

Нефтехимия, 2019, T. 59, № 6-1, стр. 690-695
АЦИЛИРОВАНИЕ О-КРЕЗОЛА ПИРОМЕЛЛИТОВЫМ ДИАНГИДРИДОМ
В. П. Нехорошев 1, *, А. С. Князев 2, И. Э. Нифантьев 3, 4
1 Сургутский государственный университет,
628400 Сургут, Россия
2 Национальный исследовательский Томский государственный университет
634000 Томск, Россия
3 Московский государственный университет имени М.В. Ломоносова, Химический факультет
Москва, Россия
4 Институт нефтехимическаого синтеза РАН
121609 Москва, Россия
* E-mail: nvp.atact@mail.ru
Поступила в редакцию 04.04.2019
После доработки 29.05.2019
Принята к публикации 02.07.2019
Аннотация
В статье приводятся результаты экспериментальных исследований по ацилированию о-крезола пиромеллитовым диангидридом (ПМДА). Экспериментально подобраны оптимальные условия реакции ацилирования о-крезола ПМДА. Синтез проводят в расплаве о-крезола при 120°С в течение 30 ч при следующем молярном соотношении реагентов: ПМДА: о-крезол : SnCI4 = 1.0 : 4.1 : 2.0. Выход 3,3,7,7-тетракис(3-метил-4-оксифенил)пиромеллитида составляет 62.0% от теоретического. Строение целевого продукта синтеза доказано физико-химическими методами: элементным анализом, тонкослойной хроматографией (ТСХ), УФ-, ИК- и ПМР-спектроскопией, ДСК, выскоэффективной жидкостной хроматографией-масс-спектометрией (ВЭЖХ/МС). Изучение свойств 3,3,7,7-тетракис(3-метил-4-оксифенил)пиромеллитида позволяет рекомендовать его в качестве химического индикатора в промысловых трассерных исследованиях нефтяных пластов.
Продукты ацилирования фенолов фталевым ангидридом, такие как фенолфталеин, крезолфталеин (КФ), флуоресцеин и др., широко используются в качестве кислотно-основных индикаторов, антисептиков в композициях косметических средств и лекарственных препаратов, мономеров для синтеза полимеров с ценным комплексом свойств и химических маркеров для идентификации веществ, материалов и изделий [1–3]. С этой точки зрения представляют практический интерес полифункциональные тетрафенольные продукты ацилирования фенолов пиромеллитовым диангидридом, поскольку возможности их практического использования значительно расширяются. В.А. Сергеев с соавт. исследовали ацилирование фенола ароматическими тетракарбоновыми кислотами и их диангидридами [4]. Ацилирование фенола проводили в расплаве реагентов с использованием в качестве катализаторов серной кислоты и различных кислот Льюиса. Взаимодействие реагентов контролировали, определяя содержание непрореагировавшего фенола в реакционной смеси. Экспериментально показано, что наилучшим каталитическим эффектом обладает четыреххлористое олово, использование которого позволяет снизить температуру реакции до 80–100°С и уменьшить до минимума образование побочных и смолообразных продуктов. Степень конверсии фенола при взаимодействии его с ПМДА при 120°С в течение 8 ч достигала 97.3%. При ацилировании фенола ПМДА образуются четыре продукта реакции с преобладанием до 80% дилактонов, изомерных крезолпиромеллитинов (КП) или 3,3,7,7-тетракис(4-оксифенил)пиромеллитида и 3,3,5,5-тетракис(4-оксифенил)пиромеллитида с низким выходом 17 и 13% соотвественно. Взаимодействие о-крезола с ПМДА в указанной работе не исследовали.
В работе [5] проведены поисковые исследования по синтезу пиромеллитовых индикаторов конденсацией ПМДА с фенолами различного строения с использованием в качестве катализатора безводного хлорида цинка, который успешно используется при синтезах фталеинов. В реакции с о-крезолом хлорид цинка оказался малоэффективным катализатором с низкой активностью и региоселективностью, выход целевого продукта реакции о-крезолпиромеллитина (КП) составлял всего 28.0%.
Целью настоящей работы является разработка методики синтеза нового соединения о-крезолпиромеллитина или 3,3,7,7-тетракис(3-метил-4-окси-фенил)пиромеллитида ацилированием о‑крезола ПМДА с использованием четыреххлористого олова в качестве катализатора, исследование строения и свойств целевого продукта.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
В качестве реагентов и растворителей при выполнении синтезов использовали о-крезол с содержанием основного вещества 99.0%; ПМДА с содержанием основного вещества 99.0 (MERCK-Schuchardt); безводный SnCl4 марки ч.; КОН марки ч. д. а.; 95%-ный этиловый спирт; хлористый ацетил марки ч. д. а.; пиридин 99% “Fluka”; 36%‑ная соляная кислота марки х. ч.; хроматографические пластины для ТСХ марки “sorbfil ПТС-Х-П-А” размером 10 х10 см.
Синтез о-крезолпиромеллитина. Синтез проводят в расплаве о-крезола при 120°С в течение 30 ч при следующем молярном соотношении реагентов: ПМДА : о-крезол : SnCI4 = 1.0 : 4.1 : 2.0. Используют немецкую лабораторную установку фирмы “Hidolph”, оборудованную стеклянным четырехгорлым реактором на 500 мл, электрической регулируемой мешалкой, электронагревателем с платиновой термопарой и обратным холодильником с хлоркальциевой трубкой. В реактор загружают 43.6 г (0.2 М) ПМДА, постепенно по каплям при перемешивании дозируют 104.2 г (0.4 М) SnCl4. В реакторе образуется ярко окрашенный фиолетовый комплекс. В реакционную смесь медленно дозируют при 25°С 108.14 г (0.82 М) о-крезола. Реакционную смесь нагревают до 120°С и выдерживают при интенсивном перемешивании в течение 30 ч. Реакционную смесь охлаждают до комнатной температуры, а затем медленно нейтрализуют 20%-ным водным раствором едкого натра до рН 10. Вишневый раствор охлаждают в холодильнике в течение суток. Выпавший осадок оксихлоридов олова отфильтровывают под вакуумом в воронку Бюхнера. Фильтрат из колбы Бунзена нейтрализуют соляной кислотой до рН 3, охлаждают 12 ч в холодильнике, отфильтровывают выпавший осадок, промывают его ледяной водой и сушат при 60°С. Реакция проходит по следующей схеме:
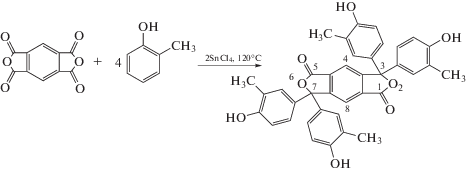
Осадок помещают в колбу Вюрца установки для перегонки с водяным паром, затем проводят отгонку с водяным паром непрореагировавшего о‑крезола. Содержание последнего в конденсате контролируют качественной реакцией с 5.0%-ным водным раствором FeCl3. Отгонку о-крезола прекращают после исчезновения фиолетового окрашивания конденсата при добавлении хлорида железа. Полученный осадок отфильтровывают, сушат и ацилируют при 120оС в течение одного часа хлористым ацетилом в ледяной уксусной кислоте с добавлением пиридина в качестве акцептора хлористого водорода. В реактор загружают 98.6 г осадка, полученного после отгонки о-крезола, добавляют 45.7 мл (0.64 М) ацетилхлорида и 49.6 мл пиридина. Реакционную смесь перемешивают и охлаждают в бане со льдом до 20°С, приливают 200 мл ледяной уксусной кислоты, нагревают до кипения (118°С) и выдерживают при этой температуре в течение 1 ч. Реакционную смесь охлаждают в холодильнике до 10°С, затем отфильтровывают выпавший серый осадок и промывают его охлажденным ацетоном. Выход 3,3,7,7-тетракис(3-метил-4-ацетоксифенил)-пиромеллитида составляет 97.0 г (62.0%). При охлаждении до 10°С фильтрата, разбавленного 150 мл воды, осадок второго изомера ацильного замещенного 3,3,5,5-тетракис(3-метил-4-ацетоксифенил)пиромеллитида не образуется, что можно объяснить стерическими препятствиями, мешающими синтезу этого изомера. Гидролиз ацетильного производного осуществляют в стеклянном реакторе объемом 250 мл, снабженном мешалкой, терморегулятором и обратным холодильником. Загружают 97.0 г 3,3,7,7-тетракис(3-метил-4-ацетоксифенил)пиромеллитида, 100 мл 20% водно-спиртового раствора (в соотношении 1 : 2 по объему) NaOH, нагревают реакционную смесь до кипения и проводят гидролиз в течение 6 ч. Фиолетовый раствор охлаждают до комнатной температуры и нейтрализуют 20% раствором соляной кислоты до рН 3–4, охлаждают в течение суток в холодильнике и отфильтровывают выпавший в колбе осадок. Полученный 3,3,7,7-тетракис(3-метил-4-оксифенил)пиромеллитид дважды перекристаллизовывают из водно-спиртового раствора (1 : 1 по объему) и высушивают. Выход – 75,5 г (61.4% в расчете на ПМДА).
Анализ. Элементный анализ синтезированного 3,3,7,7-тетракис(3-метил-4-оксифенил)пиромеллитида общей формулы С38Н30О8 выполняли с использованием системы для CHNS/О анализа типа PE2400-2 (Perkin Elmer). Результаты приведены в табл. 1.
Таблица 1.
Результаты элементного анализа о-крезолпиромеллитина
| Элемент | Содержание, мас. % | |
|---|---|---|
| найдено | вычислено | |
| C | 74.11 | 74.19 |
| H | 4.72 | 4.88 |
| O | 21.04 | 20.83 |
Полученный КП анализируют методами ТСХ, УФ-, ИК- и ПМР-спектроскопии, ДСК и ВЭЖХ/ МС, определяют интервал рН перехода цвета индикатора.
Анализ соединений методом ТСХ осуществляют следующим образом. Навеску вещества массой 0.5–1.0 мг растворяют в 0.5–1.0 мл водно-спиртового раствора (соотношение 1 : 2.3 по объему), затем наносят капилляром примерно 0.5 мкл раствора вещества на стартовую линию хроматографических пластин марки “Sorbfil ПТСХ-П-А” размерами 10 × 10 см, предварительно зачистив края по бокам пластин на 1–2 мм. В качестве стандарта использовали КФ имеющий строение сходное с КП, но с меньшей молекулярной массой. Элюирование пластин осуществляли в системе элюентов толуол−этанол−триэтиламин (9 : 1 : 1 по объему) на высоту 90 мм от линии старта, после чего пластины высушиваются в токе теплого воздуха. Детектирование хроматографических зон на полученных хроматограммах осуществляют при их осмотре в свете УФ-лампы (254 нм и 365 нм) и при помощи опрыскивания пульверизатором пластин 5%-ным спиртовым раствором щелочи. Полученные хроматограммы объектов исследования сравнивают между собой по цвету, форме и значениям Rf, выявленных хроматографических зон (табл. 2). Результаты анализа методом ТСХ показывают, что полученный КП после двух перекристаллизаций не содержит примесей других соединений. Относительный индекс удерживания КП в два раза меньше значения Rf для КФ, что объясняется большей ММ КП (614.64) и его большей функциональностью. Четыре гидроксильные группы фенольных заместителей КП сильнее адсорбируются на поверхности силикагеля, чем две гидроксильные группы КФ.
Таблица 2.
Результаты анализа продуктов синтеза методом ТСХ
| Образцы | Относительный индекс удерживания (значение Rf) | Цвет хроматографической зоны | ||
|---|---|---|---|---|
| УФ-254 нм | УФ-365 нм | 5%-ная щелочь | ||
| КП | 0.22 | Нет | Нет | Фиолетовый |
| КФ | 0.44 | Нет | Нет | Фиолетовый |
Для получения УФ-спектров синтезированного КП навеску вещества около 1 мг растворяют в 95%-ном этиловом спирте и проводят съемку в нейтральной и щелочной средах. В спиртовой раствор КП прибавляют 2 капли 5%-ного КОН и снимают окрашенные растворы. Съемку проводят при помощи двухлучевого спектрофотометра “Shimadzu UV-2600” в диапазоне от 185 до 800 нм, для снятия базовой линии использовали чистый этиловый спирт. Результаты обрабатывали при помощи программы “UVProbe”. В качестве образца сравнения сняты электронные спектры КФ (табл. 3).
Таблица 3.
Электронные спектры КП и КФ в нейтральной и щелочной среде
| Соединение | Переход | λmax, нм | D |
|---|---|---|---|
| Нейтральная среда | |||
| КФ | π → π* | 211 | 3.634 |
| n → π* | 295 | 0.854 | |
| n → π* | 375 | 0.42 | |
| n → π* | 575 | 2.514 | |
| КП | π → π* | 209.5 | 3.416 |
| n → π* | 238 | 1.803 | |
| n → π* | 285 | 0.518 | |
| Щелочная среда | |||
| КФ | π → π* | 216 | 4.4 |
| n → π* | 295 | 1.98 | |
| n → π* | 375 | 1.032 | |
| n → π* | 582.5 | 3.613 | |
| КП | π → π* | 208.5 | 3.524 |
| n → π* | 386 | 0.574 | |
| n → π* | 578.5 | 2.378 | |
При сравнении полученных электронных спектров КП со спектрами взятого в качестве стандарта КФ видно, что в нейтральной среде в спектре КП отсутствуют длинноволновые полосы поглощения с λmax 375 и 575 нм, а остальные три полосы поглощения претерпевают гипсохромный сдвиг, что объясняется отсутствием сопряжения крезольных заместителей с ароматическим кольцом. Образование хиноидных хромофорных заместителей у аниона КП в щелочной среде увеличивает цепь сопряжения в молекуле, что подтверждается появлением длинноволновых полос поглощения с λmax 386 и 578.5 нм.
ИК-спектры соединений снимали на приборе “Nicolet iS 10” Thermo Scientific методом однократно нарушенного полного внутреннего отражения на кристалле селенида цинка в диапазоне сканирования 4000–650 см–1, кривые обрабатывали при помощи программы “OMNIC”. Результаты расшифровки спектров представлены в табл. 4 [6].
Таблица 4.
Спектральные характеристики соединений
| Соединение | Характеристические полосы поглощения, см−1 | |||||||
|---|---|---|---|---|---|---|---|---|
| OH st | C=О st | C=C st (аром) | CO–O–C | C–O st | CH3 δas |
1,3,4-замещение, СН δ oop | 1,2,4,5-замещение, CH δ oop | |
| КП | 3456 3409 3296 |
1717 | 1608 | 1231 | 1120 1101 |
1451 | 884 799 695 |
873 |
| КФ | 3461 3319 |
1709 | 1612 | 1231 | 1119 1137 |
1465 | 884 799 695 |
745-1,2-замещение, ar C–C γ |
Анализ КП проводился на жидкостном хроматографе “UltiMate 3000” (колонка “Agilent Zorbax C18 4.6 × 100 мм, 5 мкм) с масс-спектрометрическим детектором API 2000 с положительной электрораспылительной ионизацией. Обработку результатов проводили с помощью программы Analyst 1,5. Хроматограмма содержала один пик КП с временем удерживания 11.26 мин, посторонних примесей не обнаружено. В масс-спектре содержатся сигналы протонированного молекулярного иона КП с m/z 615 и протонированных осколочных ионов с m/z 579, 571, 535, которые образуются при фрагментации молекулярного иона путем последовательного отщепления молекулы СО2 и двух молекул воды по следующей схеме:

Синтезированный образец КП исследован на дифференциальном сканирующем калориметре “Mettler Toledo DSC822е” в атмосфере азота в диапазоне температур от 30 до 500°С со скоростью нагрева 5°С в минуту. Полученные диаграммы ДСК обрабатывали при помощи программы “STARe”.
По данным дифференциальной сканирующей калориметрии на термограмме наблюдаются два эффекта: первый – эндотермический, связанный с внутримолекулярным отщеплением молекулы СО2 от протонированного КП; второй эффект – экзотермический, протекающий с выделением тепла реакции раскрытия циклопропаного цикла, обладающего повышенной энергией напряжения и высокой реакционной способностью. КП имеет высокую молекулярную массу 614.6, что не позволяет определить температуру его плавления на специальных приборах. Температура плавления совпадает с началом разложения образца при 354°C и окончанием эндо-эффекта при 370°C (максимум пика при 367.6°C). Экзотермический эффект начинается при 371°C и заканчивается при 379°C ( максимум – 373°C). На первой ступени деструкции масса образца уменьшается на 10.7, а на второй – на 25.3 мас. %. Приведнные результаты свидетествуют о том, что КП является чрезвычайно термостойким соединением.
Для подтверждения структуры КП снимают спектр ПМР в диметилсульфоксиде на приборе Bruker AVACE AV300 с рабочей частотой 300 МГц. В спектре 1Н-ЯМР КП обнаружены сигналы протонов 1',3',4'-тризамещенного и 1,2,4,5-тетразамещенного бензольных колец и гидроксильной группы. В ПМР-спектре регистрируются сигналы протонов метильной группы (δ = 2.05 м.д.; s; 12Н), ароматического кольца (δ = 8.20 м.д.; s; Н‑4,8; 2Н; δ = 7.00 м.д.; s; Н-2', Н-2''; 4Н; δ = 6.90 м.д.; s; Н‑6',6''; 4Н; δ = 6.67 м.д.; s; Н-5',5''; 4Н) и гидроксильной группы (δ = 9.60 м.д.; s; 4Н). Равноценность влияния заместителей на протоны 1,2,4,5-тетразамещенного бензольного кольца о-крезолпиромеллитина приводит к эквивалентности протонов у атомов углерода С4 и С8, что соответствует одному сигналу этих протонов в ПМР-спектре [7].
Интервалы перехода рН и окраску растворов в присутствии индикаторов определяли кислотно-щелочным титрованием при постоянном измерении значения pH-метром. Навеску 0.01 г КП растворяли в 50 мл дистиллированной воды и титровали 0.1 М раствором КОН до изменения окраски раствора. Навеску 0.05 г КФ растворяли в 50 мл 60% спирта, т.к. в дистиллированной воде он не растворяется. Интервал перехода КП по сравнению с КФ смещен в кислую область и находится в нейтральных значениях рН, а проявляемая окраска КП соответствует окраске фталеинов аналогичного строения (табл. 5).
Таблица 5.
Результаты кислотно-основного титрования
| Соединение | Концентрация, мас. % | Растворитель | Интервал перехода рН и окраска индикатора |
|---|---|---|---|
| КП | 0.02 | Вода | 5.7–7.3 бесцветная–фиолетовая |
| КФ | 0.05 | 60%-ный спирт | 8.2–9.8 бесцветная–фиолетовая |
КП обладает повышенной гидрофильностью и хорошо растворяется в нейтральных и кислых водных растворах, что позволяет рекомендовать его в качестве химического индикатора в промысловых трассерных исследованиях нефтяных пластов [8]. Этот вид промысловых исследований является одним из наиболее информативных методов позволяющих контролировать процесс заводнения месторождения. Он позволяет получить качественную и достоверную информацию в режиме реального времени, изучить и дать количественную оценку фильтрационно-емкостным свойствам пород-коллекторов на тот или иной период разработки месторождения. Основное требование, предъявляемое к органическим индикаторам заключается в хорошей растворимости их в воде и полной нерастворимости в нефти. Низкий предел определения КП в водных растворах определяет небольшую минимально необходимую массу закачиваемого в нагнетающую скважину индикатора.
ВЫВОДЫ
1. Экспериментально подобраны оптимальные условия реакции ацилирования о-крезола ПМДА в расплаве о-крезола при 120°С в течение 30 ч при следующем молярном соотношении реагентов: ПМДА : о-крезол : SnCI4 = 1.0 : 4.1 : 2.0. Выход 3,3,7,7-тетракис(3-метил-4-оксифенил)пиромеллитида составляет 62.0% от теоретического.
2. Строение целевого продукта синтеза доказано физико-химическими методами: элементным анализом, ТСХ, УФ-, ИК и ПМР-спектроскопией, ДСК, ВЭЖХ/МС.
3. Изучение свойств 3,3,7,7-тетракис(3-метил-4-оксифенил)пиромеллитида позволяет рекомендовать его в качестве химического индикатора в промысловых трассерных исследованиях нефтяных пластов.
Список литературы
Nekhoroshev S.V., Nekhoroshev V.P., Poleshchuk O.K. // Rus. J. of Appl. Chem. 2015. V. 88. № 4. P. 711.
Нехорошев С.В., Петрова Ю.Ю., Нифантьев И.Э. // Патент РФ № 2497860 / Б.И. 2013. № 31. С. 38.
Nekhoroshev S.V., Nekhoroshev V.P., Turov Yu.P. J. of Anal. Chem. 2010. Vol. 65. № 10. P. 988.
Сергеев В.А., Шитиков В.К., Ена А.Б., Бравкова Р.Н. // Ж. орг. химии. 1984. Т. 20. Вып. 6. С. 1284.
Гаспарян А.К., Нехорошев С.В., Князев А.С. // Орбиталь. 2017. № 1. С. 15.
Преч Э., Бюльманн Ф., Аффольтер К. Определение строения органических соединений. М.: Мир, 2006. 438 с.
Соколовский Э.В., Соловьев Г.Б., Тренчиков Ю.И. Индикаторные методы изучения нефтегазоносных пластов. М.: Недра, 1986. 157 с.
Воловенко Ю.М., Карцев В.Г., Комаров И.В., Туров А.В., Хиля В.П. Спектроскопия ядерного магнитного резонанса для химиков. М.: МБФНП, 2011. 704 с.
Дополнительные материалы отсутствуют.