

Нефтехимия, 2020, T. 60, № 1, стр. 61-75
Каталитические системы процесса селективной тримеризации этилена для получения 1-гексена
Д. Н. Чередилин 1, *, А. М. Шелоумов 1, А. А. Сенин 1, Г. А. Козлова 1, В. В. Афанасьев 1, Н. Б. Беспалова 1
1 Общество с ограниченной ответственностью “Объединенный центр исследований и разработок” (ООО “РН-ЦИР”)
119333 Москва, Россия
* E-mail: CheredilinDN@rn-rdc.ru
Поступила в редакцию 14.12.2018
После доработки 29.07.2019
Принята к публикации 09.09.2019
Аннотация
Реакцией гексагидрата хрома(III) и дифосфинов в ацетоне получены новые комплексы хрома с дифосфиновыми лигандами. Строение комплекса хрома(III) (4), имеющего в лигандном окружении молекулу воды и1,2-бис(дифенилфосфино)бензол, а также двух комплексов хрома(III) (12, 13) с 1,2-бис(дифенилфосфино)бензольными лигандами, содержащих алкенильные заместители в орто-положении одной из фенильных групп у атома фосфора, установлено методом рентгеноструктурного анализа. Изучены каталитические свойства полученных комплексов в составе каталитических систем тримеризации этилена. Показано, что ряд полученных каталитических систем проявляет высокую активность в процессе тримеризации этилена в 1-гексен, при этом селективность процесса превышает 94%. На примере комплекса хрома 13, имеющего в орто-положении одной из фенильных групп 2-метилпроп-1-енильный фрагмент, достигнуто наивысшее среди известных селективных каталитических систем тримеризации значение производительности. Определено влияние температуры, давления, а также природы используемого в составе каталитической системы растворителя на показатели процесса тримеризации этилена в 1-гексен.
Среди разработанных процессов олигомеризации этилена в линейные альфа-олефины особое место занимают селективные процессы получения 1-гексена и более тяжелых олефинов. Несмотря на значительные успехи в этом направлении, имеется немного примеров каталитических систем, обеспечивающих наряду с высокой селективностью по целевому продукту значительную производительность процесса и низкое количество побочно образующегося полимера [1–3].
Ранее нами было показано, что использование структурно модифицированных дифосфиновых лигандов в составе каталитической системы олигомеризации этилена позволяет селективно получать 1-гексен [4, 5]. Формулы модифицированных дифосфиновых лигандов в составе каталитической системы приводятся ниже по мере упоминания.
Так, раствор комплекса хрома 1 с функциональной СH2OEt- группой в орто-положении одного из фенильных заместителей у атома фосфора, активированного смесью метилалюмоксана (МАО) и триалкилалюминия, эффективно катализирует процесс тримеризации этилена. При этом при производительности процесса 33.5 кг/гCr · ч достигается селективность по 1-гексену 92.0 мас. % [4], а при оптимизации условий проведения процесса удается достичь селективности по 1-гексену 96.4 мас. % при производительности 57.9 кг/гCr · ч [5] (рис. 1).
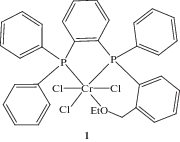
Достигнутое значение селективности по целевому продукту является достаточно высоким, однако показатель производительности по сравнению с известным процессом, разработанным компанией BP Chemicals в 2002 г. и использующей в составе каталитической системы лиганды PNP-типа (1033 кг/гCr · ч [6, 7]), более чем на порядок ниже.
В настоящем исследовании с целью повышения производительности процесса тримеризации этилена в 1-гексен нами был расширен круг используемых комплексов хрома, изучено влияние их структурных особенностей на каталитическую активность системы. Для наиболее активных каталитических систем проведена оптимизация условий проведения процесса олигомеризации: подобраны оптимальная рабочая температура и давление, изучено влияние природы растворителя на показатели процесса селективной олигомеризации этилена. Методы синтеза исходных соединений, лигандов и комплексов хрома приводятся после обсуждения результатов.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
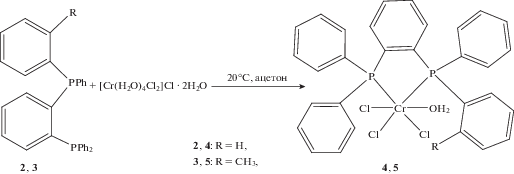
На первом этапе был значительно упрощен метод синтеза дифосфиновых комплексов хрома. Ранее синтез комплексов хрома осуществлялся реакцией трихлорида трис(тетрагидрофуран)хрома(III), CrCl3(THF)3 с соответствующими дифосфиновыми лигандами [8, 9]. Исходный металлирующий агент CrCl3(THF)3 нестабилен при длительном хранении и имеет высокую стоимость. Нами была разработана методика, позволяющая использовать в реакции синтеза дифосфиновых комплексов в качестве источника хрома доступный реагент – гексагидрат хлорида хрома(III), [Cr(H2O)4Cl2]Cl · 2H2O. Взаимодействие дифосфиновых лигандов с гексагидратом хлорида хрома(III) в ацетоне (24 ч перемешивания реакционной массы при комнатной температуре), приводит к образованию дифосфиновых комплексов хрома с выходом более 90%. Образующиеся комплексы хрома малорастворимы в среде ацетона и в процессе реакции выпадают в виде синего мелкокристаллического осадка, что облегчает процедуру их выделения и очистки. Методика была отработана при получении комплексов 4 и 5 на основе 1,2-бис(дифенилфосфино)бензола(dppb) (2) и [(2-дифенилфосфино)фенил][2-(метил)фенил]фенилфосфина (3) (рис. 2).
Строение соединения 4 было установлено рентгеноструктурным исследованием монокристалла (рис. 3). Комплекс 4 имеет искаженную октаэдрическую конфигурацию. Атом хрома содержит координированную молекулу H2O, находящуюся в транс-положении к одному из атомов хлора. Источником координационно связанной воды является используемый в реакции получения 4 гексагидрат хлорида хрома (III), [Cr(H2O)4Cl2]Cl ⋅ ⋅ 2H2O.
Рис. 3.
Молекулярная структура комплекса: [2‑(С6H5)2P(С6H4)P(С6H5)2]CrCl3(H2O) (4). Атомы водорода опущены для ясности рисунка.

Лигандное окружение атома хрома в комплексе 4, (2)CrCl3(Н2О), отличается от ранее установленного для структуры (dppb)CrCl3(THF) (6) [9], полученного в реакции 1,2-бис(дифенилфосфино)бензола с CrCl3(THF)3. В комплексе (dp-pb)CrCl3(THF) в качестве лабильного лиганда в координационной сфере атома хрома присутствует молекула тетрагидрофурана (THF), источником которой является исходный комплекс CrCl3(THF)3. Как показало исследование каталитических свойств системы олигомеризации этилена на основе комплекса 4, данная структурная модификация не влияет на производительность и селективность процесса. Результаты испытаний каталитических свойств систем на основе комплексов 4, 5, а также комплекса (dppb)CrCl3(THF) представлены в табл. 1.
Таблица 1.
Результаты испытаний каталитических систем на основе комплексов хрома 4, 5 и 6 в реакции олигомеризации этилена
| Комплекс | Производи-тельность, кг/гCr · ч | Селективность, мас. % | |||||
|---|---|---|---|---|---|---|---|
| полимер | С4 | С6 | С8 | С10+ | 1-С6 в С6-фр. | ||
| 4 | 210 | 3.8 | 0.4 | 52.8 | 27.3 | 15.7 | 88.1 |
| 5 | 450 | 0.5 | 0.4 | 84.8 | 6.9 | 7.4 | 98.7 |
| 6 | 220 | 0.4 | 6.3 | 50.3 | 29.7 | 13.3 | 86.9 |
Условия: загрузка комплексов хрома 1.9 × 10–3ммоль (концентрация в растворе 7.4 × 10–5 моль/л), растворитель метилциклогексан (25.0 мл), концентрация активатора (10% раствор метилалюмоксана в толуоле) 4.7 × 10–2 моль/л (загрузка – 1.2 ммоль), мольное соотношение [Cr] : [МАО] 1 : 630, температура 85°С, давление 20 бар, длительность эксперимента 0.5 ч.
Полученные результаты показывают, что замена лабильного лиганда в координационной сфере атома хрома (в данном случае THF на Н2О), не оказывает существенного влияния на каталитическую активность систем на основе комплексов с 1,2-бис(дифенилфосфино)бензольным лигандом. По-видимому, обработка обоих комплексов (4 и 6) раствором метилалюмоксана приводит к образованию одного и того же активированного комплекса. Ранее было показано [4–6], что введение функциональной группы, способной к координации с атомом хрома, в орто-положение одного из фенильных заместителей у атома фосфора в дифосфиновом лиганде, способствует протеканию процесса тримеризации этилена в 1-гексен. В случае комплекса 5 вводимая метильная группа не является координирующейся атомом металла, тем не менее, использование каталитической системы, включающей данный комплекс хрома, позволяет достичь повышенной селективности процесса олигомеризации этилена с одновременным увеличением производительности. Еще более значительное повышение производительности процесса олигомеризации этилена при использовании каталитической системы на основе комплекса 5, до 726 кг/гCr · ч, удается получить при рабочем давлении 40 бар. Селективность процесса по 1-гексену при этом практически не изменяется и составляет 84.5 мас. %.
На основании полученных данных по использованию комплекса 5 в реакции тримеризации этилена в 1-гексен было предположено, что введение алкенильных заместителей в орто-положение одного из фенильных заместителей у атома фосфора дифосфинового лиганда позволит приблизиться к созданию высокоактивных селективных каталитических систем. Алкенильные группы имеют потенциальную возможность для координации ненасыщенного С=С-фрагмента с каталитическим центром – атомом хрома в процессе катализа, что может способствовать повышению селективности каталитической системы. С этой целью был синтезирован ряд дифосфиновых лигандов 7–11, с различными алкенильными заместителями в орто-положении фенильного кольца. Дифосфиновые лиганды 7–11 охарактеризованы методом спектроскопии ЯМР 1Н и 31Р. Для лигандов 9 и 10 с различными заместителями R1 и R2 при двойной связи углерод–углерод зафиксировано образование цис-, транс-геометрических изомеров. Далее по разработанной методике реакцией 7–11 с гексагидратом хлорида хрома(III) были синтезированы комплексы 12–16 (рис. 4).

Комплекс 16, в ароматическое ядро которого были введены длинноцепочечные алкильные группы С10Н21, обладает хорошей растворимостью в насыщенных углеводородах, в том числе в метилциклогексане. Согласно литературным данным, улучшение растворимости предшественника катализатора может способствовать повышению активности формируемой каталитической системы. Так, производительность процесса тримеризации этилена с участием комплекса хрома на основе PNP-лиганда с алкильным С18-заместителем при атоме азота в 6.5 раз выше, чем при использовании незамещенного лиганда при одинаковых условиях их активации и использования в процессе [10].
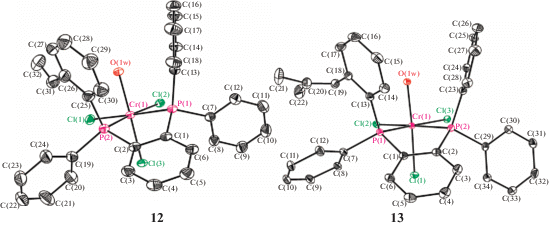
Строение соединений 12 и 13 было однозначно подтверждено данными рентгеноструктурного анализа (рис. 5). Как показало рентгеноструктурное исследование, комплексы 12 и 13 имеют искаженную октаэдрическую конфигурацию. Ориентация алкенильных фрагментов комплексов в кристалле исключает их взаимодействие с атомом хрома: алкенильные группы ориентированы относительно фенильного цикла в сторону от атома металла. Атом хрома, так же как и в структуре 4, содержит координированную молекулу H2O, находящуюся в транс-положении к одному из атомов хлора.
Рис. 5.
Молекулярная структура комплексов: [2-(С6H5)2P(С6H4)(2-(CH2=CH)С6H4)(С6H5)P]CrCl3(H2O) (12) и [2‑(С6H5)2P(С6H4)(2-(CH2=C(СН3)2)С6H4)(С6H5)P]CrCl3(H2O) (13). Атомы водорода опущены для ясности рисунка.
Отсутствие координации введенной функциональной группы у атома хрома давало основание предполагать, что применение комплексов данного типа в составе каталитической системы тримеризации этилена не позволит достичь высокой селективности процесса. Однако проведенные эксперименты по олигомеризации этилена с использованием полученных комплексов 12–16 не подтвердили это предположение. Экспериментальные данные приведены в табл. 2.
Таблица 2.
Результаты испытаний каталитических систем на основе комплексов хрома 12–16 в реакции олигомеризации этилена
| Комплекс | Т, оС | Давление, бар | Производи-тельность, кг/гCr · ч | Селективность, мас. % | |||||
|---|---|---|---|---|---|---|---|---|---|
| полимер | С4 | С6 | С8 | С10+ | 1-С6 в С6-фр. | ||||
| 12 | 85 | 40 | 640 | 0.2 | 0.3 | 91.7 | 5.6 | 2.2 | 99.5 |
| 13 | 85 | 40 | 1890 | <0.1 | 0.1 | 94.0 | 3.2 | 2.7 | 99.7 |
| 14 | 85 | 40 | 280 | 2.5 | 0.9 | 89.4 | 4.5 | 2.7 | 99.0 |
| 15 | 60 | 30 | 510 | 0.4 | 2.8 | 87.8 | 7.0 | 2.0 | 99.0 |
| 16 | 70 | 30 | 650 | 2.3 | 1.2 | 79.7 | 5.7 | 11.1 | 99.7 |
Условия: загрузка комплексов хрома 9 × 10–5 ммоль (концентрация в растворе 3.6 × 10–6 моль/л), растворитель метилциклогексан (25.0 мл), активаторы: ТМА (1.0 М раствор в метилциклогексане), МАО (0.085 М раствор в толуоле), мольное соотношение [Cr] : [МАО] : [ТМА] 1 : 300 : 1100, длительность эксперимента 0.5 ч.
Активацию комплексов осуществляли при помощи смеси растворов метилалюмоксана и триметилалюминия (ТМА). Ранее для модельных систем на основе дифосфиновых комплексов было показано, что использование смеси метилалюмоксана и триалкилалюминия в подобранном соотношении вместо индивидуального раствора метилалюмоксана способно повышать производительность процесса [11]. Аналогичное мольное соотношение комплексов хрома и активаторов было использовано при приготовлении каталитических систем на основе комплексов 12–16. Все полученные каталитические системы позволяют получать 1-гексен из этилена с высокой селективностью. Чистота получаемой С6-фракции по 1-гексену также является высокой и составляет 99.0–99.7%. Другими важными критериями эффективности каталитической системы тримеризации этилена в 1-гексен являются производительность процесса и количество побочно образующегося полимера. По этим двум показателям выделяются системы на основе комплексов 12 и 13, процесс тримеризации с их использованием проходит с селективностью более 90%. Каталитическая система на основе комплекса 16 имеет повышенную растворимость в реакционном растворе, однако это является лишь незначительным технологическим преимуществом, реализуемым при дозировании раствора комплекса хрома. Селективность процесса при использовании этой системы является самой низкой в ряду исследуемых комплексов и составляет 79.7%. Можно отметить, что при образовании высокоселективных каталитических систем тримеризации этилена с участием исследуемого ряда дифосфиновых комплексов хрома ключевую роль играют стерические факторы. Определяющее значение имеет пространственное расположение заместителей при двойной связи, способных экранировать каталитический центр и подход к нему молекул субстрата. Пространственное влияние оказывают также и заместители, более удаленные от активного центра – атома хрома, как в случае комплекса 16, алкильные С10Н21-группы в центральном ароматическом ядре. Тем не менее, нельзя исключить, что в растворе, в активированном каталитически активном комплексе имеет место взаимодействие ненасыщенного фрагмента с атомом металла, которое наряду со стерическими факторами заместителей влияет на селективность процесса тримеризации.
Для полученных высокоселективных каталитических систем тримеризации этилена в 1-гексен на основе 12 и 13 был проведен выбор оптимальных параметров процесса, таких как температура, давление, влияние природы растворителя на активность катализаторов. Температура реакции является основным фактором, определяющим стабильность каталитической системы и эффективность ее работы. Нами был изучен характер изменений показателей процесса тримеризации этилена при варьировании температуры в интервале от 30 до 140°С при постоянном давлении этилена 40 бар. Полученные экспериментальные данные приведены в табл. 3.
Таблица 3.
Влияние температуры на показатели процесса тримеризации этилена для каталитических систем на основе комплексов 12 и 13
| Т, °С | Производи-тельность, кг/гCr · ч | Селективность, мас. % | |||||
|---|---|---|---|---|---|---|---|
| полимер | С4 | С6 | С8 | С10+ | 1-С6 в С6-фр. | ||
| Каталитическая система на основе комплекса 12 | |||||||
| 41 | 690 | 1.0 | 0.2 | 72.9 | 21.7 | 4.2 | 98.6 |
| 50 | 1080 | 0.7 | 0.3 | 80.7 | 15.0 | 3.4 | 98.7 |
| 60 | 1630 | 0.2 | 0.1 | 86.0 | 10.4 | 3.3 | 98.8 |
| 70 | 1110 | 0.3 | 0.5 | 88.7 | 7.7 | 2.7 | 98.9 |
| 80 | 890 | 0.1 | 0.2 | 90.9 | 6.6 | 2.3 | 99.1 |
| 90 | 400 | 0.2 | 0.4 | 92.5 | 4.7 | 2.2 | 99.6 |
| 100 | 370 | 0.2 | 0.5 | 92.3 | 5.2 | 1.8 | 99.0 |
| 115 | 100 | 5.3 | 0.7 | 78.6 | 5.6 | 9.9 | 97.7 |
| 140 | 50 | 25.4 | 4.2 | 33.2 | 4.1 | 33.2 | 96.3 |
| Каталитическая система на основе комплекса 13 | |||||||
| 30 | 1750 | 0.1 | 0.2 | 74.0 | 21.2 | 4.5 | 98.9 |
| 50 | 2130 | 0.1 | 0.1 | 87.2 | 9.6 | 3.0 | 99.5 |
| 60 | 2510 | 0.1 | 0.1 | 89.8 | 7.0 | 3.0 | 99.7 |
| 70 | 2830 | <0.1 | 0.1 | 92.2 | 4.5 | 3.2 | 99.6 |
| 80 | 2360 | <0.1 | 0.1 | 93.3 | 3.7 | 2.9 | 99.7 |
| 90 | 1410 | <0.1 | 0.1 | 94.8 | 2.8 | 2.3 | 99.5 |
| 100 | 720 | 0.2 | 0.2 | 94.4 | 2.7 | 2.5 | 99.4 |
| 110 | 320 | 22.8 | 0.2 | 70.9 | 2.7 | 3.4 | 99.5 |
Условия: загрузка комплексов хрома 9 × 10–5 ммоль (концентрация в растворе 3.6 × 10–6 моль/л), растворитель метилциклогексан (25.0 мл), активаторы: ТМА (1.0 М раствор в метилциклогексане), МАО (0.085 М раствор в толуоле), мольное соотношение [Cr] : ТМА : МАО 1 : 1100 : 300, давление 40 бар, длительность эксперимента 0.5 ч.
Испытания каталитических систем на основе комплексов 12 и 13 при различных температурах продемонстрировали, что наибольшая производительность в процессах селективной тримеризации этилена в 1-гексен с их использованием достигается в интервале рабочих температур 50–80°С. Температурный интервал максимальной селективности процесса по 1-гексену сдвинут в сторону более высоких температур: 80–100°С. Дальнейшее повышение температуры до 140○С приводит к заметному снижению производительности катализаторов и селективности процесса, поскольку более жесткие термические условия способствуют протеканию побочных реакций образования высших олефинов С10+ и полимера. Следует отметить, что снижение температуры проведения процесса до 30–40°С приводит к росту продукта тетрамеризации этилена – 1-октена, содержание которого достигает 21%. При этом каталитические системы позволяют проводить процесс олигомеризации с достаточно высокой производительностью.
Характер изменений показателей процесса тримеризации этилена в 1-гексен в интервале давлений от 10 до 60 бар на примере использования каталитических систем на основе комплексов 12 и 13 можно проследить по экспериментальным данным, представленным в табл. 4.
Таблица 4.
Влияние давления этилена на показатели процесса селективной тримеризации этилена в 1-гексен для каталитических систем на основе комплексов 12 и 13
| Давление, бар | Производи-тельность, кг/гCr · ч | Селективность, мас. % | |||||
|---|---|---|---|---|---|---|---|
| полимер | С4 | С6 | С8 | С10+ | 1-С6 в С6-фр. | ||
| Каталитическая система на основе комплекса 12 | |||||||
| 10 | 40 | 1.0 | 1.8 | 89.7 | 2.4 | 5.0 | 97.9 |
| 20 | 210 | 0.4 | 0.5 | 92.4 | 2.9 | 3.8 | 98.4 |
| 30 | 290 | 0.8 | 0.9 | 91.0 | 3.9 | 3.4 | 99.0 |
| 40 | 400 | 0.2 | 0.4 | 92.5 | 4.7 | 2.2 | 99.6 |
| 50 | 580 | 0.3 | 0.3 | 90.3 | 6.6 | 2.5 | 99.4 |
| 60 | 850 | 9.0 | 0.6 | 80.0 | 4.6 | 5.8 | 99.3 |
| Каталитическая система на основе комплекса 13 | |||||||
| 10 | 130 | <0.1 | 0.5 | 94.3 | 2.8 | 2.4 | 99.2 |
| 20 | 400 | <0.1 | 0.3 | 94.8 | 2.7 | 2.2 | 99.4 |
| 30 | 1020 | <0.1 | 0.2 | 94.8 | 2.5 | 2.5 | 99.5 |
| 40 | 1410 | <0.1 | 0.1 | 94.8 | 2.8 | 2.3 | 99.5 |
| 60 | 1810 | 3.4 | 0.5 | 89.8 | 3.9 | 2.4 | 99.1 |
Условия: загрузка комплексов хрома 9 × 10–5 ммоль (концентрация в растворе 3.6 × 10–6 моль/л), растворитель метилциклогексан (25.0 мл), активаторы: ТМА (1.0 М раствор в метилциклогексане), МАО (0.085 М раствор в толуоле), мольное соотношение [Cr] : ТМА : МАО 1 : 1100 : 300, температура 90°С, длительность эксперимента 0.5 ч.
Производительность процесса тримеризации этилена монотонно растет с увеличением давления. При этом селективность по фракции С6 практически не изменяется, существенное ее снижение наблюдается только при давлении выше 40 бар. В отличие от температуры, давление незначительно влияет на селективность процесса. Также видно, что отсутствует четкая зависимость количества образующегося полимера от давления, в то же время с его ростом наблюдается увеличение количества С8-олефинов. При давлении 60 бар и более становятся заметными процессы полимеризации.
На примере каталитической системы на основе комплекса 12 было изучено влияние природы растворителя на показатели процесса тримеризации. Для этого нами был проведен процесс селективной тримеризации в 5 различных растворителях: метилциклогексане, изооктане, толуоле, о-ксилоле и декагидронафталине (декалине). Результаты экспериментов представлены в табл. 5.
Таблица 5.
Влияние природы растворителей на показатели процесса тримеризации при использовании каталитической системы на основе комплекса 12
| Растворитель | Производи-тельность, кг/гCr · ч | Селективность, мас. % | |||||
|---|---|---|---|---|---|---|---|
| полимер | С4 | С6 | С8 | С10+ | 1-С6 в С6-фр. | ||
| Метилциклогексан | 430 | <0.1 | 0.5 | 94.0 | 3.4 | 2.1 | 99.2 |
| Изооктан | 100 | 0.2 | 2.0 | 86.9 | 8.0 | 2.9 | 97.3 |
| Декалин | 20 | 19.1 | 13.0 | 53.9 | 10.9 | 3.1 | 95.6 |
| Толуол | 10 | 0.1 | 38.3 | 11.1 | 0.1 | 50.4 | 35.0 |
| о-Ксилол | 10 | 1.3 | 28.4 | 14.0 | 0.5 | 55.8 | 53.1 |
Условия: загрузка комплекса хрома 12 9 × 10–5 ммоль (концентрация в растворе 3.6 × 10–6 моль/л), растворитель (25.0 мл), активаторы: ТМА (1.0 М раствор в метилциклогексане), МАО (0.085 М раствор в смеси метилциклогексан/толуол), мольное соотношение [Cr] : ТМА : МАО 1 : 1100 : 300, температура 90°С, давление этилена 40 бар, длительность эксперимента 0.5 ч.
Полученные данные свидетельствуют об исключительно эффективном протекании реакции тримеризации этилена в 1-гексен в метилциклогексане по сравнению с реакциями в толуоле и о-ксилоле. Возможная причина падения производительности и селективности процесса при проведении реакции в ароматических растворителях – взаимодействие активированного комплекса с ароматическими углеводородами с образованием новых комплексов хрома с η6-координированным ароматическим кольцом молекулы растворителя, которые не способны катализировать селективный процесс получения 1-гексена [3]. Основные продукты, образующиеся при использовании толуола и о-ксилола – набор линейных альфа-олефинов и линейных алканов согласно статистическому распределению Шульца–Флори.
По мере протекания процесса тримеризации изменяется качественный состав реакционного раствора, в частности, увеличивается концентрация 1-гексена с 0 до 14 мас. %. Было исследовано влияние концентрации 1-гексена на показатели процесса тримеризации этилена путем замены части растворителя на 1-гексен. Таким образом моделировался состав реакционной смеси при различных временах протекания реакции. Результаты экспериментов представлены в табл. 6.
Таблица 6.
Влияние содержания 1-гексена в реакционной среде на показатели процесса тримеризации этилена в 1-гексен (на примере каталитической системы на основе комплекса 13)
| Начальная концентрация 1-гексена, мас. % | Производи-тельность, кг/гCr · ч | Селективность, масс. % | |||||
|---|---|---|---|---|---|---|---|
| полимер | С4 | С6 | С8 | С10+ | 1-С6 в С6-фр. | ||
| 0.0 | 1410 | <0.1 | 0.1 | 94.8 | 2.8 | 2.3 | 99.5 |
| 1.6 | 1205 | <0.1 | 0.2 | 94.0 | 3.3 | 2.5 | 99.4 |
| 4.3 | 896 | 0.1 | 0.2 | 93.0 | 3.8 | 2.9 | 99.5 |
| 6.9 | 530 | 0.1 | 0.3 | 90.9 | 4.2 | 4.5 | 98.9 |
| 10.3 | 490 | 0.2 | 0.3 | 89.5 | 4.2 | 5.8 | 99.3 |
| 13.4 | 570 | 0.5 | 0.3 | 86.5 | 4.9 | 7.8 | 98.0 |
Условия: загрузка комплекса хрома 9 × 10–5 ммоль (концентрация в растворе 3.6 × 10–6 моль/л), общий объем растворителя 25.0 мл, активаторы: ТМА (1.0 М раствор в метилциклогексане), МАО (0.085 М раствор в толуоле), мольное соотношение [Cr] : ТМА : МАО 1 : 1100 : 300, температура 90°С, давление 40 бар, длительность эксперимента 0.5 ч.
Полученные данные свидетельствуют о снижении производительности процесса тримеризации с увеличением концентрации 1-гексена в растворителе. Причиной данного факта служит падение активности каталитической системы тримеризации этилена в 1-гексен из-за конкурентных равновесных процессов координации этилена и 1-гексена у атома металла. Молекула 1-гексена является более электронодонорной по сравнению с молекулой этилена и способна образовывать с атомом хрома более прочную связь, вследствие этого повышение концентрации α-олефина способствует блокированию активных центров катализатора. Производительность процесса тримеризации этилена в 1-гексен максимальна в начальный период времени реакции, когда концентрация α-олефинов в реакционной массе минимальна. По мере увеличения концентрации образующихся олефинов, главным образом 1-гексена, эффективность работы каталитической системы, а вместе с ней и производительность процесса, снижается, процесс становится менее селективным по целевому продукту.
Таким образом, введение орто-заместителя в дифосфиновый лиганд хромового комплекса каталитической системы олигомеризации этилена способно не только повысить селективность процесса по 1‑гексену выше 90%, но и значительно увеличить производительность. При этом активация комплекса хрома 13, содержащего 2-метилпроп-1-енильный фрагмент, приводит к образованию высокоселективной каталитической системы тримеризации этилена, демонстрирующей производительность выше 2800 кг/гCr · ч (145 600 кг (олефинов)/мольCr · ч), что в несколько раз превышает аналогичный показатель для наиболее активной до настоящего времени каталитической системы тримеризации этилена на основе комплекса хрома с N-фосфиноамидиновым лигандом компании Chevron Phillips (54 670 кг (олефинов)/мольCr · ч) [12].
Резюмируя полученные результаты, можно сделать следующие выводы. Синтезирован и охарактеризован ряд новых комплексов хрома с дифосфиновыми лигандами, содержащими алкенильные заместители. Для синтеза была применена новая методика с использованием в качестве исходного соединения хрома гексагидрата хлорида хрома(III). Полученные комплексы являются основой каталитических систем селективной тримеризации этилена в 1-гексен, позволяющих проводить процесс тримеризации этилена с селективностью более 90%. Для этих систем подобран оптимальный режим проведения процесса тримеризации этилена: температура 60–90°С, давление 40 бар. Изучено влияние природы растворителя и содержания 1-гексена в реакционной среде на показатели процесса селективной олигомеризации этилена. Показано, что для изученных каталитических систем тримеризации этилена на основе дифосфиновых комплексов хрома активность и селективность по 1-гексену могут направленно регулироваться путем варьирования заместителей при двойной связи алкенильного фрагмента. Такой подход может быть применен для эффективного поиска и создания новых катализаторов селективного получения альфа-олефинов на основе металлокомплексов с полидентатными лигандами. Наиболее эффективным в настоящем исследовании оказалось использование в составе каталитической системы тримеризаци этилена комплекса хрома, содержащего 2-ме-тилпроп-1-енильный орто-заместитель в фенильном радикале дифосфинового лиганда. При этом удалось одновременно достичь решения нескольких задач: высокой селективности процесса, наивысшей производительности, минимизации образования полимера.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Используемые методы и приборы
Работы по синтезу соединений и приготовлению каталитических систем проводили в инертной атмосфере с использованием стандартной техники Шленка. В работе использовали этилен (фирма Linde Gas, 99.9 об. %), который пропускали через три последовательно соединенных колонки, заполненные активированным углем и цеолитами (3А и 13Х), и аргон высокой чистоты (ОАО Московский газоперерабатывающий завод, 99.998 об. %). Дополнительную очистку аргона проводили пропусканием через три последовательно соединенные колонки, заполненные цеолитами (3А и 13Х), CuО (восстановленный до Сu) и финишную очистку CE35KF фирмы Entegris. Это обеспечивает остаточное содержание кислорода, воды, СО и др. на уровне млрд–1. Растворители абсолютировали кипячением над подходящими осушающими реагентами с последующей перегонкой в токе аргона. Коммерческие доступные реактивы (чистотой более 97%) применяли без дополнительной очистки, за исключением дифенилфосфинхлорида (Aldrich 95%), который был перегнан в вакууме (Tкип = 124–126°С/3 мм рт. ст.). Метилалюмоксан (10%-ный раствор в толуоле, Sigma-Aldrich) перед проведением экспериментов анализировали согласно методике, описанной в работе [13]. изо-PrMgCl-LiCl был получен согласно методу, описанному в [14]. N,N-Диэтиламинофенилхлорфосфин (PhP(Cl)NEt2) был получен согласно патенту [15]. Комплексы хрома и дифосфиновые лиганды синтезировали согласно методикам, аналогичным описанным в патенте [16]. В работе использовался 1,2-бис(дифенилфосфино)бензол 2 фирмы Sigma-Aldrich чистотой 97%. Исходные соединения для синтеза дифосфиновых лигандов 14 и 15, 1-(1-пентенил)-2-бромбензол и 1-(2-фенилэтенил)-2-бромбензол, были получены в соответствии с модифицированным методом [17]. Исходное соединение для синтеза лиганда 16 – 1,2-дидецилбензол, синтезировали из 1,2-дихлорбензола и децилбромида согласно описанным в литературе методам [18–20]. Синтезы 1-бром-2-дифенилфосфинобензола и 1-бром-2-дифенилфосфино-4,5-дидецилбензола проводили, используя модифицированные методики [14, 21]. Синтезы ((2-дифенилфосфин)фенил)фенилфосфинхлорида и (2-дифенилфосфин)-4,5-дидецилфенил(фенил)фосфинхлорида проводили согласно модифицированной методике [21]. Чистоту полученных соединений определяли методами спектроскопии ЯМР 1H и 31P{1H} и масс-спектрометрии.
Спектры ЯМР 1H и 31P{1H} регистрировали на приборах BrukerAM-300, Bruker Avance-400, используя тетраметилсилан как внутренний стандарт и 85% H3PO4 как внешний стандарт, соответственно. Масс-спектры регистрировали на приборе Agilent Technologies 6890, Network Mass Selective Detector 5973. Элементный анализ выполняли в Аналитической лаборатории ООО “РН-ЦИР”.
Общая методика испытаний каталитических систем в процессе олигомеризации этилена
Общая методика испытаний, а также методика анализа жидкой фазы реакционной смеси, содержащей продукты тримеризации этилена, приведены в работе [5].
Общая схема синтеза дифосфиновых лигандов и комплексов хрома:

Рис. 6. Общая схема синтеза лигандов и комплексов хрома.
Синтез 1,2-дидецилбензола. К охлажденной до –5°С суспензии 0.3 г (0.49 ммоль) Ni(dppe)Cl2 в растворе 9.4 мл (83 ммоль) 1,2-дихлорбензола в 50 мл диэтилового эфира в токе аргона добавили по каплям 80 мл 2.4 М свежеприготовленного раствора децилмагнийбромида (2.3-кратный мольный избыток). Далее реакционную массу нагревали при перемешивании до кипения, выпадал обильный белый осадок. Смесь кипятили в течение 3 ч, затем охладили ее до 0°С, добавили к ней 50 мл 1 М раствора соляной кислоты и разделили на фракции. Отделили органическую фазу, высушили над безводным сульфатом натрия, упарили. Кубовый остаток очистили на колонке с силикагелем (Acros, 60 − 200 µm, элюент – диэтиловый эфир). После упаривания получили 28 г бесцветной маслянистой жидкости. Выход – 94%. Спек-тр ЯМР 1H (400 МГц, C6D6): δ (м. д.) 7.17 (м, 2H, Ar–H), 7.13 (м, 2H, Ar–H), 2.82 (т, 4Н, CH2), 1.71 (м, 4Н, CH2), 1.37 (м, 28Н, CH2), 0.99 (м, 6Н, CH3).
Синтез 1,2-дибром-4,5-дидецилбензола. В колбу Шленка поместили 20.3 г (56.8 ммоль) 1,2-дидецилбензола, добавили 150 мл перегнанного дихлорометана и 0.754 г (2.8 ммоль) кристаллического иода. Полученный раствор охладили до –2°С и добавили к нему по каплям 20.1 г (125.9 ммоль) брома при перемешивании, поддерживая заданную температуру. После добавления всего количества брома дают смеси нагреться до комнатной температуры и перемешивают ее 20 часов. К полученному раствору добавляют раствор гидросульфита натрия, перемешивают, отделяют органический слой, обрабатывают раствором гидрокарбоната натрия, перемешивают, дают смеси отстояться до прекращения выделения газа. Отделяют желто-коричневый органический слой, промывают водой и раствором хлорида натрия. Органическую фазу высушивают над безводным сульфатом натрия и затем упаривают. Жидкий остаток хроматографируют на колонке с силикагелем (Acros, 60–200 µm, элюент – гексан). Выход 1,2-дибром-4,5-дидецилбензола 70%. Спектр ЯМР 1Н (400 МГц, CDCl3): δ (м.д.) 7.25 (с, 2Н), 2.63 (т, J = 7 Гц, 4Н), 1.58 (уш т, 4Н), 1.40–1.10 (набор м, 28Н), 0.92 (т, J = 6 Гц, 6Н).
Синтезы 1-бром-2-дифенилфосфинобензола и 1-бром-2-дифенилфосфино-4,5-дидецилбензола. Раствор 5.38 г (22.8 ммоль) 1,2-дибромбензола в 12 мл тетрагидрофуранa добавили к заранее приготовленным 30 мл 1 М раствора изо-PrMgLiCl в тетрагидрофуране и перемешивали при температуре –17...–13°С. Спустя 3 ч к раствору добавили по каплям 5.12 г (23.25 ммоль) дифенилфосфинхлорида при температуре –20...–10°С. Полученный раствор перемешивали 30 мин при температуре –10°С, а затем дали ему нагреться до комнатной температуры и перемешивали еще 16 ч, затем упарили. К остатку добавили 30 мл дихлорметана и 20 мл насыщенного водного раствора хлорида аммония. Отделили органический слой, высушили его над безводным сульфатом натрия. Остаток после упаривания перекристаллизовали с помощью смеси этанол/ацетон.
Выход 1-бром-2-дифенилфосфинобензола –65.5%. Спектр ЯМР 1Н (300 МГц, CDCl3): δ (м. д.) 7.60 (м, J = 4 Гц, 1Н), 7.41–7.26 (м, J = 3 Гц, 10Н), 7.20 (м, J = 4 Гц, 2Н), 6.75 (кв, J = 4 Гц, 1Н). Спектр ЯМР 31P {1H} (121.49 МГц, CDCl3): δ ‒5.12 м.д. (с).
Для синтеза 1-бром-2-дифенилфосфино-4,5-дидецилбензола вместо 1,2-дибромбензола был взят 1,2-дибром-4,5-дидецилбензол. Выход 1-бром-2-дифенилфосфино-4,5-дидецибензола – 30%. Спектр ЯМР 1Н (400 МГц, CDCl3): δ (м.д.) 7.45–7.25 (набор м, 11Н), 6.50 (с, 1Н), 2.57 (т, J = 7 Гц, 2Н), 2.40 (т, J = 7 Гц, 2Н), 1.58 (уш т, 2Н), 1.40–1.10 (набор м, 30Н), 0.92 (т, J = 6 Гц, 6Н). Спектр ЯМР 31P {1H} (161.98 МГц, CDCl3): δ – 6.02 м.д. (с).
Синтез ((2-дифенилфосфин)фенил)фенилфосфинхлорида {P−PCl} и (2-дифенилфосфин)-4,5-дидецилфенил(фенил)фосфинхлорида {Dec2P-PCl}. К раствору 0.6 г (1.76 ммоль) 1-бромо-2-дифенилфосфинобензола в 8 мл абсолютного тетрагидрофурана при –78°С медленно добавили 1.15 мл 1.6 М раствора н-BuLi в гексане. Раствор приобрел ярко-оранжевый цвет. Реакционную смесь перемешивали 2.5 ч при –65°С, затем при этой температуре медленно добавили раствор 0.38 г диэтиламинофенилхлорофосфина в 1.5 мл абсолютного тетрагидрофурана.Оставили нагреваться смесь до комнатной температуры при перемешивании. Перемешивали смесь при этой температуре еще 18 ч, упарили растворитель, остаток растворили в 4 мл толуола, отфильтровали. Затем барботировали через полученный фильтрат красного цвета сухой HCl в течение 3 ч (HCl получали медленным прикапыванием концентрированной серной кислоты к хлориду аммония при перемешивании, осушку хлористого водорода осуществляли пропусканием образующегося газа через колонку с P2O5). Наблюдалось мгновенное выпадение белого осадка диэтиламмонийгидрохлорида и изменение окраски раствора на светло-желтую. Перемешивали смесь 18 ч в атмосфере аргона. Отфильтровали бледно-желтый раствор от белого осадка в токе аргона, упарили растворитель. Остаток (желтое масло) сушили в вакууме.
Выход ((2-дифенилфосфин)фенил)фенилфосфинхлорида {P–PCl} – 87%. Спектр ЯМР 31P{1H} (161.98 МГц, CDCl3): δ (м.д.) 73.40 (д, 3JPP = 282 Гц, P(Ph)Cl), –19.60 (д, 3JPP = 282 Гц, PPh2).
Для синтеза (2-дифенилфосфин)-4,5-дидецилфенил(фенил)фосфинхлорида {Dec2P−PCl} вместо 1-бромо-2-дифенилфосфинобензола был использован 1-бром-2-дифенилфосфино-4,5-дидецилбензол. Выход (2-дифенилфосфин)-4,5-дидецилфенил(фенил)фосфинхлорида {Dec2P‒PCl} 74%. Спектр ЯМР 31P {1H} (161.98 МГц, C6D6): δ (м.д.) 74.8 (д, 3JPP = 275 Гц, P(Ph)Cl), –20.1 (д, 3JPP = 275 Гц, PPh2).
Синтез [(2-дифенилфосфино)фенил][2-(метил)фенил]фенилфосфина (3). В колбу Шленка объемом 50 мл поместили 0.05 г (2 ммоль) магния, прокалили в вакууме, заполнили аргоном. Добавили 7 мл абсолютного тетрагидрофурана, каталитическое количество иода. Отдельно приготовили раствор 0.30 г (1.75 ммоль) 2-бромтолуола в 10 мл тетрагидрофурана, добавили несколько капель приготовленного раствора в реакционную колбу. Нагрели смесь до кипения без перемешивания, раствор постепенно обесцветился, медленно добавили оставшийся раствор 2-бромтолуола к кипящей смеси. Добавили еще 10 мл тетрагидрофурана. Кипятили раствор с обратным холодильником при перемешивании 3 ч. Декантировали полученный раствор реактива Гриньяра в токе аргона и охладили до –30°С, прибавили к нему по каплям раствор 0.59 г (1.47 ммоль) [(2-дифенилфосфин)фенил]фенилфосфинхлорида {P−PCl} в 4 мл тетрагидрофурана. Температура поднялась до –15°С. Убрали охлаждение, оставили нагреваться реакционную смесь до комнатной температуры, затем перемешивали смесь при этой температуре в течение суток. Упарили растворитель, обработали реакционную смесь насыщенным водным раствором хлорида аммония (10 мл), добавили 10 мл хлористого метилена, органический слой отделили (водный слой экстрагировали СН2Cl2), объединенную органическую фазу сушили над сульфатом натрия, растворитель упарили. Остаток несколько раз промыли безводным метиловым спиртом, получили бесцветное кристаллическое вещество, которое сушили в высоком вакууме. Выход: 0.59 г (87%). ЯМР 31P{1H} (161.98 МГц, CDCl3): δ (м.д.) –14.0 (д, 3JPP = 160 Гц, 1 P), –21.0 (д, 3JPP = 160 Гц, 1P). Спектр ЯМР 1H (400 МГц, CDCl3): δ (м.д.) 7.82–7.52 (м, 20Н, Ar–H), 7.50–7.40 (м, 2Н, Ar–H), 7.15–7.20 (м, 1Н, Ar–H), 2.72 (с, 3H, CH3–Ar).
Синтез 2-бромстирола. В колбу Шленка поместили 19.1 г (53.5 ммоль) метилтрифенилфосфоний бромида, добавили 150 мл абсолютного тетрагидрофурана. Полученную суспензию охладили до 0°С и медленно добавили к ней 33.4 мл (53.5 ммоль) 1.6 М раствора n-BuLi в гексане. Желто-оранжевую смесь перемешивали при 0°С в течение 40 мин, затем при этой температуре медленно добавили к смеси раствор 9 г (48.61 ммоль) 2-бромбензальдегида. Образуется вязкий, трудноперемешиваемый бело-желтый осадок. Смесь перемешивали при комнатной температуре 16 ч, добавили 100 мл водного раствора NaCl, дважды экстрагировали этилацетатом; объединенные органические фазы повторно промывали раствором NaCl и экстрагировали водную фазу этилацетатом. Органическую фазу сушили над сульфатом магния, растворитель упаривали. Твердый желтый остаток нанесли на колонку с силикагелем (Acros, 60−200 мкм), смесью гексан/этилацетат = 9/1 элюировали первую бесцветную фракцию, растворитель упарили. Получили 2-бромстирол в виде желтоватой жидкости (светочувствителен, хранится при охлаждении). Выход: 7.64 г (86%). Спектр ЯМР 1H (400 МГц, CDCl3): δ (м.д.) 7.58 (м, 2H, Ar-H), 7.33–7.29 (м, 1H, Ar–H), 7.17–7.07 (м, 2H, Ar–H, CH=CH2), 5.74 (дд, 1H, JHH = 17 Гц, JHH = 1 Гц, CH=CHAHB), 5.41 (дд, 1H, JHH = 11 Гц, JHH = 1 Гц, CH=CHAHB).
Синтез изопропилтрифенилфосфоний иодида. Раствор 26.20 г (100 ммоль) трифенилфосфина и 42.70 г (250 ммоль) изопропилиодида в 50 мл толуола кипятили в течение 18 ч. Выпавший белый осадок отфильтровали, промыли толуолом и сушили в вакууме. Выход 18.14 г (42%). Тпл = 195–196°C.
Синтез гексилтрифенилфосфонийиодида. Смесь 5.12 г (19.55 ммоль) трифенилфосфина PPh3, 2.81 мл (19.55 ммоль) н-гексилиодида и 60 мл толуола нагревали до кипения при перемешивании в токе аргона в течение 2.5 ч. Далее реакционную массу охладили до комнатной температуры и перемешивали в течение 16 ч. Выпавший осадок промывали 2 раза 30 мл толуола и высушивали в вакууме. Выход 3.04 г (33%). Спектр ЯМР 1H (400 МГц, CDCl3): δ (м.д.) 7.83 (м, 6H, Ar–H), 7.73 (м, 6H, Ar–H), 7.18 (м, 3Н, CH2), 3.67 (м, 2Н, CH2), 1.68 (м, 4Н, CH2), 1.26 (м, 4Н, CH2), 0.84 (т, 3Н, J = = 7 Гц, CH3). ЯМР 31P{1H} (161,98 МГц, CDCl3): δ (м.д.) 24.3 (с).
Синтез 1-бром-2-(2-метил-1-пропенил)бензола. В колбу Шленка помеcтили 18.1 г (41.87 ммоль) изопропилтрифенилфосфоний иодида, добавили 150 мл абсолютного тетрагидрофурана. Полученную суспензию охлаждали до 0°С и медленно добавили к ней 26.2 мл (41.87 ммоль) 1.6 М раствора н-BuLi в гексане. Полученную смесь перемешивали при 0°С в течение 1 часа, затем при этой температуре медленно добавили к образовавшемуся красно-коричневому раствору раствор 7.04 г (38.06 ммоль) 2-бромбензальдегида. Реакционная смесь приобретает оранжевую окраску. Смесь перемешивали при комнатной температуре 16 ч, добавили 100 мл водного раствора NaCl, дважды экстрагировали этилацетатом; объединенные органические фазы повторно промывали раствором NaCl и экстрагировали водную фазу этилацетатом. Объединенную органическую фазу сушили над сульфатом магния, растворитель упаривали. Твердый желтый остаток нанесли на колонку с силикагелем (Acros, 60−200 µm), гексаном элюировали бесцветную первую фракцию, растворитель упарили. Получили 1-бром-2-(2-метил-1-пропенил) бензол в виде бесцветной прозрачной жидкости. Выход: 7.1 г (88.5%). Спектр ЯМР 1H (400 МГц, CDCl3): δ (м.д.) 7.53−6.45 (м, 5H, Ar–H, CH=C(СН3)2), 1.77 (с, 3Н, CH=C(СН3)2, 1.60 (с, 3Н, CH=C(СН3)2.
Синтез 2-бромстильбена. В колбу Шленка поместили 1.67 г (4.29 ммоль) бензилтрифенилфосфоний хлорида, добавили 50 мл абсолютного тетрагидрофурана. Полученную суспензию охладили до 0°С и медленно добавили к ней 4.29 ммоль 1.6 М раствора н-BuLi в гексане. Красно-коричневую смесь перемешивали при 0°С в течение 1 ч, затем при этой температуре медленно добавили к смеси раствор 0.72 г (3.90 ммоль) 2-бромбензальдегида. Смесь перемешивали при комнатной температуре 24 часа, добавили 50 мл водного раствора NaCl, дважды экстрагировали этилацетатом, объединенные органические фазы повторно промывали раствором NaCl и экстрагировали водную фазу этилацетатом. Органическую фазу сушили над сульфатом магния, растворитель упаривали. Твердый желтый остаток нанесли на колонку с силикагелем (Acros, 60–200 мкм), гексаном элюировали первую бесцветную фракцию, растворитель упаривали. Получили 2-бромстильбен в виде вязкой бесцветной жидкости (смесь цис-, транс-изомеров). Выход 0.85 г (84%). Масс-спектр: m/z = 258 [M+] (74%), 258 [M+] (26%).
Синтез 1-бром-2-(гепт-1-ен-1-ил)бензола (смесь изомеров). 3.04 г (6.41 ммоль) гексилтрифенилфосфонийиодида растворили в 50 мл тетрагидрофурана в токе аргона. К полученному раствору, охлажденному до –5°С, по каплям добавили 4 мл 1.6 М раствора н-BuLi в гексане. После добавления всего раствора н-BuLi образовавшуюся смесь перемешивали в течение часа, затем добавили по каплям раствор 0.68 мл (5.83 ммоль) 2-бромбенз-альдегида в 10 мл тетрагидрофурана. Поддерживали температуру реакционной смеси от –2 до +2°С. Раствор приобретает оранжевую окраску, выпадает белый осадок. Смесь нагревали до комнатной температуры, перемешивали 3 ч, затем добавили к ней 30 мл насыщенного раствора NaCl, 30 мл этилацетата. Разделили слои, нижний водный слой дважды проэкстрагировали 30 мл этилацетата. Объединенные органические фазы высушили над безводным сульфатом натрия, упарили. Остаток после упаривания очистили с помощью колоночной хроматографии (силикагель Acros, 60–200 мкм, элюент – гексан). Получили 1.4 г (выход 95%). Спектр ЯМР 1H (400 МГц, CDCl3) (смесь E, Z-изомеров в мольном соотношении 1 : 2.5): δ (м.д.) 7.61 (д, J = 7 Гц, 2.5H, Ar–H), 7.56 (д, J = 7 Гц, 1H, Ar–H), 7.36–7.23 (набор м, 7Н, Ar–H), 7.18–7.04 (набор м, 3.5Н, Ar–H), 6.75 (уш. дт, J = 16 Гц, 1Н, СН=CH), 6.49 (уш. дт, J = 12 Гц, 2.5Н, СН=CH), 6.21 (дт, J = 7 Гц, 1Н, СН=CH), 5.81 (дт, J = 7 Гц, 2.5Н, СН=CH), 2.29 (д кв, J = 7 Гц, J = 1.5 Гц, 2Н, =СH–CH2), 2.21 (д кв, J = 7 Гц, J = 1.5 Гц, 5Н, =СH–CH2), 1.59–1.24 (набор м, 21Н, CH2), 0.95 (т, J = 7 Гц, 3Н, CH3), 0.91 (т, J = 7 Гц, 7.5Н, CH3).
Общая методика синтеза дифосфиновых лигандов (7–11) на примере синтеза [(2-дифенилфосфино)фенил][2-(винил)фенил]фенилфосфина (7). В колбу Шленка объемом 100 мл поместили 0.28 г (11.54 ммоль) магния, прокалили в вакууме, заполнили аргоном. Добавили 50 мл абсолютного тетрагидрофурана, каталитическое количество иода. Отдельно приготовили раствор 1.92 г (10.49 ммоль) 2-бромстирола в 20 мл тетрагидрофурана, добавили несколько капель приготовленного раствора в реакционную колбу. Нагрели смесь до кипения без перемешивания, раствор постепенно обесцветился, медленно добавили оставшийся раствор 2-бромстирола к кипящей смеси. Раствор приобрел коричнево-желтую окраску. Кипятили раствор с обратным холодильником при перемешивании 4 часа. Охладили смесь до комнатной температуры. Добавили дополнительное количество тетрагидрофурана для разбавления образовавшегося густого коричневого раствора. Отфильтровали полученный раствор реактива Гриньяра в токе аргона. Охладили раствор до –30°С, прибавили к нему по каплям раствор 2.97 г (7.34 ммоль) [(2-дифенилфосфин)фенил]фенилфосфинхлорида {P–PCl} в 20 мл тетрагидрофурана. Температура поднялась до –15°С. Убрали охлаждение, оставили нагреваться реакционную смесь до комнатной температуры, затем перемешивали смесь при этой температуре в течение суток. Упарили растворитель, обработали реакционную смесь насыщенным водным раствором хлорида аммония (30 мл), добавили 50 мл хлористого метилена, органический слой отделили (водный слой экстрагировали СН2Cl2), объединенную органическую фазу сушили над сульфатом натрия, растворитель упарили. Бледно-желтое твердое вещество растворили в СН2Cl2, добавили 5 мл гексана – выпадает белый мелкодисперсный осадок. Раствор отфильтровали от осадка через стеклянный фильтр с трубкой в токе аргона, растворитель упарили, получили бледно-желтое кристаллическое вещество, которое сушили в высоком вакууме.
Выход[(2-дифенилфосфино)фенил][2-(винил)фенил]фенилфосфина (7) 92%. Спектр ЯМР 31P{1H} (161.98 МГц, CDCl3): δ (м.д.) –13.7 (д, 3JPP = 160 Гц, P(С6H4CH=СH2)Ph), –21.2 (д, 3JPP = = 160 Гц, PPh2). ЯМР 1H (400 МГц, CDCl3): δ (м.д.) 6.76–7.62 (м, 24Н, CH=CH2, Ar–H), 5.54 (дт, 1H, JHH = 17 Гц, JHH = 1 Гц, CH=CHAHB), 5.08 (дд, 1H, JHH = 12 Гц, JHH = 1 Гц, CH=CHAHB).
Для синтеза [(2-дифенилфосфино)фенил][2-(2-метил-1-пропенил)фенил]фенилфосфина вместо 2-бромстирола был взят 1-бром-2-(2-метил-1-пропенил)бензол. Выход [(2-дифенилфосфино)фенил][2-(2-метил-1-пропенил)фенил] фенилфосфина (8) 91%. Спектр ЯМР 31P{1H} (161.98 МГц, CDCl3): δ (м.д.) –14.4 (д, 3JPP = 159 Гц, P(С6H4CH=С(СН3)2)Ph), –19.7 (д, 3JPP = 159 Гц, PPh2). ЯМР 1H (400 МГц, CDCl3): δ (м.д.) 7.14–7.03 (м, 23Н, Ar–H), 6.68 (м, 1H, CH=С(СН3)2), 1.55 (с, 3Н, CH=C(СН3)2, 1.38 (с, 3Н, CH=C(СН3)2.
Для синтеза [(2-дифенилфосфино)фенил][2-(2-фенил-1-этенил)фенил]фенилфосфина вместо 2-бромстирола был взят 2-бромстильбен.
Выход [(2-дифенилфосфино)фенил][2-(2-фенил-1-этенил)фенил]фенилфосфина (9) 96%. Спектр ЯМР 31P{1H} (161.98 МГц, CDCl3): δ (м.д.) (смесь изомеров 1 : 0.7) –13.8 (д, 3JPP = 160 Гц, 1P, P(С6H4CH=СH2)Ph), –14.9 (д, 3JPP = 157 Гц, 0.7P, P(С6H4CH=СH2)Ph), –20.4 (д, 3JPP = 160 Гц, 1P, PPh2), –21.3 (д, 3JPP = 157 Гц, 0.7P, PPh2).
Для синтеза [(2-дифенилфосфино)фенил][2-(2-н-пентил-1-этенил)фенил]фенилфосфина вместо 2-бромстирола был использован 1-бром-2-(гепт-1-ен-1-ил)бензол. Выход [(2-дифенилфосфино)фенил][2-(2-н-пентил-1-этенил)фенил]фенилфосфина (10) 82%. Спектр ЯМР 31P{1H} (161.98 МГц, CDCl3): δ (м.д.) (смесь изомеров 1 : 2.5 (цис-, транс-изомеры)) –13.8 (д, 3JPP = 160 Гц, 2.5P, P(С5H11CH=СH2)Ph,), –14.2 (д, 3JPP = 158.5 Гц, 1P, P(С5H11CH=СH2)Ph), –20.4 (д, 3JPP = 158.5 Гц, 1P, PPh2), –20.5 (д, 3JPP = 160 Гц, 2.5P, PPh2).
Для синтеза [(2-дифенилфосфино)-4,5-дидецилфенил][2-(2-метил-1-пропенил)-фенил]фенилфосфина вместо 2-бромстирола был взят 1-бром-2-(2-метил-1-пропенил)бензол, а вместо [(2-дифенилфосфин)фенил]фенилфосфинхлорида {P‒PCl} – (2‑дифенилфосфин)-4,5-дидецилфенил(фенил)фос-финхлорид {Dec2P–PCl}. Выход [(2-дифенилфосфино)-4,5-дидецилфенил][2-(2-метил-1-пропенил)-фенил]фенилфосфина (11) 94%. Спектр ЯМР 31P{1H} (161.98 МГц, CDCl3): δ (м.д.) –13.8 (д, 3JPP = 156 Гц, 1P), –19.4 (д, 3JPP = 156 Гц, 1P).
Общая методика синтеза дифосфиновых комплексов хрома(III) (4, 5, 12–16). К суспензии дифосфина (2, 3, 7–11) в ацетоне (суспензия готовится из расчета 18 мл ацетона на 1 г дифосфина) добавили гексагидрат хлорида хрома(III) [Cr(H2O)4Cl2]Cl · 2H2O в 20%-ном мольном избытке по отношению к дифосфину. Реакционная смесь сразу же приобретает зеленую окраску, через 1.5 ч перемешивания цвет смеси – бледно-синий. Перемешивали смесь при комнатной температуре 24 ч. По окончании перемешивания выпавший из раствора синий кристаллический осадок отфильтровали через стеклянный фильтр с трубкой в токе аргона, тщательно промыли ацетоном и гексаном и высушивали в высоком вакууме.
Выход ([2-(дифенилфосфино)фенил]дифенилфосфин-P,P)-(аква)-трихлорохром(III) (4) 95%. Найдено (%): С, 59.06; H, 4.60. C30H26Cl3CrOP2. Вычислено (%): С, 57.85; H, 4.21.
Выход ([(2-дифенилфосфино)фенил][2-(метил)фенил]фенилфосфин-P,P)-(аква)-трихлорохром(III) (5) 96%. Найдено (%): С, 59.45; H, 4.65. C31H28Cl3CrOP2. Вычислено (%): С, 58.46; H, 4.43.
Выход ([(2-дифенилфосфино)фенил][2-(винил)фенил]фенилфосфин-P,P)-(аква)-трихлоро-хром(III) (12) 90%. Найдено (%): С, 56.42; H, 5.06. C32H28Cl3CrOP2 · H2O. Вычислено (%): С, 56.12; H, 4.71.
Выход ([(2-дифенилфосфино)фенил][2-(2-метил-1-пропенил)фенил]фенилфосфин-P,P)-(аква)-трихлорохром(III) (13) 87%. Найдено (%): С, 59.03; H, 5.08. C34H32Cl3CrOP2. Вычислено (%): С, 58.76; H, 4.89.
Выход ([(2-дифенилфосфино)фенил][2-(2-фенил-1-этенил)фенил]фенилфосфин-P,P)-(аква)-трихлорохром(III) (смесь изомеров) (14) 75%. Найдено (%): С, 64.12; H, 4.75. C38H32Cl3CrOP2. Вычислено (%): С, 62.96; H, 4.44.
Выход ([(2-дифенилфосфино)фенил][2-(2-н-пентил-1-этенил)фенил]фенилфосфин-P,P)-(аква)-трихлорохром(III) (смесь изомеров) (15) 73%. Найдено (%): С, 62.25; H, 5.75. C37H38Cl3CrOP2. Вычислено (%): С, 62.33; H, 5.37.
Выход ([(2-дифенилфосфино)-4,5-дидецилфенил][2-(2-метил-1-пропенил)фенил]фенилфосфин-P,P)-(аква)-трихлорохром(III) (16) 83%. Найдено (%): С, 69.95; H, 5.55. C54H50Cl3CrOP2. Вычислено (%): С, 69.20; H, 5.38.
Рентгеноструктурное исследование соединений 4, 12 и 13
Монокристаллы соединения 4, 12 и 13 для рентгеноструктурного исследования были получены из растворов соответствующих соединений в смеси хлористый метилен/гексан. Кристаллографические данные и основные параметры уточнения для соединений приведены в табл. 7. Методика эксперимента рентгеноструктурного исследования приведена в работе [5].
Таблица 7.
Кристаллографические данные и основные параметры уточнения для соединениий 4, 12 и 13
| 4 | 12 | 13 | |
|---|---|---|---|
| Формула | C32H28Cl9CrOP2 | C32H28Cl3CrOP2 ⋅ 0.25H2O ⋅ ⋅ 0.75C6H14 ⋅ 0.875CH2Cl2 | (С34Н32Cl3CrOP2) ⋅ (CH2Cl2) |
| Молекулярная масса | 861.53 | 792.28 | 761.81 |
| Сингония | Моноклинная | Триклинная | Триклинная |
| Пространственная группа | P21/c | P-1 | P-1 |
| a, Å | 13.6686(15) | 13.7866(5) | 10.0845(6) |
| b, Å | 18.223(2) | 15.9078(5) | 14.1067(9) |
| c, Å | 15.3665(17) | 17.5486(6) | 14.9376(13) |
| α, град | 90 | 86.7050(10) | 61.9000(10) |
| β, град | 108.397(2) | 80.5430(10) | 75.668(2) |
| γ, град | 90 | 70.0670(10) | 78.9410(10) |
| V, Å3 | 3631.8(7) | 3568.9(2) | 1482.0(4) |
| Z | 4 | 4 | 2 |
| ρcalc, г см–3 | 1.576 | 1.475 | 1.398 |
| F(000) | 1740 | 1639 | 782 |
| μ, мм–1 | 1.093 | 0.798 | 0.801 |
| Область сканирования по θ, град | 1.57–26.00 | 1.36–26.00 | 1.50–30.03 |
| Число измеренных отражений | 33 828 | 13 878 | 23 040 |
| Число независимых отражений | 7125 | 13 878 | 10 521 |
| Полнота по θ, % | 100.0 | 99.0 | 99.5 |
| Число уточняемых параметров | 414 | 703 | 402 |
| GOF (F2) | 0.877 | 1.071 | 1.023 |
| Число отражений с I > 2σ(I) | 4221 | 10976 | 7170 |
| R1(F) (I > 2σ(I))a | 0.0633 | 0.0427 | 0.0498 |
| wR2(F 2) (для всех отражений)b | 0.1640 | 0.1272 | 0.1362 |
| Остаточная электронная плотность, eÅ–3 | 1.019/–0.705 | 0.872/–0.383 | 0.963/–1.027 |
aR1 = ${{\sum {\left| {{{F}_{{\text{o}}}} - \left| {{{F}_{{\text{c}}}}} \right|} \right|} } \mathord{\left/ {\vphantom {{\sum {\left| {{{F}_{{\text{o}}}} - \left| {{{F}_{{\text{c}}}}} \right|} \right|} } {\sum {\left( {{{F}_{{\text{o}}}}} \right)} ;}}} \right. \kern-0em} {\sum {\left( {{{F}_{{\text{o}}}}} \right)} ;}}$ bwR2 = ${{\left( {{{\sum {\left[ {w{{{\left( {F_{{\text{o}}}^{2}--F_{{\text{c}}}^{2}} \right)}}^{2}}} \right]} } \mathord{\left/ {\vphantom {{\sum {\left[ {w{{{\left( {F_{{\text{o}}}^{2}--F_{{\text{c}}}^{2}} \right)}}^{2}}} \right]} } {\sum {\left[ {w{{{\left( {F_{{\text{o}}}^{2}} \right)}}^{2}}} \right]} }}} \right. \kern-0em} {\sum {\left[ {w{{{\left( {F_{{\text{o}}}^{2}} \right)}}^{2}}} \right]} }}} \right)}^{{{1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-0em} 2}}}}.$
Список литературы
Dixon J.T., Green M.J., Hess F.M., Morgan. D.H. // J. Organomet. Chem. 2004. V. 689. № 23. P. 3641.
McGuiness D.S. // Chem. Rev. 2011. V. 111. P. 2321.
Alferov K.A., Belov G.P., Meng Y. // Applied Catalisys A. General. 2017. V. 542. P. 71.
Афанасьев В.В., Беспалова Н.Б., Козлова Г.A., Сенин A.A., Чередилин Д.Н. // Патент РФ № 2525118. 2014.
Чередилин Д.Н., Шелоумов А.М., Сенин A.A., Козлова Г.A., Афанасьев В.В., Беспалова Н.Б. // Нефтехимия. 2019. №7. С. В печати.
Carter A., Cohen S.A., Cooley N.A., Murphy A., Scutt J., Wass D.F. // Chem. Commun. 2002. P. 858.
Wass D.F. (British Petroleum) // Patent WO № 2002004119. 2002.
Чередилин Д.Н., Шелоумов А.М., Козлова Г.A., Сенин А.А., Хасбиуллин И.И., Беспалова Н.Б. // Патент РФ № 2549897. 2014.
Overett M.J., Blann K., Bollmann A., de Villiers R., Dixon J.T., Killian E., Maumela M.C., Maumela H., McGuinness D.S., Morgan D.H., Rucklidge A., Slawin A.M.Z. // J. Mol. Catal. A: Chem. 2008. V. 283. № 1–2. P. 114.
Do L.H., Labinger J.A., Bercaw J.E. // ACS. Catal. 2013. V. 3. № 11. P. 2582.
Афанасьев В.В., Беспалова Н.Б., Козлова Г.A., Сенин A.A., Чередилин Д.Н. // Патент РФ № 2525118. 2014.
Sydora O.L., Jones T.C., Small S.B., Nett A.J., Fischer A.A., Carney M.J. // ACS Catal. 2012. №2. P. 2452.
Barron A.R. // Organometallics. 1995. V. 14. P. 3581.
Krasovskiy A., Knochel P. // Angewandte Chemie. Int. Ed. 2004. V. 43. № 25. P. 3333.
Ackerman L.J., Diamond G.M., Hall K.A., Longmire J.M., Murphy V.J., Nava Salgado V.O. // Patent US № 2008/0188633 A1. 2008.
Беспалова Н.Б., Чередилин Д.Н., Шелоумов А.М., Козлова Г.A., Сенин А.А., Хасбиуллин И.И. // Патент РФ № 2581052. 2015.C1.
Hibino S., Sugino E., Adachi Y., Nomi K., Sato K., Fukumoto K. // Heterocycles. 1989. V 28. № 1. P. 275.
Standley E.A., Smith S.J., Müller P., Jamison T.F. // Organometallics. 2014. V. 33. P. 2012.
Jankowiak A., Kaszyński P. // Beils. J. Org. Chem. 2012. V. 8. P. 275.
Katsuta S., Saeki H., Tanaka K., Murai Y., Kuzuhara D., Misaki M., Aratani N., Masuo S., Ueda Y., Yamada H. // J. Mater. Chem. C. 2014. V. 2. № 6. P. 986.
Reetz M.T., Gosberg A. // Tetrahedron Asymmetry. 1999. V. 10. P. 2129.
Дополнительные материалы отсутствуют.