

Нефтехимия, 2019, T. 59, № 5, стр. 529-537
ТЕОРЕТИЧЕСКИЕ АСПЕКТЫ РЕАКЦИЙ ПРЕВРАЩЕНИЯ ЭТИЛОВОГО СПИРТА И ДИМЕТИЛОВОГО ЭФИРА В 1,3-БУТАДИЕН
А. М. Гюльмалиев *, В. Ф. Третьяков, Р. М. Талышинский, А. М. Илолов, С. Н. Хаджиев
Институт нефтехимического синтеза им. А.В. Топчиев РАН
11991 Москва, Россия
* E-mail: Gyulmaliev@ips.ac.ru
Поступила в редакцию 12.03.2019
После доработки 21.04.2019
Принята к публикации 21.04.2019
Аннотация
При синтезе 1,3-бутадиена из диметилового эфира (ДМЭ) на катализаторе Zn-Al2O3 установлено, что выход целевого продукта на 20% меньше по сравнению с превращением этилового спирта. Однако результаты термодинамических расчетов показали равную вероятность превращения этилового спирта (ЭС) и ДМЭ и одинаковую температурную зависимость равновесного состава продуктов реакции. Квантово-химическим методом расчета функционала плотности DFT B3LYP/6-31(d) установлено, что дегидратация и дегидрирование в случае ДМЭ протекает с меньшей энергией активацией. Рассмотрено взаимодействие молекул этанола и ДМЭ с кластером Zn4O4. Показано, что, молекула этанола при взаимодействии с оксидом цинка в большей степени подвергается деструкции за счет полярности ОН-связи в молекуле. Установлено, что в отличие от этанола, каталитическая система на основе оксида цинка недостаточно эффективна для обеспечения эффективного протекания реакции превращения ДМЭ в 1,3-бутадиен.
Реакция Лебедева [1], химизм которой формально сводится к стехиометрическому взаимодействию двух молекул этилового спирта 2CH3CH2OH → C4H6 + 2H2O + H2 с образованием 1,3-бутадиена, позволяет расширить подобное направление, обозначив его как превращение “оксигенатов”, в частности, ДМЭ [2–5].
Результаты экспериментальных исследований показывают, что в реакциях синтеза 1,3-бутадиена из ЭС и ДМЭ на каталитической системе ZnO/Al2O3 при температуре 400°C при времени контакта 2.5 ч выход и селективность 1,3-бутадиена следующие: для ЭС 20 и 50%, для ДМЭ 10 и 35% соответственно. В случае ДМЭ в продуктах в большем по сравнению с ЭС количестве образуется кокс. Возникает вопрос, с чем связано, что два соединения с одинаковой брутто формулой существенно отличаются по реакционной способности.
Данная работа посвящена анализу теоретических аспектов реакций синтеза бутадиена из ЭС и ДМЭ с применением методов химической термодинамики и квантовой химии.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Термодинамика реакций превращения этилового спирта и диметилового эфира в 1,3-бутадиен
В табл. 1 приведены термодинамические функции – ∆H (ккал/моль) – энтальпия, ∆S (кал/моль К) – энтропия и ∆G (ккал/моль) – энергия Гиббса – реакций ДМЭ и ЭС [6], а также реакции превращения ДМЭ в ЭС:
(1)
${\text{С}}{{{\text{Н}}}_{{\text{3}}}}{\kern 1pt} --{\kern 1pt} {\text{О}}{\kern 1pt} --{\kern 1pt} {\text{С}}{{{\text{Н}}}_{{\text{3}}}} \to {\text{С}}{{{\text{Н}}}_{{\text{3}}}}{\text{С}}{{{\text{Н}}}_{{\text{2}}}}{\text{ОН}}{\text{.}}$Таблица 1.
Величины термодинамических функций молекул СН3ОСН3, СН3СН2ОН в зависимости от температуры реакции (1): ∆H, ∆G (ккал/моль) и S (кал/моль К)
| T, K | СН3ОСН3 | СН3СН2ОН | Реакции (1) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| ∆H | S | ∆G | ∆H | S | ∆G | ∆Hр | ∆Sр | ∆Gр | |
| 298 | –43.99 | 63.83 | –26.99 | –56.12 | 67.54 | –40.22 | –12.13 | 3.71 | –13.23 |
| 300 | –44.01 | 63.93 | –26.88 | –56.14 | 67.64 | –40.13 | –12.13 | 3.71 | –13.25 |
| 400 | –45.20 | 68.92 | –20.99 | –57.32 | 72.67 | –34.60 | –12.12 | 3.75 | –13.61 |
| 500 | –46.24 | 73.51 | –14.81 | –58.31 | 77.36 | –28.80 | –12.07 | 3.85 | –13.99 |
| 600 | –47.10 | 77.83 | –8.44 | –59.11 | 81.78 | –22.83 | –12.01 | 3.95 | –14.39 |
| 700 | –47.79 | 81.91 | –1.94 | –59.76 | 85.93 | –16.73 | –11.97 | 4.02 | –14.79 |
| 800 | –48.34 | 85.77 | 4.64 | –60.27 | 89.84 | –10.55 | –11.93 | 4.07 | –15.19 |
| 900 | –48.74 | 89.42 | 11.30 | –60.65 | 93.52 | –4.30 | –11.91 | 4.10 | –15.6 |
| 1000 | –49.03 | 92.89 | 17.99 | –60.98 | 97.00 | 1.98 | –11.95 | 4.11 | –16.01 |
Из данных табл. 1 видно, что в температурном интервале Т = 298–1000 K значения термодинамических функций реакции (1) существенно не меняются. По величине ∆Hр и ∆Gр реакция (1) экзотермична и направлена в сторону образования этилового спирта.
Рассмотрим реакции термического разложения ДМЭ и ЭС. Термодинамический расчет температурной зависимости равновесного состава реакций термического превращения проводился по схеме:
(2)
$\begin{gathered} \left\{ {\text{Х}} \right\} \to \{ {{{\text{H}}}_{{\text{2}}}},{{{\text{C}}}_{{\text{2}}}}{{{\text{H}}}_{{\text{4}}}},{{{\text{H}}}_{{\text{2}}}}{\text{O}}\,\,\left( {\text{г}} \right),{\text{CO}},{{{\text{O}}}_{{\text{2}}}},{\text{CO}},{\text{C}}{{{\text{O}}}_{{\text{2}}}}, \\ {{{\text{C}}}_{{\text{4}}}}{{{\text{H}}}_{{\text{6}}}}\,\,\left( {{\text{1}},{\text{3 - бутадиен}}} \right),{{{\text{C}}}_{{\text{2}}}}{{{\text{H}}}_{{\text{6}}}}{\text{O}}\,\,\left( {{\text{ДМЭ}}} \right), \\ {{{\text{C}}}_{{\text{2}}}}{{{\text{H}}}_{{\text{6}}}}{\text{O}}\,\,{\text{(ЭС),C}}{{{\text{H}}}_{{\text{2}}}}{\text{O}}\,\,\left( {{\text{формальдегид}}} \right),{{{\text{C}}}_{{\text{2}}}}{{{\text{H}}}_{{\text{6}}}}, \\ {{{\text{C}}}_{{\text{2}}}}{{{\text{H}}}_{{\text{2}}}},{{{\text{C}}}_{{\text{2}}}}{{{\text{H}}}_{{\text{4}}}}{\text{O}}\,\,{\text{(ацетальдегид}}),{{{\text{H}}}_{{\text{2}}}}{\text{O}},{\text{С}}\} ,~ \\ \end{gathered} $Расчеты проводили [7] в два этапа. На первом этапе в состав продуктов включали углерод С (модель полного термолиза), во втором – углерод исключали (модель частичного термолиза). Результаты расчетов приведены на рис. 1. Как следует из рис. 1, температурная зависимость равновесных концентраций ДМЭ и ЭС практически не отличаются.
Рис. 1.
Температурная зависимость равновесного состава реакции (1) для случаев Х = СН3ОСН3 и СН3СН2ОН в присутствии (а) и в отсутствие в продуктах углерода (б).

В модели полного термолиза 1,3-бутадиен незначительно образуется при температуре ниже 300°С, а в модели частичного термолиза до температуры 450°С является основным продуктом.
Квантово-химический анализ индексов реакционной способности ДМЭ и ЭС. Исследование электронной структуры ДМЭ и ЭС проводили квантово-химическим методом функционала плотности DFT B3LYP/6-31G(d) [8] с полной оптимизацией геометрических параметров и расчетом частот нормальных колебаний молекул. Вычисляли следующие энергетические характеристики молекул: E0 = Eelec + ZPE, E = E0 + Evib + Erot + Etransl, H = E + RT, G = H – TS, где Eelec – электронная энергия молекулы, ZPE – энергия нулевых колебаний, Evib – колебательная энергия молекулы при температуре Т = 298 K, Erot – вращательная энергия, Etransl – поступательная энергия, Н – энтальпия, S – энтропия, G – cвободная энергия Гиббса, R – универсальная газовая постоянная.
В табл. 2 приведены результаты расчета электронной структуры молекул ДМЭ и ЭС, а также молекулы Н2О (для сравнительного анализа): малликеновские эффективные заряды на атомах, дипольные моменты и энергетические характеристики, энтальпии Н, энергии Гиббса G, энергии верхней занятой En и нижней вакантной En + 1 молекулярных орбиталей.
Таблица 2.
Результаты квантово-химических расчетов электронной структуры ДМЭ и ЭС
| СН3ОСН3 | СН3СН2ОН |
|---|---|
 |
 |
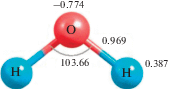
| μ =1.30 Дебай Eelec = –155.025 а.е. E0 = –154.945 а.е. H = –154.939 а.е. G = –154.969 а.е. En = –6.85 эВ, En + 1 = 2.51 эВ, ΔE = 9.36 эВ |
μ = 1.08 Дебай Eelec = –155.034 а.е. E0 = –154.953 а.е. H = –154.948 а.е. G = –154.979 а.е. En = –7.12 эВ, En + 1 = 2.09 эВ, ΔE = 9.21 эВ |
 |
|
| μ = 2.09 Дебай En = –7.92 эВ, En + 1 = 1.70 эВ, ∆E = 9.62 эВ | |
| Контурные карты изопотенциалов | |
 |
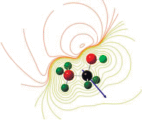 |
Молекулярный электростатический потенциал (МЭП). Для интерпретации электрофильных и нуклеофильных свойств молекулы МЭП вычисляется по формуле:
(3)
$U(r) = - \int {\frac{{\rho ({{r}_{i}})}}{{\left| {r - {{r}_{i}}} \right|}}d{{r}_{i}}} + \sum\nolimits_j {\frac{{\left| e \right|{{Z}_{j}}}}{{\left| {r - {{r}_{j}}} \right|}},} $В табл. 2 МЭП молекул ДМЭ и ЭС изображен в виде карты с контурными линиями равных потенциалов (изолинии). Области с отрицательным МЭП соответствуют зонам высокой плотности электронов, представляющим сильное притяжение между протоном и точками на поверхности молекулы, и имеют яркий красный цвет. Положительные регион – районы с низкой плотностью электронов, указаны зеленым цветом. Изолинии вокруг атомов кислорода показывают благоприятные направления их протонирования по неподеленным электронным парам.
Электростатический потенциал наглядно показывает области реакционной способности молекул к электрофильным и нуклеофильным атакам, которые существенно отличаются у ДМЭ и ЭС.
Проведем сравнительный анализ индексов реакционной способности молекул ДМЭ и ЭС, которые определяются по энергиям граничных МО. Так как энергия верхней занятой молекулярной орбитали (ВЗМО) En определяет донорную способность, а энергия нижней вакантной молекулярной орбитали (НВМО) En + 1 – акцепторную, то согласно данным табл. 3, молекула ДМЭ по значениям En и En + 1 лучший донор и худший акцептор электрона, чем молекула ЭС. В табл. 3 приведены значения различных индексов реакционной способности [9, 10] молекул ДМЭ и ЭС:
Таблица 3.
Индексы реакционной способности ДМЭ и ЭС
| Индекс | ДМЭ | ЭС |
|---|---|---|
| En, эВ | –6.85 | –7.12 |
| En + 1, эВ | 2.51 | 2.09 |
| ∆Е = EНВМО – EВЗМО, эВ | 9.36 | 9.21 |
| χ = –(EНВМО + EВЗМО)/2, ккал/моль | 50.04 | 58.01 |
| η = (EНВМО – ЕВЗМО)/2, ккал/моль | 107.98 | 106.11 |
| ω = μ2/2η, ккал/моль | 11.59 | 15.86 |
– ширина “запрещенной зоны” –∆Е указывает, что ДМЭ более стойка к возбуждению;
– по величине абсолютной электроотрицательности – χ молекула ЭС более способна притягивать к себе электронную плотность;
– по индексу абсолютной химической жесткости η молекула ДМЭ более стойка к химическим воздействиям;
– индекс электрофильности ω у ЭС больше, чем у ДМЭ.
Квантово-химический расчет энергии потенциальной поверхности элементарных актов превращения реагентов в продукты. Для сравнительного анализа реакций превращения молекул ДМЭ и ЭС проводили квантово-химический расчет энергии потенциальной поверхности элементарных актов превращения реагентов в продукты [10]. Поиск энергии переходного состояния осуществляли методами расчета переходного состояния (TS), линейного транзита (QST2) и квадратичного синхронного транзита (QST3).
Рассмотрели следующие первичные реакции превращения ДМЭ и ЭС (рис. 2):
(4)
${\text{С}}{{{\text{Н}}}_{{\text{3}}}}{\text{ОС}}{{{\text{Н}}}_{{\text{3}}}} \to {\text{С}}{{{\text{Н}}}_{{\text{3}}}}{\text{С}}{{{\text{Н}}}_{{\text{2}}}}{\text{ОН,}}$(5)
${\text{С}}{{{\text{Н}}}_{{\text{3}}}}{\text{ОС}}{{{\text{Н}}}_{{\text{3}}}} \to {{{\text{C}}}_{{\text{2}}}}{{{\text{H}}}_{{\text{4}}}} + {{{\text{H}}}_{{\text{2}}}}{\text{O,}}~$(6)
${\text{С}}{{{\text{Н}}}_{{\text{3}}}}{\text{ОС}}{{{\text{Н}}}_{{\text{3}}}} \to {\text{С}}{{{\text{Н}}}_{{\text{3}}}}{\text{СНО}} + {{{\text{H}}}_{{\text{2}}}},$(7)
${\text{С}}{{{\text{Н}}}_{{\text{3}}}}{\text{С}}{{{\text{Н}}}_{{\text{2}}}}{\text{ОН}} \to {{{\text{C}}}_{{\text{2}}}}{{{\text{H}}}_{{\text{4}}}} + {{{\text{H}}}_{{\text{2}}}}{\text{O,}}$(8)
${\text{С}}{{{\text{Н}}}_{{\text{3}}}}{\text{С}}{{{\text{Н}}}_{{\text{2}}}}{\text{ОН}} \to {\text{С}}{{{\text{Н}}}_{{\text{3}}}}{\text{СНО}} + {{{\text{H}}}_{{\text{2}}}}.$Рис. 2.
Профиль поверхности потенциалов реакций превращения: (а) – диметилового эфира в этиловый спирт; (б) и (в) – реакции отщепления воды и водорода от диметилового эфира; (г) и (д) – от этилового спирта соответственно.

Для реакции (4) с профилем поверхности потенциала на рис. 3 в газовой фазе, температурную зависимость констант скоростей прямой k1(T) и обратной k–1(T) мономолекулярных реакций вычисляли по формуле Эйрига [9]:
(9)
$k(T) = \frac{{{{k}_{{\text{B}}}}T}}{h}\exp \left( {\frac{{ - \Delta G}}{{{{k}_{{\text{B}}}}T}}} \right),\,\,{1 \mathord{\left/ {\vphantom {1 {{\text{c,}}}}} \right. \kern-0em} {{\text{c,}}}}$(11)
${{k}_{{ - 1}}}(T) = 2.08329 \times {{10}^{{10}}}Te\frac{{ - 56{\kern 1pt} {\kern 1pt} {\kern 1pt} 308.7}}{T}.$
На рис. 3а и 3б приведены поверхности потенциальных энергий, реакции превращения ДМЭ и ЭС, с использованием масс-взвешенных координат (внутренних координат реакции (IRC)), кратчайшим путем соединяющих реагенты с продуктами через переходные состояния. В обоих случаях продуктами реакций являются формальдегид и метан. Однако, в случае ДМЭ энергия активации составляет Е = 15 ккал/моль, а в случае ЭС – Е = 69 ккал/моль.
Отметим, что интерес исследователей к проблеме взаимодействия пропилена и формальдегида с целью получения 1,3-бутадиена неуклонно возрастает, что определяет актуальность и практическую значимость этой реакции [11].
Исходя из результатов термодинамических и квантово-химических расчетов, были исследованы следующие реакции получения 1,3-бутадиена:
(12)
${\text{2}}{{{\text{С}}}_{{\text{2}}}}{{{\text{Н}}}_{{\text{4}}}} \to {{{\text{С}}}_{{\text{4}}}}{{{\text{Н}}}_{{\text{6}}}} + {{{\text{Н}}}_{{\text{2}}}},$(13)
${{{\text{С}}}_{{\text{3}}}}{{{\text{Н}}}_{{\text{6}}}} + {\text{С}}{{{\text{Н}}}_{{\text{2}}}}{\text{О}} \to {{{\text{С}}}_{{\text{4}}}}{{{\text{Н}}}_{{\text{6}}}} + {{{\text{Н}}}_{{\text{2}}}}{\text{О}},$(14)
${\text{С}}{{{\text{Н}}}_{{\text{3}}}}{\text{СНО}} + {{{\text{С}}}_{{\text{2}}}}{{{\text{Н}}}_{{\text{4}}}} \to {{{\text{С}}}_{{\text{4}}}}{{{\text{Н}}}_{{\text{6}}}} + {{{\text{Н}}}_{{\text{2}}}}{\text{О}}.$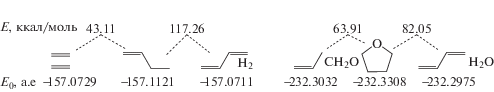
Судя по энергиям активации второй вариант, по реакции (13) более предпочтителен.
На рис. 4 приведена поверхность потенциальной энергии реакции
Рис. 4.
Поверхность потенциальной энергии реакции СН3СНО + С2Н4 → С4Н6 + Н2О (слева) и 3СО + 3Н2 → → СН3ОСН3 + СО2 (справа).
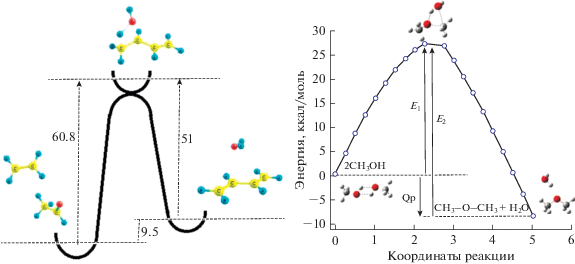
Интерес представляет также представленная на рис. 4 поверхность потенциальной энергии реакции синтеза ДМЭ из метилового спирта 2СH3ОН → СН3ОСН3 + Н2О с энергией активации 26 ккал/моль; энергия активации обратной реакции составляет 33 ккал/моль. Эта реакция показывает, что в продуктах превращения ДМЭ образуются различные соединения (например, СН3ОН), которые могут быть промежуточными при синтеза 1,3-бутадиена.
Возникает вопрос, может ли ZnO-содержащий катализатор реакции синтеза из ЭС 1,3-бутадиена, быть также эффективным катализатором для синтеза 1,3-бутадиена из ДМЭ? Чтобы ответить на этот вопрос, исследовали электронную структуру комплексов CH3OCH3⋅⋅⋅Zn4O4 и CH3CH2ОН⋅⋅⋅Zn4O4 (рис. 5):
Рис. 5.
Электронная структура комплекса [CH3OCH3-Zn4O4] и [CH3CH2OH-Zn4O4]. Расчеты проводили в приближении “супермолекулы” с оптимизацией всех геометрических параметров комплексов и проверкой соответствия полученного решения к минимуму энергии.

Согласно данным рис. 5, комплекс CH3CH2ОН⋅⋅⋅Zn4O4 на –25.3 ккал/моль устойчивее, чем комплекс CH3OCH3⋅⋅⋅Zn4O4. В обоих случаях, молекулы через атом кислорода с кластером образуют связь O–Zn. Эффективные малликеновские заряды Q на молекулах и на кластере следующие: (Zn4O4)–0.17(CH3OCH3)+0.17, (Zn4O4)–0.04(CH3CH2OH)+0.04, то есть ДМЭ более электронадонорна Q = –0.17, чем ЭС Q = –0.04. По данным рис. 5 при E0(Zn4O4) = –7417.451 а.е., энергия взаимодействия ∆E0 ДМЭ и МС с кластером Zn4O4 составляют:
Отметим, что результаты исследования электронной структуры комплекса CH3OCH3⋅⋅⋅Mg4O4 оказались такими же, как и CH3OCH3⋅⋅⋅Zn4O4. Следовательно, катализаторы ZnO и MgO не могут быть эффективными катализаторами реакции синтеза 1,3-бутадиена из ДМЭ. Экспериментальные исследования превращения ДМЭ проводились в работах [2, 4].
Энергию стабилизации комплекса CH3CH2ОН⋅⋅⋅Zn4O4 удобно анализировать в базисе естественных связовых орбиталей (natural bond orbitals – NBO), которые строятся на основе делокализованных МО [12]. Вклады в энергии стабилизации всех возможных взаимодействий между донорными и акцепторными орбиталями определяются с применением теории возмущения второго порядка. Энергия стабилизации вычисляется с учетом недиагональных элементов матрицы Фока в базисе NBO:
Приведенные данные показывают возможные взаимодействия между “заполненными” (донорными) НБО орбиталями Льюисовского типа и “пустыми” (акцепторными) не Льюиского НБО типа. В результате таких взаимодействий за счет делокализации электронов квантово-химическая система получает дополнительную стабилизацию. Обычно в литературе приводятся энергии взаимодействия, превышающие 0.5 ккал/моль. В табл. 4 мы привели значения энергии больше 10 ккал/моль.
Таблица 4.
Вклады в энергии стаблизации комплекса [CH3CH2OH-Zn4O4] взаимодействия донорных и акцепторных NBO-связей (приведены значения Е(2) > 10 ккал/моль)*
| [CH3CH2OH-Zn4O4] | ||||
|---|---|---|---|---|
| донорные NBO(i) | акцепторные NBO(j) | E(2), ккал/моль | E(j) – E(i), a.u. | F(i,j), a.u. |
| BD(1)Zn11–O14 | LP*(6)Zn13 | 10.97 | 0.58 | 0.072 |
| BD(1)Zn11–O17 | LP*(6)Zn13 | 12.27 | 0.57 | 0.076 |
| BD(1)Zn11–O17 | BD*(2)Zn12–O15 | 13.24 | 0.54 | 0.076 |
| BD(2)Zn12–O15 | LP*(6)Zn11 | 59.15 | 0.60 | 0.169 |
| BD(1)Zn12–O16 | LP*(6)Zn13 | 10.60 | 0.58 | 0.071 |
| BD(1)Zn12–O17 | LP*(6)Zn13 | 10.90 | 0.57 | 0.071 |
| BD(1)Zn13–O14 | LP*(7)Zn11 | 11.65 | 0.51 | 0.070 |
| BD(1)Zn13–O17 | LP*(7)Zn11 | 10.52 | 0.51 | 0.066 |
| LP(2)O8 | LP*(6)Zn10 | 14.38 | 0.41 | 0.069 |
| LP*(6)Zn10 | LP*(6)Zn11 | 11.89 | 0.04 | 0.050 |
| LP*(6)Zn10 | BD*(2)Zn12–O15 | 10.86 | 0.01 | 0.029 |
| LP*(6)Zn11 | BD*(1)Zn12–O15 | 32.24 | 0.01 | 0.048 |
| LP*(7)Zn11 | LP*(6)Zn13 | 10.84 | 0.05 | 0.055 |
| LP*(7)Zn11 | BD*(1)Zn11–O14 | 43.26 | 0.04 | 0.112 |
| LP*(7)Zn11 | BD*(1)Zn11–O17 | 23.70 | 0.06 | 0.106 |
| LP*(7)Zn11 | BD*(2)Zn12–O15 | 30.33 | 0.02 | 0.056 |
| LP*(6)Zn13 | BD*(1)Zn13–O14 | 96.71 | 0.01 | 0.093 |
| LP*(6)Zn13 | BD*(1)Zn13–O16 | 82.03 | 0.01 | 0.092 |
| BD*(2)Zn12–O15 | BD*(1)Zn12–O15 | 25.99 | 0.04 | 0.095 |
| BD*(2)Zn12–O15 | BD*(1)Zn12–O17 | 24.26 | 0.04 | 0.092 |
В табл. 4 приведены следующие одноцентровые и двухцентровые естественные связевые орбитали донорного и акцепторного типа:
– BD – двухцентровая связывающая орбиталь;
– BD* – двухцентровая разрыхляющая орбиталь;
– LP – заполненная одноцентровая орбиталь;
– LP*– незаполненная одноцентровая орбиталь.
Как видно из табл. 4, в энергию стаблизации комплекса большой вклад (E(2) = 96.71 ккал/моль) вносит донорно-акцепторное взаимодействие орбиталей LP*(Zn13) и BD*(Zn13–O14).
Отметим, что результаты исследования электронной структуры комплекса [CH3OCH3–Mg4O4] оказались такими же, как и [CH3OCH3–Zn4O4]. Следовательно, индивидуальные оксиды ZnO и MgO не могут быть эффективными катализаторами реакции синтеза 1,3-бутадиена из ДМЭ.
Таким образом, результаты исследования структуры и свойств диметилового эфира и этилового спирта показали:
– температурная зависимость равновесного состава продуктов превращения диметилового эфира и этилового спирта, при равных термодинамических условиях, практически не отличаются;
– согласно результатам квантово-химических расчетов диметиловый эфир реакционноспособнее этилового спирта. Однако энергия взаимодействия диметилового эфира с поверхностью кластера ZnO в два раза меньше, чем этилового спирта. Поэтому, диметиловый эфир в отличие от этилового спирта на кластере ZnO более склонен к термическому разложению и образованию кокса, приводя к снижению селективности процесса по 1,3-бутадиену.
Работа выполнена в рамках Государственного задания ИНХС РАН.
Список литературы
Лебедев С.В. Жизнь и труды. ОНТИ ХИМ. ТЕОРЕТ. Л., 1938. 878 с.
Khadzhiev S.N., Maksimov A.L., Tret’yakov V.F.1, Talyshinskii R.M., Ilolov A.M. // Petrol. Chemistry. 2018. V. 58. № 8. P. 405.
Tret'yakov V.F., Talyshinskii R.M., Ilolov A.M., Maksimov A.L., Khadzhiev S.N. // Petrol. Chemistry. 2014. V. 54. № 3. P. 195
Хаджиев С.Н., Маганов Н.У., Максимов А.Л., Бабынин А.А., Третьяков В.Ф., Илолов А.М. // Патент РФ № 2666591. Заявл. 17 окт. 2017, № 2017136568, опубл. 12 окт. 2018. Заявитель ИНХС РАН.
Цодиков М.В. Каталитические превращения продуктов биомассы в энергоносители. Форум “Открытые инновации” 31 октября–02 ноября 2012, Москва.
Сталл Д., Вестрам Э., Зинке Г. Химическая термодинамика органических соединений. М.: Мир, 1971. 807 с.
HSC Chemistry 6. http://www.hsclchemistry.net/.
Granovsky A.A. GAMESS. V. 7.1. http://classic.chem.msu.su/gran/gamess/index.html.
Shubin L. // J. Chem. Sci. V. 117. № 5. September 2005. P. 477.
Pearson R.G. // Proc. Natl. Acad. Sci. USA. 1986. V. 83. P. 8440.
Tret’yakov V.F., Ilolov A.M., Talyshinskii R.M., Gyul’maliev A.M., Khadzhiev S.N. // Petrol. Chemistry. 2017. V. 57. № 8. P. 686.
Welnhold F. Landis C.R. // Chem. Edu. Res. Pract. 2001. V. 2. № 2. P. 91.
Дополнительные материалы отсутствуют.