

Нефтехимия, 2020, T. 60, № 2, стр. 172-182
Комплексообразование тиофеновых соединений с переходными металлами – ключ к пониманию механизмов обессеривания нефтепродуктов (обзор)
А. Л. Максимов 1, 2, *, А. И. Нехаев 1
1 Институт нефтехимического синтеза им. А.В. Топчиева РАН
119991 Москва, Россия
2 Московский государственный университет имени М.В. Ломоносова. химический факультет
119991 Москва, Россия
* E-mail: max@ips.ac.ru
Поступила в редакцию 28.08.2019
После доработки 12.09.2019
Принята к публикации 14.10.2019
Аннотация
Обобщены результаты исследований в активации связей С–Н и С–S тиофенов и их производных соединениями переходных металлов. Рассмотрены разнообразные способы координации тиофена с металлом, которые зависят от конкретного металла и окружающих его лигандов, а также трансформация тиофеновых соединений под действием комплексов переходных металлов.
Гетерогенные катализаторы на основе сульфидов никеля, молибдена и кобальта довольно эффективны для удаления большинства соединений серы из нефтепродуктов в процессе гидрообессеривания [1–8]. Тиофены (Т) довольно инертны, поскольку атом серы в них является очень слабым донором и они становятся еще менее активны, когда есть заместители рядом с серой. Тиофеновые соединения, особенно алкилзамещенные бензотиофены (БТ) и дибензотиофены (ДБТ), представляют собой группу сернистых соединений, которые трудно удаляются из нефтепродуктов [9, 10]. Две метильные группы в положениях 4 и 6 ДБТ стерически препятствуют взаимодействию металла со связями C–S. Скорости обессеривания 4,6-Ме2ДБТ на промышленных Co–Mo–Al-катализаторах в 10–15 раз меньше, чем ДБТ [11, 12].
Понимание механизма активации тиофеновых соединений может привести к открытию новых каталитических систем для гидрообессеривания. Однако трудно добиться понимания, что именно происходит при обессеривании на молекулярном уровне, поскольку в промышленности применяются гетерогенные катализаторы [13]. Поэтому большое количество работ посвящено исследованию роли соединений переходных металлов в активации связей С–Н и С–S тиофенов и их производных.
Если раньше исследования ограничивались синтезом комплексных соединений производных тиофена и установлением их структуры и свойств, то в последнее время с появлением мощных компьютеров и программного обеспечения стало возможным проводить моделирование процесса комплексообразования, главным образом, с помощью теории функционала плотности (DFT).
В последнее время появляются сообщения о возможном практическом применении комплексных соединений, содержащих тиофеновый фрагмент. Например, комплексы меди проявляют биологическую активность и имеют перспективу в лечении, в частности, рака [14, 15] и птичьего гриппа [15]. Получены пористые органические полимеры, содержащие Т, для удаления ионов Cu(II) [16], тиофенсодержащие вещества для селективного определения ионов Ag+, Cu2+ и Fe3+ в присутствии ионов Na+, K+, Ca2+ и Mg2+ в природной воде [17]. Электроактивную устойчивую полимерную пленку, полученную из комплекса Ru, можно использовать в качестве датчиков в соответствии с ее окислительно-восстановительными свойствами [18].
Ранее опубликован ряд обзоров, обобщающих достижения в области взаимодействия сернистых соединений, в том числе производных Т, с комплексами переходных металлов [19–28].
В настоящем обзоре мы систематизировали способы взаимодействия комплексов переходных металлов с тиофеновыми соединениями не по способу координации, а по металлу.
ЖЕЛЕЗО И РУТЕНИЙ
Связь С–S в БТ и ДБТ расщепляется при действии трехъядерного пентагидридного комплекса рутения {(η5-C5Me5)Ru}3(µ-H)3(µ3-H)2 [29]. Конечные продукты реакции с БТ – µ3-сульфидотрирутениевый комплекс и этилбензол. Как правило, весьма трудно зафиксировать возникновение металлациклов из-за активации связи С–S и повышения донорной координационной способности атома серы. Тем не менее удалось получить промежуточный темно-зеленый рутенатиациклогексадиеновый комплекс (рис. 1 ), который образуется в результате окислительного присоединения связи С–S в БТ к одному из трех рутениевых центров. Через 90 мин его выход достигает 90%. Интермедиат был выделен из реакционной смеси охлаждением до –30°С и охарактеризован спектрами 1Н и 13С ЯМР.
Однако в реакции с ДБТ никаких интермедиатов выделить не удалось, только конечные продукты – µ3-сульфидотрирутениевый комплекс и дифенил. Очевидно, это связано с тем, что стерические препятствия вокруг атома серы в ДБТ гораздо больше, чем в БТ.
Fe3(CO)12 реагирует с Т и БТ, но не с ДБТ [30–32]. Фотохимическая реакция Fe(CO)5 с ДБТ тоже не происходит. Карбонилы железа и рутения разрывают связи С–S в ДБТ, если он имеет азотсодержащие заместители. В то время как для проведения реакции с карбонилом рутения достаточно нагревания до 100°С, пентакарбонил железа требует УФ-облучения [33–35].
Из Т и дигидридного комплекса рутения [RuH2(η2-H2)2(PCy3)2] при комнатной температуре образуется η4-тиоаллильное соединение [RuH(η4(S,C)–SC4H5)(PCy3)2], которое под действием Н2 снова дает исходный дигидридный комплекс. Вещество [RuH2(η2-H2)2(PCy3)2] может использоваться в качестве предшественника катализатора гидрирования Т до тетрагидротиофена, 2-МеТ и БТ – до 2,3-дигидропроизводных в мягких условиях (80°C, 3 атм H2). ДБТ в этих условиях не восстанавливается, давая S-координированный комплекс [RuH2(η2-H2){η1(S)–C12H8S}(PCy3)2] [36]. В катионе [Ru(NH3)5(H2O)]2+ ДБТ и в меньшей степени 4,6-Ме2ДБТ способны замещать координированную молекулу воды [37]. Хиральный комплекс рутения позволяет проводить энантиоселективное гидрирование замещенных тиофенов и ДБТ. При этом не наблюдаются реакции внедрения металла в связи С–S, гидрогенолиза и гидрообессеривания [38].
ДБТ выполняет роль мостикового лиганда в комплексе [CpRu(CO)2(µ2-η1-S:η6-ДБT)RuC5Me5]2+ [39]. ДБТ связывается с рутением прочнее, чем БТ или замещенные тиофены [40, 41].
Свойства η4-бис- и η4-монотиофеновых комплексов железа, которые трудно получить экспериментально, исследованы методом DFT [42]. Расчеты показывают, что могут существовать устойчивые комплексы железа с тиенильными мостиками, имеющие одномерную супермолекулярную архитектуру. Определены частоты поглощения комплексов в инфракрасной, видимой и ультрафиолетовой областях. Тиофеновые комплексы железа устойчивее, чем никелевые и кобальтовые.
КОБАЛЬТ, РОДИЙ И ИРИДИЙ
Исследовательская группа Джонса (W.D. Jones) из Рочестерского университета (США) провела подробное изучение реакций внедрения по связи С–S в T, БТ и ДБТ фрагментов, содержащих Rh, Co и Ir в состоянии 1+ [43–48]. Авторы полагали, что результаты экспериментов с дейтерированным комплексом [44] дают доказательства для возникновения интермедиата с координацией T через серу в качестве непосредственного предшественника расщепления связи С–S за счет последующей миграции α-углерода к металлу. Кроме того, η2-координация по двойной связи T приводит к получению продуктов активации связей С–Н, которые термодинамически менее выгодны, но кинетически значимы. Поскольку сера может быть извлечена из T после этого начального этапа, не следует пренебрегать возможностью такого пути в качестве дополнительного механизма для гидрообессеривания нефтепродуктов.
Тиофен образует комплексы с плоским шестичленным кольцом, в то время как металлациклы из алкилзамещенных тиофенов, БТ и ДБТ изогнуты. Во всех случаях комплексы тиофеновых производных содержат диеновый фрагмент между металлом и серой (рис. 2 ). В работе [46] приведены рентгеноструктурные данные для 14 комплексов родия с T, БТ, ДБТ и их производными. Электронное влияние заместителей в тиофеновых производных незначительно, а стерические эффекты велики. Так, например, связь С–S в тетраметилтиофене, 4,6-Me2- и 4,6-Et2ДБТ не расщепляется. Получаются лишь лабильные комплексы со связью М–S.
Выполненные через 11 лет расчеты по методу DFT взаимодействия фрагмента [(C5Me5)Rh(PMe3)] с тиофеном [47] изменили ранее предложенный механизм, по которому координация η1(S) приводит непосредственно к активации связей C–S. Авторы пришли к выводу, что как η1(S)-координация, так и η2(C,C)-координация приводит к интермедиату η2(C, S) который непосредственно предшествует расщеплению связи C–S. Расчетная энергия связывания η1-интермедиата, определенная в более ранней работе [49], на 15.5 ккал/моль меньше, чем для η2-интермедиата.
Интересно, что переходное состояние, найденное для [C5Me5Rh(PMe3)], похоже на таковое для комплекса Pt [50] двумя свойствами. Во-первых, интермедиат η2(C, S)-тиофен предшествует разрыву связи C–S, во-вторых, формирование связей металл–сера и металл–углерод в значительной степени завершено (в пределах 4% конечных расстояний).
Проведено сравнение теории (метод DFT) и экспериментальных данных для реакции фрагмента [(C5Me5)Rh(PMe3)] с 2- и 3-замещенными тиофенами [48]. Показано, что в случае 2-МеТ образование продукта в результате активации незамещенной связи C–S предпочтительнее на 5.8 ккал/моль. Это совпадает с экспериментальными результатами. Для 3-замещенных тиофенов соотношение продуктов внедрения металла в связи C–S составляет 1 : 1. Расчеты объясняют такой результат небольшой разницей между энергиями основного состояния двух продуктов внедрения (0.4–0.8 ккал/моль). В переходных состояниях происходит значительная делокализация электронов в тиофеновых кольцах, среднее значение углов изгиба металлацикла составляет 61°, что существенно выше, чем в конечных продуктах (до 21°). Селективности всех продуктов соответствуют термодинамическому контролю процесса.
Реакция Т с фрагментом [(C5Me5)Rh(PMe3)] привлекла внимание и авторов работы [51], которые для расчетов применили гибридный метод IMOMM, сочетающий квантовую механику и молекулярную механику. Этот метод показал хорошую сходимость с ранее выполненными расчетами геометрии на основе только квантовой механики. Однако есть отличия в энергиях возможных путей реакции, связанные с изменением окислительного состояния атома родия. Рассчитано влияние метильных заместителей на каждой стадии реакции с определением доли стерического и электронного фактора.
При действии [Rh(μ-Cl)(CO)2]2 на ДБТ с пиридильным заместителем первоначально возникает моноядерный комплекс родия (I) цис-[RhCl(CO)2(η1(N)-PyДБT)], который затем при 100°С в течение 3 дней превращается в тетраядерный комплекс родия [Rh (I)/Rh (III)/Rh (III)/Rh (I)] с хелатным тридентатным лигандом – дианионом 3'-(2''-пиридил)-1,1'-дифенил-2-тиола – [{Rh(μ-PyBPT-κ3-N,C,S)}(μ-Cl)2{Rh(CO)2}]2 – продуктом расщепления связи C–S в ДБТ [33
Димерный комплекс родия [RhP(CH2)2P(µ-H)]2 при обработке ДБТ (100°C, 10 дней), 4-МеДБТ (135°С, 7 дней) и 4,6-Ме2ДБТ (135°С, 8.5 дней) дает продукты расщепления связи C–S с мостиковым атомом серы [52]. В случае 4,6-Ме2ДБТ это [Rh2{P(CH2)2P}2(µ-SMe2-ДБT)(µ-H)]. Однако первоначально в качестве основного продукта образуется [Rh2{P(CH2)2P}2(µ-SH)(µ-H)], но, когда исходный комплекс полностью расходуется, соотношение продуктов составляет 50 : 50. В фенокситиине, содержащем два фрагмента о-С6Н5, соединенных мостиковыми атомами S и O, расщепляется только связь C–S, в то время как связь С–О не затрагивается.
Из Т и [Ir(циклооктадиен)(PMe3)3]Cl в результате внедрения металла в связь C–S образуется продукт с шестичленным иридатиациклом, фосфиновыми и хлорным лигандами [53]. Авторы утверждают, что чередование длин связей по всему кольцу является наглядным свидетельством отсутствия ароматичности. Для комплекса, имеющего только фосфиновые лиганды, в работе [54] сделан вывод об ароматичности иридатиабензола с оговоркой, что эта ароматичность “довольно хрупкая”. Структурные характеристики иридатиабензольного комплекса с фосфиновыми и тиолатным лигандами определены в работе [55]. Расчеты электронной структуры в иридатиабензоле показали [56], что сильная связь М–S нарушает кольцевую π-систему и приводит к некоторой локализации π-связей кольца. Замена одного из фосфиновых лигандов на тиолатный еще больше нарушает кольцевую π-систему. Тем не менее, спектр 1Н ЯМР подтверждает ароматичность иридатиабензольного фрагмента. Кроме того, его “хрупкой” ароматичности оказывается достаточно для замещения п-ксилола с “крепкой” ароматичностью в комплексе [Mo(η6-п-ксилол)(CO)3] [54, 57].
Гидридный комплекс иридия [Ir(C5Ме5)(μ-H)Cl]2 в атмосфере водорода не только разрывает обе связи C–S в 2,5-Ме2Т, но и гидрируeт двойные связи [58]. Однако, если реакцию проводить без водорода и в присутствии акцептора водорода – трет-бутилэтилена, то ход реакции резко изменяется. Происходит двойная активация связи С–Н 2,5-Ме2Т и возникает необычное соединение [Ir(Cl)(C5Me5)(μ-H){μ-(η1:η3-C6H6S)}(IrC5Me5)] [59], в котором имеется η3-аллильное взаимодействие бывшего 2,5-Ме2Т с одним из атомов иридия за счет атомов С2 и С3 тиофенового ядра и бывшей метильной группой в положении 2, один из атомов водорода которой превратился в водородный мостик. Своеобразным мостиком, связывающим оба атома иридия, стал и атом С3 тиофенового ядра.
При гидрировании [Ir(циклооктадиен)(PPh3)2]PF6 в присутствии Т, БТ или ДБТ с высокими выходами образуются комплексы [IrH2(η1(S)T*)2(PPh3)2]PF6, где Т* – Т, БТ или ДБТ [60], для которых определены геометрические параметры. Структура комплексов представляет искаженный октаэдр с цис-расположением тиофенов, гидридных атомов водорода и транс-трифенилфосфиновыми лигандами. Авторы полагают, что такие структуры могут использоваться в качестве моделей для начальной стадии хемосорбции тиофеновых производных на твердых катализаторах.
Выполнены теоретические расчеты по методу DFT для комплексов Со(η4-Т) и Со(η2-Т)2 [42], которые показали, что тиофеновые комплексы кобальта должны иметь заметное поглощение в ИК-области при 155.8 см–1 и менее устойчивы, чем комплексы железа и никеля.
НИКЕЛЬ, ПАЛЛАДИЙ И ПЛАТИНА
В литературе имеются примеры активации связи C–S в 4,6-Ме2ДБТ. В частности, на это способны гидридофосфиновые комплексы никеля (90°С, 21 ч, 1 атм Н2) и платины (120°С, 10 ч). В случае никеля происходит процесс обессеривания и с выходом 23% выделяется 3,3'-диметилдифенил. В случае платины с выходом 40% образуется комплекс, содержащий шестичленный тиаплатиновый цикл [61].
Этими же авторами получен и структурно охарактеризован ряд серосодержащих комплексов никеля из тиофеновых соединений [62]. В 2-цианотиофене никель внедряется по связи C–S, соседней с нитрильной группой [63]. Имеется обзор по активации разнообразных связей, в том числе C–S, низковалентными комплексами никеля [64].
Трис(триэтилфосфин)платина внедряется в связи C–S в Т, БТ и ДБТ [65], а также 2-МеБТ, 3-МеБТ, 4-МеДБТ, 1,9-Ме2ДБТ [66]. Взаимодействие [Pt(PEt3)3] с 4,6-Ме2ДБТ высокой чистоты вместо активации связи C–S активирует связь С–Н предположительно в положении 3 бензольного кольца. Лишь обработка 4,6-Ме2ДБТ комплексом цис-[PtCl2(PЕt3)2] в присутствии металлического натрия и водорода приводит к внедрению платины по связи C–S (рис. 3 ).
Обнаружена интересная и важная возможность активации связей C–S в БТ и ДБТ, в том числе алкилированных, за счет предварительной координации карбоциклического кольца с электрофильными фрагментами [Mn(CO)3]+, FeCp+, RuCp+, [Ru(C6Me6)]2+, Cr(CO)3 [67–69] (рис. 4 ).
Так, реакция мягкого нуклеофила Pt(PPh3)2(C2H4) с комплексами алкилированных БТ и ДБТ, содержащими фрагмент [Mn(CO)3]+, координированный с карбоциклическим кольцом, приводит при комнатной температуре к быстрому внедрению платины в связи C–S с образованием металлатиациклических комплексов [69]. Для сравнения, [Pt(PPh3)2(C2H4)] не реагирует со свободным БТ [65].
Авторы [69] полагают, что с комплексами БТ, не имеющими заместителей в положениях 2 или 3, начальная быстрая координация платины по связи C=C в гетероциклическом кольце происходит перед внедрением в связь C(винил)–S. Когда в этих положениях есть заместитель, образование η2-(C=C)-интермедиата блокируется, скорость реакции замедляется, и внедрение в связи C(арил)–S становится возможным или даже преобладающим. В этих случаях предполагается возникновение η1(S)-интермедиатов. Внедрение в C–S-связь, которая ближе к координированному кольцу в дибензотиофеновых комплексах, даже алкилированных в положениях 4 и 6, происходит быстро и со скоростями, близкими к тем, которые наблюдаются для комплексов алкилированных БТ. Даже в обычно малоактивном 4,6-Me2ДБТ становится возможным быстрый разрыв связи C–S платиной после предварительной координации с фрагментом [Mn(CO)3]+. Продукты внедрения платины в 4,6-диалкил-ДБТ из реакционной смеси выделить не удалось, получены лишь спектральные данные in situ. Не обнаружено никаких промежуточных продуктов, в том числе типа η2-(С=С).
На основе наблюдаемой региоселективности, данных низкотемпературного ИК-исследования и изучения кинетики при комнатной температуре методом “остановленной струи”, предложен механизм внедрения платины в координированные БТ и ДБТ. Установлено, что палладиевый комплекс [Pd(PPh3)2(C2H4)] способен внедряться в связи C–S в координированных тиофенах [69], однако металлатиациклы палладия менее устойчивы и образуются не так быстро, как платиновые.
Взаимодействие тиофена с [Pt2{Ph2P(CH2)3PPh2}H3]ClO4 приводит к активации связи C–S и частичному гидрированию тиофена. С чистым тиофеном образуется смесь моноядерного [Pt{Ph2P(CH2)3PPh2}(SC4H4–C,S)] и биядерного [Pt2{Ph2P(CH2)3PPh2}2(μ-SC4H5–C,S)]ClO4 комплексов в мольном соотношении 2 : 3. При использовании в качестве растворителя бензола или толуола это соотношение резко меняется на 1 : 9 [70]. В то время как действие HBF4 на моноядерный комплекс приводит к раскрытию тиофенового кольца и образованию [Pt2{Ph2P(CH2)3PPh2}2(µ-SC4H5)2](BF4)2, реакция HBF4 с биядерным комплексом дает соединение [Pt2{Ph2P(CH2)3PPh2}2(μ-SC4H6)](BF4)2, для которого выполнен рентгеноструктурный анализ (рис. 5).
Благодаря хелатному эффекту можно выделить обычно неустойчивые η2-тиофеновые комплексы. Так, селективная активация связи С–Н в тиофеновом фрагменте, а также появление η2-тиофенового интермедиата наблюдалось при взаимодействии [Pt(CH3)2(SMe2)]2 с монохинолиновыми производными Т [71]. С бис(хинолиновыми) производными получены и структурно охарактеризованы биядерные η2-тиофеновые комплексы с сэндвичевой структурой [72].
Методом DFT изучено превращение ДБТ в дифенил и H2S при действии Н2 на монослойных сульфидных кластерах NiMo18 [73]. Энергия активации для критических стадий разрыва двух связей C–S в ДБТ на никелевом активном центре оценена в примерно 32 ккал/моль.
Методом DFT показано, что тиофеновые комплексы никеля занимают промежуточное положение между стабильными комплексами железа и наименее устойчивыми комплексами кобальта. Бис(η2-тиофеновые) комплексы никеля склонны к распаду на монотиофеновое соединение и свободный тиофен. Расчетная частота асимметричных валентных колебаний тиофен – Ni – тиофен для Ni(η2-тиофен)2 составляет 383 см–1, а валентных колебаний Ni – тиофен в Ni(η5-тиофен) – 150 см–1 [42].
Образование устойчивого тиаплатинового кольца происходит при действии на T [Pt{i-Pr2P(CH2)2P-i-Pr2}H]2 в ТГФ (>60°C) [74]. Методами DFT и МО исследовано аналогичное гипотетическое соединение [Pt{Me2P(CH2)2P-Me2}-η2(C,S)C4H4] [50]. Расчеты показывают, что реакция является в целом экзотермической и, кроме того, предсказывают, что начальная η2-координация T через двойные связи C=C энергетически более выгодна, чем координация через атом серы (ΔG 9.3 ккал/моль). В присутствии ТГФ величина ΔG возрастает до 11.4 ккал/моль.
ХРОМ, МОЛИБДЕН И ВОЛЬФРАМ
Ультрафиолетовым фотолизом гексановых растворов карбонилов М(CO)6 (М = Мо, W, Cr) с избытком 2,5-Me2T, БТ и ДБТ получены малоустойчивые комплексы типа η1(S) соответствующего тиофенового лиганда с фрагментом М(CO)5 (рис. 6 а) [75]. Только комплексы хрома и вольфрама с ДБТ существовали несколько дней, поэтому для них удалось сделать рентгеноструктурный анализ. Из этого можно сделать вывод, что ДБТ связывается с металлами прочнее, чем БТ или 2,5-Me2T, что было показано ранее для ру-тения в работах [40, 41]. С ДБТ и карбонилом молибдена наряду с вышеуказанным получается π-комплекс Mo(CO)3(η6-ДБТ), в котором металл координирован с бензольным кольцом (рис. 6 б).
В комплексах W и Cr атом серы находится в центре тригональной пирамиды (псевдо-sp3-гибридизация). Нечто подобное наблюдается в [FeCp(CO)2(η1(S)ДБТ)] [76], [Ir(C5Me5)Cl2(η1(S)ДБТ)] [77], [Мо{Me2Si(C5Me4)2}(η1-ДБТ)] [78] и других η1-тиофеновых комплексах (рис. 7 ). Пирамидальная геометрия серы в η1-тиофеновых комплексах – характерный признак уменьшения антисвязывающих взаимодействий между неподеленной электронной парой атома серы и заполненными d-орбиталями металла. Связь М–С существенно короче для транс-СО, чем для цис-СО. Угол θ между связью М–S и вектором от атома серы к середине расстояния между атомами С6 и С7 бензольного кольца ДБТ для вольфрамового комплекса чуть меньше, чем для хромового (118.8° и 121.8° соответственно). На основе расчетов по методу молекулярных орбиталей [79, 80] полагают, что величина угла θ в комплексах металл-тиофен зависит от способности металла к обратному π-допированию на тиофеновый лиганд. Чем больше электронная плотность на металле, тем сильнее обратное π-связывание с тиофеном и тем больше угол θ.
Сравнение реакционной способности Т и тетрагидротиофена при взаимодействии с [W(CO)5(цикло-С6Н12)], синтезированным лазерным фотолизом раствора W(CO)6 в циклогексане, показало, что более реакционноспособным является тетрагидротиофен, поскольку он более сильный донор электронов [81].
Для комплекса [W(NO)(PMe3){гидридотрис(пиразолил)борат}(η2-T)], полученного с выходом 31–40% из [W(NO)(PMe3){гидридотрис(пиразолил)борат}Br] и T, осуществлены алкилирование, окисление, протонирование и гидрирование некоординированной двойной связи [82].
Гидридные соединения вольфрама способны расщеплять связи C–S в Т, БТ и ДБТ с образованием комплексов, которые при нагревании в присутствии водорода выделяют продукты обессеривания: бутен-1, этилбензол и дифенил, соответственно. Например, [W(PMe3)4(η2-CH2PMe2)H] с ДБТ дает соединение [W(κ2-С12Н8)(PMe3)]-(μ‑S)(μ-CH2PMe2)(μ-PMe2)[W(PMe3)3] с дибензометаллациклопентадиеновым фрагментом [83]. Буква κ – греческая буква “каппа”.
Важно знать энергии адсорбции для всех промежуточных продуктов реакции, так как они определяют термодинамику реакции. Поэтому в работе [84] рассчитали энергии адсорбции для 12 различных серосодержащих молекул, координированных по типу η1 на 5 различных структурах Co/MoS2.
Показано, что анса-молибденоценовые гидриды [Мо{Me2Si(C5Me4)2}H2], в молекулах которых атомы в п-положениях ароматического кольца связаны цепью атомов –(СН2)n–, и [Mo{Me2Si(C5Me4)2}(Ph)H] расщепляют связи C–S в T и БТ [78] в отличие от незамещенных молибденоценов Cp2MoH2 [85]. Ключевая роль в осуществлении этой реакции принадлежит анса-мостику SiMe2, связывающему два циклопентадиенильных кольца. По предварительным расчетам, продукты разрыва связи C–S термодинамически более выгодны для анса-молибденоценовой системы, чем продукты активации связей С–Н для циклопентадиенильной системы. Увеличение стабильности продукта расщепления связи C–S, как считают авторы [78], является следствием смещения к η3,η3-координации циклопентадиенильных колец, чему способствует мостик SiMe2. Количество электронов у атома молибдена в [Мо{Me2Si(C5Me4)2}η2(C, S)(T)] меньше 18, и будет формально 14 в предельном случае, если циклопентадиенильные лиганды будут η3-координированы в чистом виде. В таком случае донирование неподеленной электронной пары атома серы обеспечит дополнительную возможность стабилизации продукта расщепления связи C–S.
В конкурирующей реакции между T и БТ при действии [Mo{SiMe2(C5Me4)2}(Ph)H] БТ кинетически более восприимчив к разрыву связи C–S (селективность 3 : 1). ДБТ дает только аддукт [Мо{Me2Si(C5Me4)2}(η1-ДБТ)] [78] (рис. 7 ).
Авторы постулируют, что возникает интермедиат [Мо{Me2Si(C5Me4)2}], первоначально присоединяющий тиофеновое соединение по типу η1(S). Затем в T и БТ η1(S)-интермедиатах металл внедряется в связь C–S, в то время как в ДБТ η1(S)-комплексе это не происходит. Показано, что ДБТ-лиганд в комплексе [Мо{Me2Si(C5Me4)2}(η1-ДБТ)] довольно слабо связан с молибденом: так, он легко замещается водородом и бензолом, T и БТ. Тем не менее, удалось выполнить рентгеноструктуный анализ не только соединения [Мо{Me2Si(C5Me4)2}η2(C, S)(T)], но и аддукта [Мо{Me2Si(C5Me4)2}(η1-ДБТ)] [78]. По сравнению с другими η1-тиофеновыми комплексами угол θ (М–S – центр тиофенового ядра) в [Мо{Me2Si(C5Me4)2}(η1-ДБТ)] меньше и составляет 26°, в то время как в других η1-тиофеновых комплексах величина этого угла находится в пределах 37–61°.
В отличие от молибденовых комплексов [Мо{Me2Si(C5Me4)2}η2(C,S)(T)] и [Мо{Me2Si(C5Me4)2}η2(C,S)(БТ)] вольфрамовый родственник [W(Cp2) η2(C,S)(T)] реагирует с бензолом, давая [W(Cp2)(Ph)H] [85].
Реакция Т с [Мо(PMe3)6] изучена методом DFT [86]. Сравнение различных возможных путей реакции выявило, что наиболее каталитически активные частицы – пяти- и четырехкоординированные фрагменты, возникающие при последовательной диссоциации двух групп PMe3 от [Мо(PMe3)6]. Авторы полагают, что частица [Mo(PMe3)4] с Т первоначально дает [Mo(PMe3)4(η1(S)Т)], перестраивающийся в [Mo(PMe3)4(η2-(C2,S)Т]. Затем происходит разрыв связи C–S тиофена и возникает интермедиат [Mo(PMe3)4(κ2-C2,S)(T)] (А). Бутадиентиолатный комплекс [Mo(η5-C4H5S) (PMe3)2(η2-CH2PMe2)] (Б) может получаться путем ухода одной группы PMe3, переноса водорода и структурных преобразований (рис. 8 ). Аддукт [Мо(PMe3)3(η5-Т)] может получаться из [Мо(PMe3)4(η2(C2,C3)Т)] в результате удаления одной группы PMe3 и изменения η2-координации в η5-координацию. По сравнению с [Мо(PMe3)3(η5-Т)] бутадиентиолатный комплекс Б более стабилен на 3.5 ккал/моль. Вычислены энергетический барьеры образования [Мо(PMe3)3(η5-Т)] и [Mo(η5-C4H5S) (PMe3)2(η2-CH2PMe2)], которые составляют 20 и 10.4 ккал/моль, соответственно. Такие барьеры легко преодолеваются, так что эти два интермедиата могут экспериментально наблюдаться. Таким образом, по результатам расчетов превращение интермедиата А в бутадиентиолатный комплекс Б термодинамически и кинетически может происходить очень легко.
Т, как показано in situ методом ИК-спектроскопии с Фурье-преобразованием и подтверждено расчетами [87], может адсорбироваться на поверхности сульфидированного катализатора Mo/γ-Al2O3 различными способами: через координацию по атому серы, по двойной связи С=С либо по связи С–С с ненасыщенными центрами Моd+, расположенными по краям граней структур MoS2. Бóльшая энтальпия образования комплекса с координацией по η2-типу свидетельствует о предпочтительности координации T с Мо-катализатором по двойной связи С=С. В координированном T уменьшается ароматичность и ослабляется связь С–S.
МАРГАНЕЦ И РЕНИЙ
Комплекс [Re(C5Me5)(CO)2БT] существует в виде равновесной смеси изомеров, в которой БТ координируется с рением по двойной связи (η2-2,3) или по атому серы (η1-S) [88, 89].
В соединении [MnCp(CO)3] при ультрафиолетовом фотолизе в гексане в присутствии избытка 2,5-Me2T, БТ или ДБТ один из карбонильных лигандов замещается на тиофеновый. Из тиофеновых продуктов только ДБТ-производное [MnCp(CO)2(η1(S)-ДБT)] более устойчиво и существует несколько дней, что позволило получить для него структурные характеристики [75]. Структура этого соединения подобна определенной для комплекса иридия [IrCpМе5Cl2(η1S-DBT)] [77] с син-ориентацией ДБТ-лиганда относительно циклопентадиенильного кольца.
Угол θ между связью М–S и вектором от атома серы к центроиду тиофенового фрагмента в комплексе [MnCp(CO)2(η1(S)ДБT)] составляет 125.6°, что сравнимо с вышеупомянутым соединением иридия (128°), но выше, чем в ДБТ-производных хрома (121.8°), вольфрама (118.8°) [75] и железа (119.4°) [76]. На основе расчетов по методу молекулярных орбиталей полагают, что величина угла θ увеличивается с ростом электронной плотности на металле и усилением за счет этого обратного π-связывания с тиофеном [79, 80].
Взаимодействие Re2(CO)10 с тиофеновыми соединениями в условиях ультрафиолетового фотолиза протекает по-разному [90, 91]: ДБТ дает два вещества – [Re2(CO)9(η1(S)ДБT] (В) и [Re2(CO)8(µ-C12H7S)(µ-H)] (Г), а 2,5-Me2T – продукт внедрения металла по связи С–S – [Re2(CO)7(µ-2,5-Me2T)] (Д) (рис. 9 ) [90]. В таких же условиях БТ и его метильные производные образуют комплексы [Re2(CO)7(µ-МехБT] (Е) (рис. 10 ) [91], в которых сохраняется связь Re–Re. Соединения [Re2(CO)9(µ1(S)МеБT)] (Ж) (рис. 10 ) получаются из [Re2(CO)9(ТГФ)] и БТ и его метильных замещенных при комнатной температуре. Под дейтвием УФ-излучения комплексы (Ж) легко и с выходами 40–60% превращаются в (Е). Фотолиз 3,5-Me2БТ и Re2(CO)10 в атмосфере водорода дает вещество (Е) и комплекс с частично гидрированным тиофеновым кольцом [Re2(CO)7(µ-3,5-Me2БT-H)(µ-H)]. В соединении (В) угол θ довольно мал (113°). Механизм реакции включает как отрыв СО, так и гомолитический распад связи Re–Re.
ЗАКЛЮЧЕНИЕ
Комплексообразование тиофеновых соединений с переходными металлами может происходить как за счет координации с π-системой Т или атомом серы (рис. 11 ), так и в результате внедрения атома металла в тиофеновое кольцо с разрывом связи С–S.
Комплексы типа η1(S) образуют рутений [RuH2(η2-H2){η1(S)-C12H8S}(PCy3)2] [36], [CpRu(CO)2{µ2-η1(S):η6-ДБT}RuC5Me5]2+ [39], иридий [IrH2(η1(S)T*)2(PPh3)2]PF6, где Т* – Т, БТ или ДБТ [60], [IrCpМе5Cl2(η1S-DBT)] [77], хром, молибден и вольфрам [η1(S)T*M(CO)5] [75], [Мо{Me2Si(C5Me4)2}(η1-ДБТ)] [78], марганец [MnCp(CO)2(η1(S)-ДБT)] [75], рений (рис. 9, 10 ) [90, 91].
Координация по двойной связи тиофенового кольца наблюдается в комплексе [W(NO)(PMe3){гидридотрис(пиразолил)борат}(η2-T)] [82], в [Re(C5Me5)(CO)2БT], который существует в виде равновесной смеси η2(С2=С3)- и η1(S)-изомеров [88, 89]. С двойной связью и атомом серы тиофенового цикла связывается рений и в комлексе (Е) [91] (рис. 10 ). Известен комплекс 2,5-Ме2Т, содержащий два атома рения, один из которых координирован по типу η5, а другой – по типу η2(C,S) [90] (рис. 9 , Д). η4-Тиоаллильная координация Т обнаружена в [RuH(η4(S,C)-SC4H5)(PCy3)2] [36].
Устойчивость η1(S)-комплексов невелика. В комплексе [Мо{Me2Si(C5Me4)2}(η1-ДБТ)] ДБТ-лиганд легко замещается водородом и бензолом, а также T и БТ [78]. Соединение [MnCp(CO)2(η1(S)-ДБT)] существует несколько дней, время жизни подобных комплексов других тиофеновых производных еще меньше [75]. Комплексы типа η1-S тиофенового лиганда с фрагментом М(CO)5 (М = Cr, Mo, W) (рис. 6 а) малоустойчивы [75].
Начальная η2-координация T через двойные связи C=C энергетически более выгодна, чем координация через атом серы [50, 87]. η2-Тиофеновые комплексы обычно неустойчивы. Стабильность тиофеновых комплексов возрастает в ряду: кобальт < никель < железo [42]. Бис(η2-тиофеновые) комплексы никеля склонны к распаду на монотиофеновое соединение и свободный тиофен [42].
По сравнению с [Мо(PMe3)3(η5-Т)] бутадиентиолатный комплекс более стабилен на 3.5 ккал/моль. Энергетический барьер образования [Мо(PMe3)3(η5-Т)] составляет 20 ккал/моль [86].
Установлено, что как η1(S)-координация, так и η2(C,C)-координация приводит к интермедиату η2(C,S) который непосредственно предшествует расщеплению связи C–S, поскольку в координированном T уменьшается ароматичность и ослабляется связь С–S.
Никель, палладий и платина весьма активно взаимодействуют с тиофеновыми соединениями, что обычно приводит к внедрению металла по связи C–S и последующему разрушению тиофенового цикла.
Реакция тиофеновых соединений с комплексами металлов является в целом экзотермической.
Электронное влияние заместителей в тиофеновых производных незначительно, а стерические эффекты велики. Так, например, связь С–S в Ме4Т, 4,6-Ме2- и 4,6-Et2ДБТ не расщепляется. Получаются лишь лабильные комплексы со связью М–S.
Для расщепление связи C–S в БТ металл-координационно ненасыщенных нуклеофильных фрагментов, как правило, внедряется в связь C(винил)–S, а не в более сильную связь С(арил)–S свободного БТ.
η6-Координация БТ и ДБТ на промышленных сульфидных Мо/Со-катализаторах гидрообессеривания может быть действенным механизмом активации связей C–S до внедрения металла и последующего гидрирования.
Сравнение соединений со связями С–S и С–О в конкурирующих условиях показывает, что расщепляются только связи С–S.
Рис. 1.
Интермедиат из реакции {(h5-C5Me5)Ru}3(µ-H)3(µ3-H)2 с БТ [29] – рутенатиациклогексадиеновый комплекс.

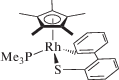
Рис. 3.
Продукт внедрения платины по связи C–S в 4,6-диметил-ДБТ с образованием: цис-[Pt(PEt3)2(η2-C,S-C14H12S)] [66].



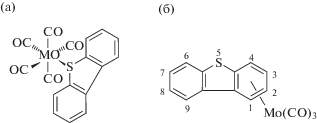

Рис. 8.
Вероятная схема превращения комплекса [Mo(PMe3)4(κ2-C2,S-тиофен)] (А) в бутадиентиолатный комплекс [Mo(η5-C4H5S) (PMe3)2(η2-CH2PMe2)] (Б) [86].
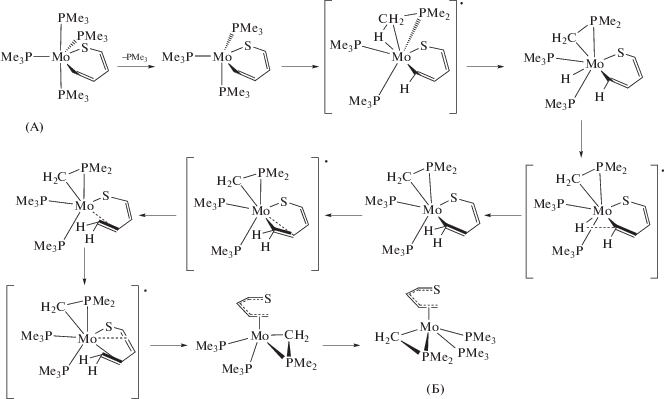
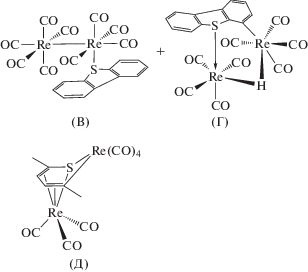

Рис. 11.
Возможные способы связывания T с переходным металлом [28]: 1 – М[η1(S)]; 2 – M(η2-T); 3 – М2(η2-T); 4 – M(η4-Т); 5 – M(η2-T)M(η2-T), 6 – M(η4-Т)М'[η1(S)], 7 – M(η4-Т)${\text{M}}_{{\text{2}}}^{{\text{'}}}$[η1(S)], 8 –M(η5-Т).

Список литературы
Pawelec B., Navarro R.M., Campos-Martin J.M., Fierro J.L.G. // Catal. Sci. Technol. 2011. V. 1. P. 23.
Srivastava V.C. // RSC Adv. 2012. V. 2. P. 759.
Stanislaus A., Marafi A., Rana M.S. // Catal. Today. 2010. V. 1. P. 1.
Wang L., He W., Yu Z. // Chem. Soc. Rev. 2013. V. 42. P. 599.
Pashigreva A.V., Bukhtiyarova G.A., Klimov O.V., Chesalov Yu.A., Litvak G.S., Noskov A.S. // Catal. Today. 2010. V. 149. № 1–2. P. 19.
Klimov O.V., Pashigreva A.V., Fedotov M.A., Kochubey D.I., Chesalov Yu.A., Bukhtiyarova G. A., Noskov A.S. // J. Mol. Catal. A: Chem. 2010. V. 322. № 1–2. P. 80.
Pashigreva A.V., Klimov O.V., Bukhtiyarova G.A., Kochubey D.I., Prosvirin I.P., Chesalov Yu.A., Zaikovskii V.I., Noskov A.S. // Catal. Today. 2010. V. 150. P. 164.
Leonova K.A., Klimov O.V., Gerasimov E.Yu, Dik P.P., Pereyma V.Yu., Budukva S.V., Noskov A.S. // Adsorption. 2013. https://doi.org/10.1007/s10450-013-9500-0
Shih S.S., Mizahi S., Green L.A., Sarli M.S. // Ind. Eng. Chem. Res. 1992. V. 31. P. 1232.
Topsøe H., Gates B.C. // Polyhedron. 1997. V. 16. P. 3212.
Houalla M., Broderic D.H., Sapre A.V., Nag N.K., de Beer V.H.J., Gates B.C., Kwart H. // J. Catal. 1980. V. 61. P. 523.
Ma X., Sakanishi K. Mochida I. // Ind. Eng. Chem. Res. 1995. V. 34. P. 748.
Старцев А.Н., Захаров И.И. // Успехи химии. 2003. Т. 72. № 6. С. 579.
Su J.-C., Chang J.-H., Huang J.-W., Chen P.P.-Y., Chen K.-F., Tseng P.-H., Shiau C.-W. // Chem.-Biol. Interactions. 2015. V. 228. P. 108.
Mobin S.M., Tauqeer M., Mohammad A., Mishra V., Kumari P. // J. Coord. Chem. 2016. V. 69. P. 2015.
He Y., Liu Q., Liu F., Huang C., Peng C., Yang Q., Wang H., Hu J., Liu H. // Micropor. Mesopor. Mater. 2016. V. 233. P. 10.
Hammud H.H., El Shazly S., Sonji G., Sonji N., Bouhadir K.H. // Spectrochim. Acta. Part A. 2015. V. 150. P. 94.
González B., del Valle M.A., Díaz F.R., Espinosa-Bustos C., Ramírez A.M.R., Hernández L.A. // J. Appl. Polym. Sci. 2016. V. 133. P. 43547.
Angelici R.J. // Coord. Chem. Rev. 1990. V. 105. P. 61.
Rauchfuss T.B. The coordination chemistry of thiophenes // In: Progress in Inorganic Chemistry. V. 39. Ed. Lippard S.J. Ch. 5. Hoboken, NJ, USA: J. Wiley & Sons, Inc., 1991. P. 259.
Sánchez-Delgado R. A. // J. Mol. Catal. 1994. V. 86. № 1. P. 287.
Angelici R.J. // Bull. Soc. Chim. Belges. 1995. V. 104. № 4–5. P. 265.
Angelici R.J. // Polyhedron. 1997. V. 16. № 18. P. 3073.
Harris S. // Polyhedron. 1997. V.16. № 18. P. 3219.
Angelici R.J. // In: Transition Metal Sulphides. NATO ASI Series. Springer Netherlands.1998. V. 60. P. 89.
Angelici R.J. // Organometallics. 2001. V. 20. № 7. P. 1259.
Sánchez-Delgado R.A. // In: Organometallic Modeling of the Hydrodesulfurization and Hydrodenitrogenation Reactions. Catalysis by Metal Complexes. Springer Netherlands. 2002. V. 24. P. 95.
Wang L., He W., Yu Z. // Chem. Soc. Rev. 2013. V. 42. P. 599.
Matsubara K., Okamura R., Tanaka M., Suzuki H. // J. Amer. Chem. Soc. 1998. V. 120. № 5. P. 1108.
King R.B., Treichel P.M., Stone F.G.A. // J. Amer. Chem. Soc. 1961. V. 83. P. 3600.
Hübener P., Weiss E. // J. Organomet. Chem. 1977. V. 129. P. 105.
Ogilvy A.E., Draganjac M., Rauchfuss T.B., Wilson S.R. // Organometallics. 1988. V. 7. P. 1171.
Shibue M., Hirotsu M., Nishioka T., Kinoshita I. // Organometallics. 2008. V. 27. № 17. P. 4475.
Hirotsu M., Tsuboi C., Nishioka T., Kinoshita I. // Dalton Trans. 2011. V. 40. P. 785.
Hirotsu M., Santo K., Hashimoto H., Kinoshita I. // Organometallics. 2012. V. 31. № 21. P. 7548.
Borowski A F., Sabo-Etienne S., Donnadieu B., Chaudret B. // Organometallics. 2003. V. 22. № 23. P. 4803.
McKinley S.G., Angelici R.J. // Energy & Fuels. 2003. V. 17. № 6. P. 1480.
Urban S., Beiring B., Ortega N., Paul D., Glorius F. // J. Amer. Chem. Soc. 2012. V. 134. № 37. P. 1524.
Vecchi P.A., Ellern A., Angelici R.J. // Organometallics. 2005. V. 24. № 15. P. 3725.
Benson J.W., Angelici R.J. // Organometallics. 1992. V. 11. P. 922.
Benson J. W., Angelici R. J. // Organometallics. 1992. V. 12. P. 680.
Ding Y., He M., Niu Y., Wang D., Cui Y., Feng S. // J. Phys. Chem. A. 2009. V. 113. № 38. P. 10291.
Jones W.D., Dong L. // J. Amer. Chem. Soc. 1991. V. 113. № 2. P. 559.
Dong L., Duckett S.B., Ohman K.F., Jones W.D. // J. Am. Chem. Soc. 1992. V. 114. № 1. P. 151.
Jones W.D., Chin R.M. // J. Amer. Chem. Soc. 1994. V. 116. № 1. P. 198.
Jones W.D., Vicic D.A., Chin R.M., Roache J.H., Myers A.W. // Polyhedron. 1997. V. 16. № 18. P. 3115.
Ateşin T.A., Jones W.D. // Organometallics. 2008. V. 27. № 15. P. 3666.
Ateşin T.A., Jones W.D. // Inorg. Chem. 2008. V. 47. № 23. P. 10 889.
Sargent A.L., Titus E.P. // Organometallics. 1998. V. 17. № 1. P. 65.
Ateşin T.A., Jones W.D. // Organometallics. 2008. V. 27. № 1. P. 53.
Maresca O., Maseras F., Lledós A. // New. J. Chem. 2004. V. 28. P. 625.
Oster S.S., Grochowski M.R., Lachicotte R.J., Brennessel W.W., Jones W.D. // Organometallics. 2010. V. 29. № 21. P. 4923.
Grieb A.L., Merola J.S. // J. Organomet. Chem. 2012. V. 713. P. 163.
Bleeke J.R., Hinkle P.V. // J. Amer. Chem. Soc. 1999. V. 121. № 3. P. 595.
Bleeke J.R., Hinkle P.V., Shokeen M., Rath N.P. // Organometallics. 2004. V. 23. № 17. P. 4139.
Welch W.R.W., Harris S. // Inorg. Chim. Acta. 2008. V. 361. P. 3012.
Bleeke J.R. // Acc. Chem. Res. 2007. V. 40. № 10. P. 1035.
Grochowski M.R., Brennessel W.W., Jones W.D. // Organometallics. 2009. V. 28. P. 2661.
Grochowski M.R., Brennessel W.W., Jones W.D. // J. Chem. Crystallogr. 2011. 1007/s10870-011-0006-x
Sánchez-Delgado R.A., Herrera V., Bianchini C., Masi D., Mealli C. // Inorg. Chem. 1993. V. 32. № 17. P. 3766.
Vicic D.A., Jones W.D. // Organometallics. 1998. V. 17. P. 3411.
Vicic D.A., Jones W.D. // J. Amer. Chem. Soc. 1997. V. 119. P. 10855.
Grochowski M.R., Li T., Brennessel W.W., Jones W.D. // J. Amer. Chem. Soc. 2010. V. 132. № 35. P. 12 412.
Arévalo A., García J. J. // Eur. J. Inorg. Chem. 2010. № 26. P. 4063.
Garcia J.J., Mann B.E., Adams H., Bailey N.A., Maitlis P.M. // J. Amer. Chem. Soc. 1995. V. 117. № 8. P. 2179.
Arévalo A., Bernés S., García J.J., Maitlis P.M. // Organometallics. 1999. V. 18. № 9. P. 1680.
Dullaghan C.A., Zhang X., Greene D.L., Carpenter G.B., Sweigart D.A., Camiletti C., Rajaseelan E. // Organometallics. 1998. V. 17. P. 3316.
Li H., Carpenter G.B., Sweigart D.A. // Organometallics. 2000. V. 19. P. 1823.
Yu K., Li H., Watson E.J., Virkaitis K.L., Carpenter G.B., Sweigart D.A. // Organometallics. 2001. V. 20. № 16. P. 3550.
Nova A., Novio F., González-Duarte P., Lledós A., Mas-Ballesté R. // Eur. J. Inorg. Chem. 2007. № 36. P. 5707.
Tan R., Song D. // Organometallics. 2011. V. 30. № 6. P. 1637.
Tan R., Song D. // Inorg. Chem. 2010. V. 49. № 5. P. 2026.
Weber T., van Veen J.A.R. // Catal. Today. 2008. V. 130. P. 170.
Ateşin. T.A., Oster S.S., Skugrud K., Jones W.D. // Inorg. Chim. Acta. 2006. V. 359. P. 2798.
Reynolds M.A., Guzei I.A., Logsdon B.C., Thomas L.M., Jacobson R.A., Angelici R.J. // Organometallics. 1999. V. 18. № 20. P. 4075.
Goodrich J D., Nickias P.N., Selegue J.P. // Inorg. Chem. 1987. V. 26. P. 3426.
Rao K.M., Day C.L., Jacobson R.A., Angelici R.J. // Inorg. Chem. 1991. V. 30. P. 5046.
Churchill D.G., Bridgewater B.M., Parkin G. // J. Amer. Chem. Soc. 2000. V. 122. № 1. P. 178.
Harris S. // Organometallics. 1994. V. 13. P. 2628.
Harris S. // Polyhedron. 1997. V. 16. P. 3219.
Schultz R.H. // Organometallics. 2004. V. 23. № 19. P. 4349.
Delafuente D.A., Myers W.H., Sabat M., Harman W.D. // Organometallics. 2005. V. 24. № 8. P. 1876.
Sattler A., Parkin G. // J. Amer. Chem. Soc. 2011. V. 133. № 11. P. 3748.
Joshi Y.V., Ghosh P., Venkataraman P.S., Delgass W.N., Thomson K.T. // J. Phys. Chem. C. 2009. V. 113. № 22. P. 9698.
Jones W.D., Chin RM., Crane T.W., Baruch D.M. // Organometallics. 1994. V. 13. P. 4448.
Liao C., Wang J., Li B. // J. Organomet. Chem. 2014. V. 749. P. 275.
Liu D., Li Z., Sun Q., Kong X., Zhao A., Wang Z. // Fuel. 2012. V. 92. № 1. P. 77.
Choi M.G., Angelici R.J. // Organometallics. 1992. V. 11. № 10. P. 3328.
Rudd J. A. II, Angelici R.J. // Inorg. Chim. Acta. 1995. V. 240. № 1–2. P. 393.
Reynolds M.A., Guzei I.A., Angelici R.J. // Organometallics. 2001. V. 20. № 6. P. 1071.
Reynolds M.A., Guzei I.A., Angelici R.J. // J. Amer. Chem. Soc. 2002. V. 124. № 8. P. 1689.
Дополнительные материалы отсутствуют.