

Нефтехимия, 2019, T. 59, № 5, стр. 538-543
Дегидратация глицерина в акролеин на сульфатированных железооксидных катализаторах
П. Г. Мингалев 1, А. Г. Асланлы 1, Г. В. Лисичкин 1, *
1 Московский государственный университет имени М.В. Ломоносова,
Химический факультет
119234 Москва, Россия
* E-mail: Lisich@petrol.chem.msu.ru
Поступила в редакцию 09.06.2018
После доработки 02.04.2019
Принята к публикации 13.05.2019
Аннотация
Статья посвящена исследованию проблемы утилизации глицерина – основного отхода при производстве биодизельного топлива. Показано, что дешевые катализаторы на основе оксида трехвалентного железа могут служить катализаторами превращения глицерина в акролеин – важный полупродукт промышленного органического синтеза. Исследовано влияние внешних факторов на протекание процесса дегидратации глицерина в акролеин.
Как известно, основной способ производства биодизельного топлива [1] в качестве ключевой стадии включает переэтерификацию растительных масел метиловым спиртом на катализаторах основной или кислотной природы [2]. Получающиеся метиловые эфиры жирных кислот представляют собой биодизель. Побочный продукт глицерин образуется в значительных количествах – на 1 т биодизеля около 100 кг глицерина.
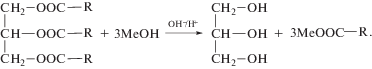
Основной барьер, сдерживающий широкое использование биодизельного топлива, – необходимость выведения из сельскохозяйственного оборота значительных площадей и высокая стоимость [3]. Поэтому важно как можно более полно использовать все продукты, образующиеся в процессе производства биодизеля, в частности, глицерин. Однако количество глицерина, получаемое в качестве отхода производства биодизеля, многократно превышает потребность в нем. Соответственно, актуальной задачей становится разработка химических процессов, использующих глицерин в качестве исходного сырья.
Одно из наиболее ценных веществ, синтезируемых из глицерина, – акролеин – важный полупродукт современной химической промышленности [4]. Из него получают акриловую кислоту, нитрил и эфиры акриловой кислоты, необходимые для производства разнообразных пластиков и полимерных волокон, а также незаменимую аминокислоту D,L-метионин, используемую в качестве добавки к кормам, и ее аналог 2-гидрокси-4-метилтиомасляную кислоту (гидроксианалог метионина, ГАМ).
Исследованию методов превращения глицерина в акролеин посвящено большое количество работ [5–11]. Для осуществления этой реакции используют разнообразные кислотные катализаторы. Однако хорошие выходы акролеина, как правило, достигаются либо в жестких условиях (высокие давление и температура), либо с применением катализаторов, содержащих достаточно дефицитные элементы (цирконий, вольфрам и т.п.). В то же время, как было отмечено выше, наибольшую актуальность имеет не просто разработка путей утилизации глицерина, а поиск наиболее дешевых методов такой утилизации. Соответственно, перед исследователями стоит задача максимального удешевления каждого этапа схемы переработки глицерина, в частности, поиск наиболее дешевых катализаторов этой переработки.
Превращение глицерина в акролеин происходит на катализаторах, имеющих кислую природу. Один из наиболее дешевых и недефицитных кислотных катализаторов – оксид железа(III), дополнительно обработанный кислотами, например серной кислотой [12]. В настоящей работе мы исследовали применимость нескольких таких катализаторов для реакции дегидратации глицерина в акролеин.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Синтез оксида железа(III). FeCl3 · 6H2O (16.9 г) растворяли в 50 мл дистиллированной воды при постоянном перемешивании магнитной мешалкой. Затем из капельной воронки по каплям прибавляли раствор 15.75 г NaHCO3 в 190 мл H2O. После этого состаривали выпавший осадок кипячением в течение 1.5 ч и оставляли на ночь. Осадок 6 раз промывали дистиллированной водой и центрифугировали при 10 000 об./мин. Полученный образец сушили в печи в течение 5 ч при 200°С. После этого катализатор размалывали в порошок в ступке. Пылеобразную фракцию отделяли путем седиментации в дистиллированной воде.
Синтез оксида никеля(II). NiCl2 · 6H2O (18 г) растворяли в 50 мл дистиллированной воды при постоянном перемешивании электрической мешалкой. Затем при помощи капельной воронки по каплям прибавляли раствор 11 г NaOH в 25 мл H2O. Смесь непрерывно перемешивали в течение 1 ч и оставляли на ночь. Для очистки от солей осадок 5 раз промывали дистиллированной водой и центрифугировали при 10 000 об./мин. Полученный образец сушили в печи в течение 6 ч при 230°С. После этого катализатор был размолот в порошок в ступке. Пылеобразную фракцию отделяли путем седиментации в дистиллированной воде.
Определение удельной поверхности образцов производили на приборе “Сорбтометр-М” методом тепловой десорбции азота. Для расчета удельной поверхности использовали уравнение БЭТ.
Определение кристаллической структуры исходного носителя и приготовленных катализаторов методом рентгено-фазового анализа (РФА) получали на приборе DRON-3M (Kα-излучение кобальта, длина волны 0.179021 нм).
Кислотные свойства образцов изучали методом термопрограммированной десорбции аммиака (ТПД NH3) на сорбционном анализаторе УСГА-101 (“Унисит”). Образец прокаливали в токе осушенного гелия при температуре 400 или 600°С, затем охлаждали до 60°С. Адсорбцию аммиака проводили в течение 30 мин при 60°C , аммиак разбавляли азотом в соотношении 1 : 1. Физически сорбированный аммиак отдували в токе осушенного гелия при 100°С в течение 1 ч. Эксперименты по ТПД NH3 проводили в интервале температур от 60 до 800°С в токе осушенного гелия (скорость 30 мл/мин). Скорость нагрева составляла 8°С/мин.
Определение общего объема пор капельным методом. К мелкоизмельченному осадку при помощи пипетки по каплям прикапывали дистиллированную воду встряхивая образец и добиваясь разрушения образующихся вначале комочков влажного носителя. Прибавление проводили до тех пор, пока комочки носителя не переставали разрушаться от потряхивания, что свидетельствовало о полном насыщении пор водой. По добавленному объему воды судили об объеме пор.
Приготовление катализаторов. На основе железооксидных носителей (Sуд= 53 м2/г; Vпор= 0.6 мл/г и Sуд = 364 м2/г; Vпор = 0.83 мл/г) были получены катализаторы Fe2O3 · H2SO4 (табл. 1 и 2). Их готовили путем смешивания навески оксида с разбавленной кислотой. Концентрацию кислоты выбирали так, чтобы объем добавляемой кислоты соответствовал объему пор навески оксида. После этого катализатор сушили 2 ч при 160°С, а затем проводили нагрев катализатора со скоростью 5°C/мин до соответствующей температуры и оставляли при ней на нужное время.
Таблица 1.
Условия приготовления катализаторов на основе оксида железа с удельной поверхностью 53 м2/г
| Порядковый № образца | Число молекул кислоты на 1 нм2 поверхности | Температура прокаливания, °С | Время прокаливания, ч |
|---|---|---|---|
| 1 | 10 | 200 | 6 |
| 2 | 10 | 400 | 6 |
| 3 | 10 | 600 | 6 |
| 4 | 5 | 200 | 2 |
| 5 | 5 | 200 | 4 |
| 6 | 5 | 200 | 6 |
| 7 | 5 | 400 | 2 |
| 8 | 5 | 400 | 4 |
| 9 | 5 | 400 | 6 |
| 10 | 5 | 600 | 2 |
| 11 | 5 | 600 | 4 |
| 12 | 5 | 600 | 6 |
| 13 | 3 | 200 | 6 |
| 14 | 3 | 400 | 6 |
| 15 | 3 | 600 | 6 |
Таблица 2.
Условия приготовления катализаторов на основе оксида железа с удельной поверхностью 364 м2/г
| Порядковый № образца | Число молекул кислоты на 1 нм2 поверхности | Температура прокаливания, °С | Время прокаливания, ч | Масса навески катализатора, мг |
|---|---|---|---|---|
| 1 | 3 | 200 | 2 | 120 |
| 2 | 3 | 600 | 6 | 120 |
| 3 | 3 | 200 | 2 | 20 |
| 4 | 3 | 600 | 6 | 20 |
| 5 | 6 | 200 | 2 | 120 |
| 6 | 6 | 200 | 2 | 20 |
На основе железооксидного носителя с Sуд = = 364 м2/г и Vпор = 0.83 мл/г были приготовлены катализаторы Fe2O3 · H2SO4 (табл. 2)
На основе никельоксидного носителя (Sуд = = 150 м2/г; Vпор = 0.8 мл/г) были приготовлены катализаторы дегидратации NiO · H2SO4 (табл. 3). Способ приготовления катализаторов был таким же, как в случае железооксидных носителей.
Таблица 3.
Условия приготовления катализаторов на основе оксида никеля
| Порядковый № образца | Число молекул кислоты на 1 нм2 поверхности | Температура прокаливания, °С | Время прокаливания, ч |
|---|---|---|---|
| 1 | 3.0 | 400 | 6 |
| 2 | 3.0 | 600 | 6 |
| 3 | 1.5 | 400 | 6 |
| 4 | 1.5 | 600 | 6 |
Проведение экспериментов по дегидратации. К катализатору, находящемуся в колбе, прибавляли глицерин; массовое отношение катализатор : глицерин составляло 1 : 20. Затем эту смесь перегоняли в колбе Кляйзена, собирая выделяющийся дистиллят. В колбе после перегонки остается твердая смола.
Газохроматографический анализ. Анализ проводили на газовом хроматографе “Кристалл-Люкс” с пламенно-ионизационным детектором. Использовали капиллярную колонку длиной 20 м и внутренним диаметром 0.22 мм. Фаза FFAP (полиэтиленгликоль, этерифицированный нитротерефталевой кислотой). Толщина слоя неподвижной фазы 0.25 мкм. Газ-носитель гелий. Элюирование первые 2 мин вели при 30°С, затем производили нагрев колонки до 200°С со скоростью 20°/мин. Продолжительность одного анализа составляла 15 мин.
Анализировали полученные продукты в виде 10%-ных растворов в этиловом спирте. Для количественных измерений в хроматограф микрошприцем подавали модельную смесь, состоящую из 80 мас. % этанола, 10 мас. % глицерина и 10 мас. % акролеина. Для определения характеристик гидроксиацетона использовали 10%-ный раствор гидроксиацетона в этаноле. По полученным хроматограммам определяли время удерживания глицерина, гидроксиацетона и акролеина, а также относительную чувствительность детектора к глицерину, гидроксиацетону и акролеину по сравнению с этанолом. Чувствительность детектора к акролеину составляла 0.67 чувствительности к этанолу, к гидроксиацетону – 1.5 чувствительности к этанолу, к глицерину – 1.6 чувствительности к этанолу.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Дегидратация на железооксидных катализаторах
По данным РФА, все исследованные образцы представляют собой чистый гематит (α-Fe2O3), другие фазы не обнаружены ни в одном из случаев.
Влияние величины удельной поверхности катализаторов на протекание реакции дегидратации. Для того, чтобы исследовать, как влияет величина удельной поверхности катализатора на протекание реакции дегидратации, были приготовлены два образца носителей, с удельной поверхностью в 53 и 364 м2/г.
Было обнаружено, что активность и селективность образцов с высокой удельной поверхностью заметно выше – при прочих равных выход акролеина может достигать 21%. Следует, однако, отметить, что синтез образцов оксида железа с настолько высоким значением удельной поверхности (364 м2/г) отличается низкой воспроизводимостью. Поэтому до отработки воспроизводимых методов синтеза таких носителей мы не можем их рекомендовать для реакции дегидратации глицерина. Дальнейшее обсуждение будет касаться только образцов с удельной поверхностью в 53 м2/г, характеризующихся высокой воспроизводимостью.
Влияние количества кислоты, нанесенной на катализаторы, на выход акролеина. Для выяснения этого влияния была синтезирована серия образцов с числом молекул кислоты на 1 нм2 поверхности от 3 до 10. Полученные катализаторы испытаны в реакции дегидратации, результаты представлены в табл. 4. Время прокаливания образцов 6 ч.
Таблица 4.
Влияние количества кислоты на результаты дегидратации глицерина (железооксидные катализаторы, Sуд 53 м2/г)*
| Число молекул кислоты на 1 нм2 поверхности | Температура прокаливания катализатора, °С | Yd, % | Ya, % | Yh, % |
|---|---|---|---|---|
| 10 | 200 | 77.0 | 2.3 | 0.3 |
| 10 | 400 | 51.0 | 7.8 | 1.1 |
| 10 | 600 | 65.0 | 0.2 | 0.3 |
| 5 | 200 | 32.0 | 6.2 | 0.3 |
| 5 | 400 | 38.0 | 3.2 | 0 |
| 5 | 600 | 68.0 | 2.5 | 0.3 |
| 3 | 200 | 17.0 | 0.2 | 0.2 |
| 3 | 400 | 16.0 | 0.2 | 0.1 |
| 3 | 600 | 20.0 | 0.2 | 0.1 |
Как видно из табл. 4, практически во всех случаях наилучшими свойствами обладают образцы с формальной поверхностной концентрацией кислоты в 5 молекул на 1 нм2. Образцы с более низкой концентрацией кислоты оказались практически неактивными во всех случаях, по-видимому, из-за слишком низкой кислотности. Что касается образцов с более высокой кислотностью, то выход дистиллята в этом случае также выше, однако он содержит в основном продукты олигомеризации акролеина. Поэтому дальнейшее обсуждение будет касаться только образцов с поверхностной концентрацией кислоты в 5 молекул на 1 нм2 поверхности носителя.
Влияние температуры и времени прокаливания катализаторов. Результаты проведения реакции дегидратации на катализаторах, полученных в различных условиях, представлены в табл. 5.
Таблица 5.
Влияние температуры и времени прокаливания железооксидных катализаторов на результаты дегидратации глицерина*
| Температура прокаливания, °С | Время прокаливания, ч | Yd, % | Ya, % | Yh, % |
|---|---|---|---|---|
| 200 | 2 | 74.0 | 2.7 | 0.4 |
| 200 | 4 | 54.0 | 0.2 | 0.2 |
| 200 | 6 | 32.0 | 6.2 | 0.3 |
| 400 | 2 | 49.0 | 8.1 | 1.4 |
| 400 | 4 | 54.0 | 11 | 2.3 |
| 400 | 6 | 38.0 | 3.2 | 0 |
| 600 | 2 | 28.0 | 4.5 | 1.2 |
| 600 | 4 | 21.0 | 7.5 | 0.1 |
| 600 | 6 | 680 | 2.5 | 0.3 |
В результате исследований было выяснено, что зависимость активности и селективности катализатора от температуры прокаливания проходит через максимум, то есть, увеличивается выход как дистиллята, так и акролеина. Наибольший выход наблюдается при температуре прокаливания в 400°С. Для этой температуры зависимость активности и селективности катализатора также проходит через максимум. Исследование катализаторов с помощью ТПД аммиака показывает, что наилучшие выходы достигаются для катализаторов с высоким относительным содержанием сильных кислотных центров. На образцах же с высоким относительным содержанием слабых кислотных центров выходы низки. Связано это, по-видимому, с тем, что для дегидратации глицерина требуются достаточно сильные кислотные центры [4], однако побочные реакции (типа полимеризации) слабые кислотные центры ускорять вполне могут.
Было также обнаружено, что весьма важным является также наличие на поверхности катализатора сульфата железа. Это соединение не было обнаружено методом РФА, однако, можно полагать, что разложение этого соединения ответственно за пик на спектрах ТПД при температурах около 600°С. На катализаторах, не содержащих сульфата железа, выходы целевого продукта (а также гидроксиацетона) крайне низки, несмотря на наличие достаточного количества сильных кислотных центров в образце.
Для объяснения этого эффекта можно предположить, что активные центры катализатора являются бренстедовскими, а сульфат железа в данном случае выступает как кислота Льюиса, образуя донорно-акцепторные комплексы с молекулой глицерина. По стерическим причинам, по-видимому, образуются преимущественно комплексы сульфата железа с концевыми ОН-группами молекул глицерина. Это благоприятствует получению акролеина, так как к его образованию приводит протонирование молекулы глицерина по среднему гидроксилу, который в таких комплексах остается свободным. Заметим, что глицерин является более сильным основанием Льюиса, чем акролеин. Поэтому образование комплексов глицерина с льюисовскими кислотными центрами должно предотвращать адсорбцию акролеина на поверхности катализатора, и, соответственно, затруднять протекание нежелательных побочных реакций с участием акролеина. Для проверки этого предположения были приготовлены и исследованы образцы на основе оксида никеля (см. ниже).
Следует также отметить, что выходы гидроксиацетона на наших образцах довольно низки – они всегда ниже выходов акролеина. Вероятно, это связано с неустойчивостью гидроксиацетона в кислой среде – он может конденсироваться по альдольно-кротоновому типу, выступая в качестве как карбонильной, так и метиленовой компоненты. Акролеин же в этой реакции может выступать только в качестве карбонильной компоненты, соответственно, для конденсации по этому типу ему требуются сторонние молекулы.
Таким образом, для образования кислотных центров нужной силы на поверхности требуется отжиг образцов, однако не слишком сильный.
Дегидратация на никельоксидных катализаторах
По данным РФА, все исследованные образцы представляют собой чистый бунзенит (α-NiO), другие фазы не обнаружены ни в одном из случаев.
Влияние концентрации кислоты и температуры прокаливания на активность и селективность катализатора. Для исследования влияния концентрации кислоты на протекание реакции дегидратации были приготовлены две серии катализаторов с количеством кислоты в 3 и 1.5 молекул на поверхности (табл. 6). Катализатор с количеством кислоты на поверхности в 3 г/нм2 оказался относительно более селективным по акролеину. Также было выяснено, что повышение концентрации кислоты в составе катализатора приводит к резкому уменьшению выхода дистиллята. Полученные в этом случае образцы дистиллята, ввиду их малого количества, не подвергали хроматографическому анализу.
Таблица 6.
Влияние температуры и времени пркаливания катализаторов на основе никельоксидного носителя (Sуд = 150 м2/г; Vпор = 0.8 мл/г) на результаты дегидратации глицерина*
| Число молекул кислоты на 1 нм2 | Т прокаливания, °С | Время прокаливания, ч | Yd, % | Ya, % | Yh, % |
|---|---|---|---|---|---|
| 3.0 | 400 | 6 | 30.0 | 2.7 | 0.2 |
| 3.0 | 600 | 6 | 31.0 | 6.9 | 0.6 |
| 1.5 | 400 | 6 | 33.0 | 4.0 | 0.4 |
| 1.5 | 600 | 6 | 50.0 | 4.9 | 0.5 |
Для выяснения влияния указанных факторов на протекание реакции были проведены две серии опытов (см. табл. 6) – одна серия при 400, а вторая – при 600°С. В результате исследований было выяснено, что активность и селективность катализатора увеличивается с повышением температуры прокаливания.
Весьма важно было получить ответ на вопрос о роли льюисовских кислотных центров на поверхности. Как было сказано выше, для железооксидных катализаторов наличие сульфата железа на поверхности является необходимым условием протекания целевой реакции. Этот факт может быть объяснен предположением, что сульфат железа в данном случае выступает как кислота Льюиса (см. выше). Для проверки этого предположения с помощью ТПД аммиака были исследованы катализаторы на основе оксида никеля, допированного серной кислотой. Никель является химическим аналогом железа, эти элементы весьма близки по химическим свойствам – однако никель, в отличие от железа, наиболее устойчив в двух-, а не в трехвалентном состоянии. Радиус иона Ni2+ больше радиуса иона Fe3+ (0.069 нм против 0.064), а заряд – меньше. Соответственно, сульфат никеля(II) является заметно более слабой кислотой Льюиса, чем сульфат железа(III).
Было обнаружено, что количество и сила кислотных центров на никельоксидных катализаторах примерно соответствуют таковым для железооксидных катализаторов. Однако выходы акролеина и гидроксиацетона на этих катализаторах заметно ниже, чем в случае железооксидных. Из этого можно сделать вывод о том, что, действительно, наличие на поверхности катализатора, наряду с сильными бренстедовскими, льюисовских кислотных центров является необходимым условием успешного протекания реакции. Кроме того, было показано, что для образца, содержащего большее количество сильных кислотных центров (прокаливание при 600°С) выход акролеина выше, что находится в согласии с результатами, полученными для железооксидных катализаторов.
Таким образом, сульфатированный оксид железа(III) проявляет активность в реакции дегидратации глицерина. Выход акролеина на воспроизводимо получаемых образцах составляет около 11 мас. %. Он может быть увеличен путем повышения удельной поверхности катализатора. Проблема, однако, заключается в низкой воспроизводимости синтеза образцов оксида железа с высокоразвитой удельной поверхностью. Это объясняется тем, что текстура оксида железа зависит от большого числа параметров синтеза, влияние которых слабо изучено.
Найдено, что увеличение концентрации кислоты на поверхности ведет к росту активности катализатора в реакции дегидратации глицерина, однако при этом падает селективность по целевому продукту. Показано, что для дегидратации глицерина в акролеин важно наличие на поверхности катализатора сильных кислотных центров (по-видимому, бренстедовского типа), тогда как наличие слабых кислотных центров ведет к снижению селективности катализаторов. Образование сильных центров на поверхности требует прокаливания катализатора.
Наличие сульфата железа на поверхности железооксидных катализаторов ведет к увеличению выхода продукта. В его отсутствие селективность катализаторов, несмотря на наличие сильных кислотных центров, крайне низка.
Список литературы
Ma F.R., Hanna M.A., Wilson E.K. // Chem. Eng. News. 1999. V. 70. P. 1.
Meher L.C., Sagar D.V., Naik S.N. // Renewable and Sustainable Energy Reviews. 2006. V. 10. P. 248.
Арутюнов В.С., Лисичкин Г.В. // Усп. химии. 2017. Т. 86. № 8. С. 777.
Tsukuda E., Sato S., Takahashi R., Sodesawa T. // Catalysis Communications. 2007. V. 8. № 9. P. 1349.
Niu S., Zhu Y., Zheng H., Zhang W., Li Y. // Chinese J. of Catalysis. 2011. V. 32. P. 345.
Kim Y. T., Jung K.-D., Park E.D. // Applied Catalysis A: General. 2011. V. 393. № 1–2. P. 275.
Данов С.М., Есипович А.С., Белоусов А.С., Рогожин А.Е. // Химическая промышленность сегодня. 2014. № 2. С. 15.
Tao L.-Z., Chai S.-H., Zuo Y., Zheng W.-T., Liang Y., Xu B.-Q. // Catalysis Today. 2010. V. 158. № 3–4. P. 310.
Ramayya S., Brittain A., De Almeida C., Mok W., Antal M.J. // Fuel. 1987. V. 66. P. 1364.
Takanori A., Masayuki Y. // Patent Japan. № 2009292773. 2009.
de Oliveira A.S., Vasconcelos S.J.S., de Sousa J.R., de Sousa F.F., Filho J.M., Oliveira A.C. // Chem. Eng. J. 2011. V. 168. P. 765.
Крылов О.В. Гетерогенный катализ. М.: ИКЦ Академкнига, 2004. 270 с.
Дополнительные материалы отсутствуют.