезактивация происходит в СЛР. При этом во всех реакторах катализатор демонстрирует высокую селективность превращения в целевые продукты – олефины состава С2=–С4= (рис. 2б). Данные по интенсивности гидридного переноса, как основной реакции, ответственной за дезактивацию катализатора (рис. 2в) полностью согласуются с данными по скорости дезактивации – для РПСК данный процесс имеет наименьшую интенсивность, для СЛР – наибольшую.
Рис. 2.
Данные каталитических исследований образца SAPO-34 в конверсии метанола в олефины в различных типах реакторов: (а) конверсия метанола; (б) селективность по целевым продуктам – алкенам С2=–С4=; (в) индекс гидридного переноса. Условия эксперимента: 400°С, MeOH/N2 = 1/5 мольн., скорость подачи сырья: 2 г/(гкат ч).
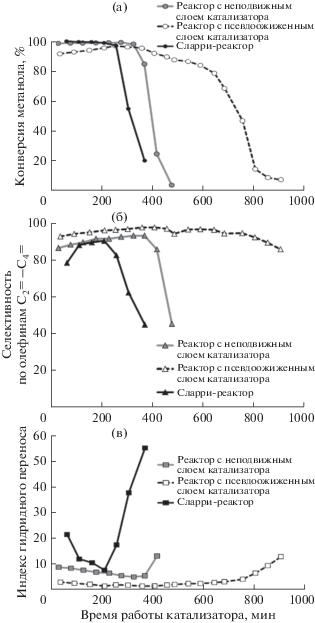
Следует отметить, что полученные результаты не согласуются с работой [6], в которой отмечалось, что в РНСК и РПСК деактивация происходит с приблизительно одинаковой скоростью. Это, по всей видимости, связано с иными условиями проведения эксперимента. Для изучения превращения метанола в олефины в работе [6] использовали разбавленный в воде метанол, что существенно замедляет дезактивацию в обоих типах реакторов. Кроме того, существенное влияние на коксообразование оказывает более высокая температура проведения процесса в работе [6] (450–500°С), по сравнению с нашей работой (400°С). Отметим, что, согласно результатам [12], для цеолитного катализатора структуры MFI дезактивация в случае реактора с кипящим слоем, как и в нашей работе, происходит значительно медленнее.
Методом термогравиметрического анализа была изучена кинетика образования коксовых отложений на катализаторе в реакторах различного типа в зависимости от времени их работы в токе метанола. Результаты экспериментов представлены на рис. 3. Общая кинетика накопления коксовых отложений (рис. 3а) находится в соответствии с индексом гидридого переноса (рис. 2в): в реакторах с большей скоростью накопления коксовых отложений гидридный перенос протекает интенсивнее. Линейное накопление коксовых отложений в РНСК со временем полностью соответствует модели “горящей сигары” [9] и сложившимся представлениям о постоянном качественном составе коксовых отложений в дезактивированной зоне катализатора. В СЛР и РПСК рост содержания кокса во времени имеет нелинейный характер, что указывает на изменение качественного состава коксовых отложений и постепенную эволюцию полизамещенных бензолов, находящихся в порах катализатора, в более тяжелые молекулы нафталинов и антраценов.
Рис. 3.
Данные термогравиметрического анализа образцов после частичной дезактивации в реакторах различного типа: (а) количество коксовых отложений на катализаторе в зависимости от времени его работы; (б) профили ДТА образцов после 50 и 120 мин работы в реакторах различного типа. Условия дезактивации: 400°С, MeOH/N2 = 1/5 мольн., скорость подачи сырья: 2 г/(гкат ч).
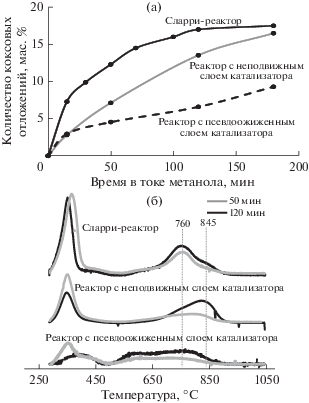
Тип продуктов уплотнения может быть оценен по температуре их выгорания в токе воздуха. Кривые ДТА образцов после 120 мин работы в токе метанола в реакторах различных типов представлены на рис. 3б. Согласно этим данным, коксовые отложения, образованные в РНСК, имеют наибольшую температуру выгорания, что соответствует полной дезактивации верхних слоев катализатора с образованием в порах тяжелых коксовых отложений неизменного состава (нафталинов и антраценов). В РПСК и СЛР коксовые отложения выгорают при более низкой температуре, что соответствует образованию более легких ароматических УВ (алкилбензолов и нафталинов) в этих условиях. Такой состав продуктов уплотнения в СЛР и в РПСК свидетельствует о том, что их превращение еще не завершено.
Таким образом, анализ полученных результатов показывает, что дезактивация катализатора существенным образом зависит от типа реактора. В РНСК происходит послойная дезактивация по типу “горящей сигары”: коксовые отложения формируются в активной зоне катализатора, которая перемещается вниз по слою. При этом в активной зоне накапливаются тяжелые коксовые отложения, а качественный состав кокса не зависит от времени эксперимента. В РПСКи СЛР гранулы катализатора подвергаются равномерной дезактивации, а качественный состав продуктов уплотнения изменяется во времени.
Сравнение дезактивации в РНСК и в РПСК показывает, что послойная дезактивация катализатора происходит быстрее, чем равномерная. Высокую скорость послойной дезактивации можно объяснить автокаталитической природой процесса [13]: высокая концентрация метанола в зоне активной работы катализатора способствует интенсивному протеканию процесса коксообразования, скорость которого может быть не прямо пропорциональна концентрации метанола. Сравнение РПСК и СЛР, в случае которых дезактивация происходит равномерно во всех гранулах катализатора, показывает, что среда проведения реакции оказывает существенное влияние на процесс. На это указывают как данные по скорости дезактивации, так и по кинетике коксообразования в СЛР и РПСК, различающихся лишь наличием среды для диспергирования катализатора. Согласно данным [7, 8], в ПДМС концентрация олефинов выше, чем в газовой фазе. Из-за этого в СЛР интенсифицируются процессы коксообразования и деактивации, связанные с процессамиалкилирования ароматических углеводородов олефинами в порах катализатора. В РНСК и РПСК адсорбция обусловлена равновесием газ–твердое тело и время контакта катализатора с олефинами значительно ниже, а дезактивация связана с процессами алкилирования алкилароматических соединений метанолом.
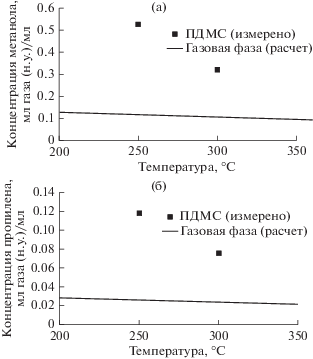
Концентрация молекул метанола и олефинов в ПДМС определяется их растворимостью в данном полимере. Однаков литературе отсутствуют какие-либо данные о растворимости перечисленных соединений в ПДМС при высоких температурах, поэтому нами был разработан новый метод измерения растворимости газов в жидкостях, основанный на масс-спектрометрии потока газов, выходящих из реактора. Результаты эксперимента по измерению растворимости метанола и пропилена представлены на рис. 4. Измерение при более высоких температурах оказалось невозможно, поскольку это вызывает частичное разложение ПДМС. Показано, что при высоких температурах концентрация, как метанола, так и пропилена в ПДМС значительно выше, чем в газовой фазе независимо от разбавления азотом. Таким образом, было показано, что различия скоростей дезактивации катализатора в СЛР и в РПСК обусловлены различием в концентрациях реагентов и продуктов реакции, ответственных за образование коксовых отложений вблизи частиц катализатора.
ЗАКЛЮЧЕНИЕ
При комплексном сравнении каталитических свойств силикоалюмофосфатного катализатора со структурой CHA (SAPO-34) в процессе конверсии метанола в низшие олефины в трех различных типах реакторов (РНСК, РПСК и СЛР) показано, что дезактивация катализатора существенным образом зависит от типа реактора. В РНСК происходит послойная дезактивация по типу “горящей сигары”: коксовые отложения формируются в активной зоне катализатора, которая перемещается вниз по слою; при том в активной зоне накапливаются тяжелые коксовые отложения, а качественный состав кокса не зависит от времени эксперимента. В РПСК и СЛР гранулы катализатора подвергаются равномерной дезактивации, а качественный состав продуктов уплотнения изменяется во времени.
Сравнение свойств катализатора в СЛР и РПСК выявило, что среда проведения реакции оказывает существенное влияние на процесс дезактивации. Проведение реакции в ПДМС способствует увеличению времени контакта гранул катализатора с метанолом и олефинами, что сопровождается большей скоростью накопления продуктов уплотнения и ускоренной дезактивацией в СЛР. Это связано с высокой растворимостью метанола и олефинов в ПДМС и концентрированием продуктов реакции вокруг гранул катализатора. Напротив, в РПСК газовая среда способствует быстрому уносу продуктов из зоны реакции, что объясняет более высокую устойчивость к дезактивации в этом реакторе.
БЛАГОДАРНОСТИ
Работа выполнена в рамках Государственного задания ИНХС РАН.
Список литературы
Chang C.D., Silvestri A.J. // J. of Catalysis. 1977. V. 47. P. 249.
Stöcker M. // Microporous and Mesoporous Materials. 1999. V. 29. P. 3.
Tian P., Wei Y., Ye M., Liu Zh. // ACS Catalysis. 2015. V. 5. P. 1922.
Bleken F., Bjørgen M., Palumbo L., Bordiga S., Svelle S., Lillerud K.-P, Olsbye U. // Topics in Catalysis. 2009.V. 52. № 3. P. 218.
Hack M., Koss U., König P., Rothaemel M., Holtmann H.D. // Patent WO № 0192190. 2001.
Yuan Xi., Li H., Ye M., Liu Zh. // Chemical Engineering J. 2017. V. 329. P. 35.
Konnov S.V., Pavlov V.S., Ivanova I.I., Khadzhiev S.N. // Petrol. Chemistry. 2016. V. 56. № 12. P. 1154.
Konnov S.V., Pavlov V.S., Kots P.A., Zaytsev V.B., Ivanova I.I. // Catalysis Science and Technology. 2018. V. 8. P. 1564.
Rojo-Gama D.,Etemadi S., Eliot Kirby E., Ellerud K.-P., Beato P., Svelle S.,Olsbye U. // Faraday Discussions. 2017. V. 197. P. 421.
Knyazeva E.E., Konnov S.V., Shutkina O.V., Dobryakova I.V., Ponomareva O.A., Ivanova I.I. // Petrol. Chemistry. 2014. V. 54. № 4. P. 288.
Rostami R.B., Ghavipour M., Di Zu., Wang Y., Behbahani R.M. // RSC Advances. 2015. V. 5. P. 81965.
Müller S., Liu Y., Vishnuvarthan M., Sun X., van Veen A.C., Haller G.L., Sanchez-Sanchez M., Lercher J.A. // J. of Catalysis. 2015. V. 325. P. 48.
Chen N.Y., Reagan W.J. // J. of Catalysis 1979. V. 59. № 1. P. 123.
Дополнительные материалы отсутствуют.