

Нефтехимия, 2020, T. 60, № 4, стр. 464-475
Химическая переработка отходов полимеров в моторные топлива и нефтехимическое сырье (обзор)
М. Л. Грингольц 1, К. И. Дементьев 1, *, Х. М. Кадиев 1, А. Л. Максимов 1, Е. Ш. Финкельштейн 1
1 Институт нефтехимического синтеза им. А.В. Топчиева РАН
119991 Москва, Россия
* E-mail: kdementev@ips.ac.ru
Поступила в редакцию 03.01.2020
После доработки 21.02.2020
Принята к публикации 11.03.2020
Аннотация
Рассмотрены проблемы переработки отходов полимеров основных типов. Особое внимание уделяется работам, связанным с использованием современных способов конверсии отходов, позволяющих получить моторные топлива и ценные нефтехимические продукты. Подробно обсуждаются процессы термического и каталитического крекинга, гидроконверсии, а также метатезиса полимеров.
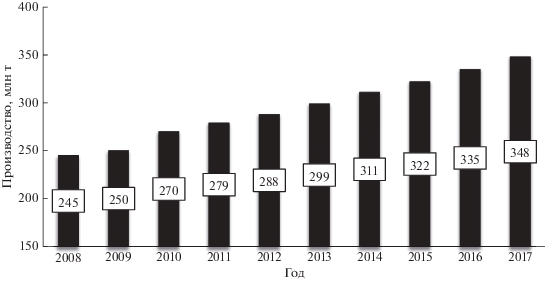
Полимеры и материалы на их основе давно стали важной и неотъемлемой частью нашей жизни. Их популярность обусловлена уникальным набором ценных свойств – полимеры устойчивы к различным факторам окружающей среды и не разлагаются при дальнейшей переработке. Часто они заменяют традиционные материалы – дерево, металл, стекло, а во многих случаях у полимеров нет аналогов. По этой причине объем производства полимерных материалов значительно увеличился за последние 60 лет и в настоящее время превышает 340 млн т (рис. 1). Среднегодовой прирост объемов производства в последние 10 лет составляет около 3.5–4.0% [1].
Основными областями применения полимерных материалов являются изготовление упаковочных изделий (39.9%), строительство зданий и сооружений (19.7%), автомобилестроение (8.9%), электроника (5.8%), сельское хозяйство (3.3%) и другие (22.4%), в которые входят продукты потребления, бытовые продукты, мебельные изделия, изделия для спорта, здоровья и т.д. [2]. Наиболее массовые виды полимеров – полиэтилен низкой и высокой плотности (ПЭНП, ПЭВП), полиэтилентерефталат (ПЭТФ), полипропилен (ПП), полистирол (ПС) и поливинилхлорид (ПВХ) [3]. По некоторым оценкам, до 70% всей массы отходов пластиков составляет использованная упаковка, состоящая из перечисленных полимеров [4], причем основным компонентом отходов является полиэтилен [5].
Широкое использование полимеров в качестве упаковочного материала – основной фактор, угрожающий экологическому благополучию окружающей среды. Жизненный цикл упаковки не превышает месяца, после чего она выбрасывается [6]. На данный момент большинство полимерных изделий не перерабатываются из-за сложностей со сбором отходов и выделения из них полимеров [7]. За последние годы, вместе с мировым ростом производства пластмасс увеличилось и количество пластиковых твердых отходов, которые в развивающихся странах составляют уже 8–13% от общей массы твердых бытовых отходов [8]. Так, например, только в России ежегодное накопление полимерных отходов составляет примерно 1 млн т [9, 10].
На производство полимеров ежегодно тратится в общей сложности около 7–8% добываемых углеводородных ресурсов. Так как эти ресурсы относится к классу невозобновляемых, то вопрос утилизации полимерных отходов с получением ценных продуктов стоит достаточно остро [11].
Традиционные способы переработки полимерных отходов – захоронение, сжигание, пиролиз – характеризуются массой недостатков (высокие энергозатраты, невозможность получения ценных продуктов и др.) и, вследствие этого, низкой экономической эффективностью, а вторичное использование имеет крайне ограниченное применение [12]. Поскольку для производства полимеров используется невозобновляемое углеводородное сырье, химические методы переработки с получением топлив и/или продуктов нефтехимии выглядят гораздо более перспективными.
ТЕРМИЧЕСКИЙ И КАТАЛИТИЧЕСКИЙ КРЕКИНГ
Термическая деструкция полимеров является объектом изучения с даты их первого коммерческого использования. Будучи изначально направленной на выяснение пределов условий эксплуатации изделий из полимеров, в настоящее время термическая деструкция вызывает интерес с точки зрения применения для переработки отходов полимеров.
Термическая деструкция большинства полимеров в отсутствие катализатора становится заметной при температурах свыше 250°С и при дальнейшем повышении до 400–500°С протекает с достаточной полнотой, однако, для большинства полимеров неселективно. Основной продукт деструкции полиолефинов при 500°С – жидкий продукт широкого фракционного состава, выход газа невелик и не превышает 5–7% [13]. При деструкции ПВХ и ПЭТФ, помимо жидкой фракции и газа образуется смоляной остаток в количестве свыше 50% на исходный полимер, что происходит вследствие интенсивного протекания реакций циклизации и ароматизации в процессе деструкции [14, 15].
Газ деполимеризации состоит, в основном, из метана и насыщенных углеводородов (УВ) С2–С4. Повышение температуры до 600–700°С приводит к увеличению выхода газа, однако, состав его меняется слабо. Жидкий продукт деполимеризации представляет собой смесь большого числа соединений, причем его состав зависит не только от типа, но и от структуры соответствующего полимера [16]. Деполимеризат полиэтилена и полипропилена содержит, в основном, неразветвленные и слаборазветвленные алканы и некоторое количество олефиновых УВ С6–С20 [13], продукты деструкции полистирола, напротив, представляют собой концентрат моноароматических УВ с примесью димеров и тримеров стирола [17]. В жидком продукте деполимеризации ПЭТФ превалирует бензойная кислота и ряд ее эфиров [18].
Содержание мономера в продуктах деполимеризации зависит от типа соответствующего полимера. Наибольший выход мономера достигается в случае полиметилметакрилата, политетрафторэтилена и полиметакрилонитрила (свыше 90%), в случае полистирола выход составляет составляет 40–45%, а при разложении полиэтилена и полипропилена – менее 1% (табл. 1). По всей видимости, высокий выход мономера зависит от наличия четвертичных атомов углерода и заместителей с отрицательным индуктивным эффектом в структуре полимера, что определяет особый механизм деструкции полимерной цепи.
Таблица 1.
Выход мономера при деструкции полимеров различного типа [19].
| Полимер | Выход мономера, мас. % |
|---|---|
| Полиметилметакрилат | 91–98 |
| Политетрафторэтилен | 95 |
| Полиметакрилонитрил | 90 |
| Полистирол | 42–45 |
| Полиизобутилен | 18–25 |
| Полиэтилен | 0.03 |
| Полипропилен | 0–17 |
| Полибутадиен | 1 |
| Поливинилхлорид | 0–0.07 |
| Полиакрилонитрил | 5 |
Таким образом, термическая деполимеризация не позволяет получить ценные продукты из отходов полимеров в одну стадию, что обуславливает интерес к проведению деполимеризации в присутствии катализаторов.
Чаще всего в качестве катализаторов деполимеризации применяют аморфные алюмосиликаты и цеолиты, использование которых приводит к существенному углублению деполимеризации и изменению селективностей по продуктам. Крекинг полиэтилена при 470–500°C в присутствии цеолитов Y и Beta протекает практически количественно до газа, при этом выход жидких и твердых продуктов составляет не более 5–7% [20]. Газ крекинга при этом содержит, в основном, УВ С3–С4, а жидкий продукт обогащен ароматическими УВ. Снижение температуры крекинга до 450°С приводит к уменьшению выхода газа до 19–27% за счет увеличения выхода жидкого продукта [21]. Распределение продуктов крекинга и их состав в присутствии фожазита, морденита и аморфного алюмосиликата отличается слабо, в то время, как в присутствии ZSM-5 выход газовой фракции составляет 45–60%, а жидкая фракция содержит свыше 50% ароматических УВ С6–С9, что очевидно, объясняется различием в размере пор катализаторов [22]. Перспективным видом катализаторов являются также мезопористые материалы. В [23] изучался крекинг полистирола в присутствии MCM-41; показано, что MCM-41 проявляет высокую каталитическую активность и высокую селективность к углеводородам C5–C12, а наибольший выход стирола (50.7%) достигается при соотношении сырье/катализатор около 200.
Для деполимеризации полистирола активно изучаются также катализаторы на основе оксидов металлов [24]. По всей видимости, наилучшими катализаторами конверсии полистирола являются цинк, медь и магний [25]. Для изучения влияния состояния цинка на деполимеризацию полистирола реакцию проводили в присутствии массивных катализаторов на основе Zn (Zn, ZnO, ZnCl2) [26]. Наилучшие показатели были достигнуты при использовании цинка в металлической форме при 450°C в течение 120 мин: выход стирола составил 47.9% при почти количественном выходе жидких продуктов. Для сравнения при термическом пиролизе без катализатора выход стирола в тех же условиях составляет 39.3%.
Влияние носителя на процесс деполимеризации полистирола исследовалось в работе [27]. Изучалось разложение вспененного полистирола (EPS), полученного из отходов, в реакторе периодического действия при 450–500°C в течение 30–60 мин с применением следующих катализаторов: металлическая медь, Cu–Al2O3, Cu–Mmn (Mmn – монтмориллонит) и Cu–AC (AC – активированный уголь). При применении катализаторов на носителях было зафиксировано повышение селективности по низкомолекулярным ароматическим УВ по сравнению с массивной медью. Наибольший выход мономера стирола 60.5% был достигнут в присутствии 20% Cu–Al2O3.
Shah и соавт. в [28] изучали переработку отходов полистирола в ценные продукты с использованием катализаторов на основе магния. Реакция проводилась с вспененным полистиролом в реакторе периодического действия при 450–500°C в течение 30 мин с катализаторами Mg, 15% Mg–Al2O3, 20% Mg–Mmn, 20% Mg–AC (активированный уголь). Наибольший выход жидких продуктов и мономера стирола, как и в случае меди, был получен при использовании катализатора на основе оксида алюминия – 95.5% и 56.2%, соответственно.
Тем не менее, несмотря на возможность практически полной деполимеризации отходов основных полимеров, состав получаемых продуктов обуславливает их низкую ценность. Газ деполимеризации, как правило, содержит большое количество “сухих” компонентов (водород, метан), а также предельных УВ. Что касается жидкого продукта, то он может быть использован как топливо, однако, из-за высокого содержания олефинов (а в случае деполимеризатов полистрола, стирола и α-метилстирола) его термоокислительная стабильность очень низкая. Также, применение продукта разложения полистирола с высоким содержанием ароматических УВ сопровождается повышенным нагарообразованием в двигателе при использовании его в качестве автомобильного топлива.
Кроме того, при деполимеризации полимера, находящегося в твердом состоянии или в расплаве, существует ряд проблем, которые мешают повысить эффективность реакции: трудность обеспечения контакта между полимером и теплоносителем и, как следствие, неравномерный подвод тепла к молекулам полимера, интенсификация побочных реакций (в т.ч. реакций межмолекулярной конденсации) при высокой температуре и большом времени контакта. Поэтому важным фактором любого процесса термокаталитической переработки отходов полимеров является выбор реакционной системы. На сегодняшний день для организации процесса разложения отходов полимеров изучены практически все типы реакторных систем: автоклавы различных систем строения [29–31], реактор с псевдоожиженным слоем полимера и теплоносителя [32–34], реактор с фонтанирующим слоем [35], реактор с движущимся слоем [36]. Процесс разложения ведут как под давлением [31], так и под вакуумом; в ряде работ предложен микроволновой пиролиз [37, 38]. Наибольший выход газообразных и жидких продуктов наблюдается в реакторах с псевдоожиженным и фонтанирующим слоем. Наличие мелкозернистого теплоносителя при этом улучшает подвод тепла к гранулам полимера, а малое время пребывания паров снижает долю вторичных реакций.
В рамках другого подхода к решению проблемы предложено проводить разложение полистирола в среде углеводородов. Так, Puente c соавт. [39] в качестве сырья крекинга использовали раствор полистирола в бензоле, а в качестве реакционной системы – реактор с псевдоожиженным слоем теплоносителя при температуре 550°C и времени пребывания паров 3–9 с. Выход стирола при этом составил 75.6%, что близко к достигнутому максимуму. Выход газа в процессе деполимеризации крайне низок (не более 1%), твердых продуктов не образуется вовсе. В качестве среды для конверсии полимеров в присутствии катализатора изучались также толуол [40], легкий газойль [41] и вакуумный дистиллят [42].
Наибольший интерес в качестве сред для деполимеризации полистирола вызывают фракции жидких продуктов каталитического крекинга с Ткип > 200°С – легкий и тяжелый газойли (ЛГКК и ТГКК). Они обладают следующими преимуществами:
1) высокая растворяющая способность по отношению к полистиролу вследствие преимущественного содержания ароматических УВ;
2) стабильность к термическим превращениям при температурах вплоть до 550°С;
3) широкая доступность и низкая стоимость.
Осуществление деполимеризации полистирола в среде ЛГКК и ТГКК позволил получить выход стирола свыше 84% при полной конверсии полимера, что является наивысшим значением, достигнутым сегодня [43]. Высокая концентрация стирола в жидком продукте обуславливает возможность его экономически эффективного выделения и очистки с последующим использованием в качестве мономера.
При крекинге полиолефинов в среде нефтяных фракций селективное получение мономера затруднено, а потому более интересной является возможность получения моторных топлив и нефтехимического сырья. Изучена деполимеризация растворов полистирола [44], полиэтилена [45], полипропилена [46] в вакуумном дистилляте в условиях FCC с целью получения максимального количества жидкой фракции с применением коммерческих катализаторов [47]. Показано, что в этих условиях полиолефины претерпевают полное превращение, причем селективность по бензиновой фракции с октановым числом 92 п. составляет 50–60 мас. % [48].
ГИДРОКРЕКИНГ И ГИДРОКОНВЕРСИЯ
Не менее интересным процессом, позволяющим перерабатывать отходы полимеров в моторные топлива, является совместная гидроконверсия с нефтяными фракциями. В качестве модельных отходов полимеров изучались полиэтилен низкой и высокой плотности [49–52], полипропилен, полистирол, бакелит [49–53], полиэтилентерефталат [51] и поливинилхлорид [51, 52]. Кроме того, в [54] и [55] использовали образцы муниципальных пластиковых отходов. Работы в основном проводятся в автоклавах под давлением водорода 3.4–8.3 МПа при температурах 400–430°С с использованием промышленных катализаторов нефтепереработки. Представляет определенный интерес более подробное изложение отдельных публикаций, например [49, 51], где переработку пластиков вели соответственно в среде мазута и гудрона.
Так, в [49] рассматривали каталитические превращения полиэтилена плотностью d = 0.920–0.922 г/см3 (ПЭНП) и d = 0.948 г/см3 (ПЭВП), а также полипропилена (ПП; d = 0.918 г/см3) и полистирола (ПС; d = 1.050 г/см3). Растворителем служил мазут легкой Арабской нефти, содержавшийся в ней в количестве ~45%. Соотношение мазут/пластик было 3 : 2 по массе. В рабочую смесь вводили катализатор в количестве 1–3% в расчете на сырьевой пластик. Информация о катализаторах приведена в табл. 2.
Таблица 2.
Катализаторы гидроконверсии смесей полиолефинов и мазута арабской нефти
| № кат. | Катализатор | Состав и свойства |
|---|---|---|
| 1 (Н) | Ni,W/аморфн. алюмосиликат | Носитель – аморфн. Al–Si; уд. поверхность S = 239 м2/г, уд. объемом пор V = 0.3 см3/г; размер частиц d = 0.2–0.6 мм |
| 2 (Z) | Цеолит ZSM-5 | Мольное отношение SiО2/Al2O3 = 30; S = 390 м2/г; d = 2 мкм |
| 3 (F) | Катализатор процесса FCC (металл на цеолите Y) | Содержание Al 34%; S = 195 м2/г; d = 22–120 мкм |
| 4 (N) | NiMo/γ-Al2O3 | S = 210 м2/г; V = 0.38 см3/г; d = 0.2–0.6 мкм |
О положительной роли нефтяной фракции как среды гидроконверсии полимера свидетельствует следующий факт: в каталитическом процессе без мазута при типичных условиях переработки (430°С; 8.2 МПа; содержание катализатора 1%; τ = 1 ч) конверсия ПЭВД была 42.2–55.0%, тогда как в среде мазута она повысилась до 79.1–84.4% соответственно. Для ПЭНД без мазута, но в присутствии различных катализаторов, конверсия составляла 43.6–57.8%, а в среде мазута она достигла соответственно 78.4–86.9%. В случае полипропилена наблюдали повышение конверсии с 64.2–73.1% в чистом виде до 85.4–89.8% в среде мазута. В отличие от полиэтилена обоих типов и ПП, полистирол превращается практически количественно как в чистом виде, так и в среде мазута. При этом наибольшие значения конверсии были зафиксированы при использовании NiMo/γ-Al2O3 (табл. 3).
Таблица 3.
Влияние типа катализатора на конверсию (%) пластиков в среде мазута арабской нефти (при 430°С; 8.2 МПа; τ = 1 ч; 1% КТ, соотношении мазут/пластик 3 : 2 по массе)
| Катализатор (по табл. 2) | Пластик | |||
|---|---|---|---|---|
| ПЭНП | ПЭВП | ПП | ПС | |
| 1 (Н) Ni,W/аморфн. алюмосиикат | 79.1 | 79.7 | 85.4 | 96.0 |
| 2 (Z) Цеолит ZSM-5 | 81.3 | 78.4 | 87.3 | 95.3 |
| 3 (F) Катализатор процесса FCC | 81.5 | 85.0 | 88.0 | 96.3 |
| 4 (N) NiMo/γ-Al2O3 | 84.4 | 86.9 | 89.8 | 97.4 |
Роль количества вводимого катализатора с 1 до 5% установили на примере повышения конверсии полиэтилена LDPE с 84 до 88.2%, причем в основном за счет прироста масел (продуктов, растворимых в н-гексане) с 70.7 до 72.2%.
Влияние давления показано на примере ПЭНП. При варьировании давления от 3.42 до 8.2 МПа конверсия полиэтилена низкой плотности увеличилась с 79.1 до 84.4% (при промежуточном значении давления 6.84 МПа она составила 82.4%).
В работе [51] исследовали термическое разложение полиэтилена двух типов (ПЭНП и ПЭВП), полипропилена (ПП), полистирола (ПС), полиэтилентерефталата (ПЭТ) и поливинилхлорида (ПВХ). Изучение их проводили методом ТГА и установили, что за исключением ПВХ, кривые ТГА остальных пластиков однотипны, в них после начала разложения проявляется резкий рост потери массы вплоть до практически полного разложения без формирования твердого остатка. Особняком расположена кривая ТГА для ПВХ, который уже при Т < 300°С подвергается быстрому разложению с выделением HCl и низкомолекулярных хлорорганических соединений. К моменту Т ~ 350°С, когда остальные пластики еще не вступили в процесс разложения, потеря массы ПВХ уже достигает 65%. После прохождения участка пониженной активности в области 325–425°С для ПВХ наступает второй участок повышения потери массы вплоть до окончательного разложения. Для остальных пластиков при Т > 425°С открывается основной период интенсивного разложения, и ∫-образные кривые ТГА в соответствии с относительной реакционной способностью пластиков располагаются вдоль оси температур почти параллельно в ряду, обратном ряду их активности: ПС < ПЭТ < ПП ≈ ПЭНП < ПЭВП.
На примере ПП аналогичные опыты в режиме ТГА провели в среде вакуумного остатка нефти при соотношении 1 : 1. При этом в сырьевую смесь вводили катализаторы (табл. 4) в количестве 3–8% в расчете на пластик.
Таблица 4.
Сведения о катализаторах гидроконверсии пластиков
| Обозначение | Название | Фирма-изготовитель | Показатели * | ||
|---|---|---|---|---|---|
| S, м2/г | V, см3/г | Ktot, мМ/г | |||
| C-1 | NiMo/TiO2** | Ун-т Карачи (лаб. синтез) | 359 | 0.42 | 1.48 |
| C-2 | KC-2710 | AKZO Nobel | 182 | 0.23 | 0.85 |
| C-3 | Z-713 | ZeolistIntrnational | 221 | 0.34 | 1.15 |
| C-4 | HC-100 | UOP | 231 | 0.25 | 1.20 |
| C-5 | RCD-8 | UOP | 210 | 0.21 | 1.50 |
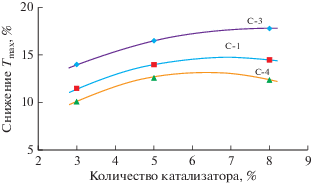
Изучая влияние катализаторов на превращения пластиков (отдельно и в смеси с вакуумным остатком нефти) методом ТГА, фиксировали температуру начала разложения Tin, достижения максимальной скорости реакции Tmax и ее завершения Tend, а также потерю массы Δm. В случае полипропилена и смеси ПП с гудроном авторы [51] отметили, что значения указанных температур понижались, а значения Δm возрастали при использовании всех катализаторов из табл. 4. Однако абсолютные значения показателей ТГА зависели от типа катализатора. Наибольшей активностью обладал контакт С-3 (катализатор гидрокрекинга Z-713), а наименьшей – контакт С-5 (катализатор каталитического крекинга RCD-8 разработки компании Юниверсал Ойл Продактс). Никельмолибденовый контакт С-1 на оксиде титана, синтезированный в лаборатории авторов, занимал по активности промежуточное положение между С-3 и С-4 (рис. 2).
Рис. 2.
Зависимость величины относительного (%) снижения температуры максимальной скорости превращения полипропилена в смеси (1 : 1) с гудроном от концентрации трех наиболее активных катализаторов (график по данным [51]).
Анализ приведенных литературных данных показывает, что весь предыдущий опыт каталитической переработки пластиков основан на применении твердых контактов, на которых стабилизированы активные каталитические частицы. Технологии прямой гидрогенизации твердых углеродсодержащих отходов практически отсутствуют, гидрогенизации преимущественно подвергались жидкие продукты термической деструкции твердых пластиков. Это было обусловлено отчасти и тем, что не были разработаны методы эффективного контакта твердых углеродсодержащих отходов с катализатором и водородом. Ситуация изменилась с разработкой метода гидрогенизационной переработки различных твердых органических материалов в жидкие УВ в присутствии наноразмерных катализаторов без традиционного твердого носителя, синтезируемых из предварительно диспергированных в углеводородной среде прекурсоров [56].
В разработанной в ИНХС РАН им. А.В. Топчиева технологии гидроконверсии тяжелого органического сырья использован водоростворимый прекурсор – парамолибдат аммония, вводимый в процесс в форме обратной эмульсии водного раствора в жидкой углеводородной среде. В результате термохимического превращения капель эмульсии прекурсора в восстановительной среде в присутствии серы формируется суспензия наноразмерных частиц MoS2, стабилизированных высокомолекулярными компонентами сырья. Этот метод положен в основу синтеза катализаторов для гидрогенизационной переработки остатков дистилляции нефтей, тяжелых высоковязких нефтей, битумов [57–59].
В работе [60] авторами представлены результаты гидрогенизационной переработки твердых пластиков (шииной резины и полиэтилена низкого давления) в смеси с тяжелыми нефтяными остатками в присутствии наноразмерных частиц катализатора, формируемых в реакционной среде из прекурсоров катализатора, содержащих Мо, (Zr + Al2О3), (Zr + Mo).
Следует отметить, что по данным термогравиметрических анализов (ТГА) шинной резины, полиэтилена и гудрона параметры их термической деструкции существенно разнятся: интервал температуры терморазложения составляет 275–470°С, 309–492°С и 264–598°С, потеря массы 66.4, 99.4 и 82.7 мас. %, а температура максимальной скорости разложения – 375, 465 и 420°С соответственно. Для ПЭВП данные по гидроконверсии и термической деструкции хорошо согласуются. При гидроконверсии суммарный выход газа, жидких продуктов и растворимых в толуоле продуктов составляет 99.8 мас. %, при термодеструкции в автоклаве 98.3 мас. %, а потеря массы по ТГА равна 99.4 мас. %. При гидроконверсии с применением прекурсора молибденового катализатора достигается практически полная конверсия ПЭВП с образованием газа и жидких продуктов (табл. 5). Образующийся нерастворимый в толуоле твердый остаток характеризуется высоким содержанием водорода, свидетельствует о глубоком протекании реакций гидрирования, как и в случае с резиной.
Таблица 5.
Гидроконверсия полиэтилена высокой плотности: Р = 7 МПа, Т = 450°С, ${{v}_{{{{{\text{H}}}_{{\text{2}}}}}}}$ = 18–20 нл/ч, время опыта на режиме 2 ч; прекурсор – парамолибдат аммония (Mo = 0.05 мас. %) [60].
| Показатели | Катализатор | ||
|---|---|---|---|
| Прекурсор | – | ПМА | |
| Газ, мас. % | 25.7 | 29.8 | |
| Жидкие углеводороды, мас. % | 72.1 | 70.0 | |
| Непревращенный остаток, мас. % | 2.2 | 0.2 | |
| В т.ч. нерастворимые в толуоле, мас. % | 1.7 | 0.16 | |
| Элементный состав нерастворимых в толуоле, мас. % | C | 85.7 | 90.7 |
| H | 2.6 | 6.2 | |
| Н/С (атомное) | 0.36 | 0.82 | |
Результаты исследования гидроконверсии смеси пластиков и тяжелым остатком нефти (табл. 6) показали принципиальную возможность и целесообразность переработки твердых полимерных отходов в смеси с тяжелыми нефтяными остатками. Отмечено положительное влияние использования линейных полимеров на результаты совместной гидрогенизации с гудроном.
Таблица 6.
Гидроконверсия гудрона и смеси его с резиной и/или полиэтиленом высокой плотности (содержание добавки – 33.5%); Р = 7 МПа, Т = 450°С, ${{v}_{{{{{\text{H}}}_{{\text{2}}}}}}}$ = 18–20 нл/ч, время опыта на режиме 2 ч. Прекурсор катализатора – парамолибдат аммония [60]
| Показатели | Параметры | |||
|---|---|---|---|---|
| Сырье | гудрон 100% | полимер 33.5% : гудрон 66.5% | ПЭВП + резина (1 : 1) : гудрон 66.5 мас. % | |
| резина | ПЭВП | |||
| Газ, мас. %. | 24.3 | 24.9 | 29.8 | 35.8 |
| Жидкие углеводороды, мас. % | 48.2 | 44.9 | 47.1 | 40.9 |
| Непревращенный остаток, мас. % | 27.5 | 30.2 | 23.1 | 23.3 |
| в т.ч. нерастворимые в толуоле, мас. % | 18.87 | 27.63 | 21.3 | 22.5 |
| Элементный состав, мас. %: | ||||
| C | 89.96 | 88.83 | 90.58 | 90.64 |
| H | 4.96 | 3.24 | 4.10 | 2.65 |
| N | 1.64 | 1.16 | 1.57 | 1.17 |
| S | 2.60 | 1.8 | 1.63 | 1.41 |
Важным условием эффективного протекания реакции гидрогенизации твердых пластиков в смеси с нефтяными остатками является растворение в них полимеров. В твердом состоянии молекулы полиэтилена имеют большие линейные размеры и переплетены. В работе [61] на примере ПЭВП показано, что для растворения твердого полимера необходимо разорвать связь С–С. Энергии сольватации для этой цели недостаточно. Отмечено, что растворение полиэтилена протекает через стадию набухания полимера с переходом в стадию образования истинного раствора. Поэтому растворение полиэтилена необходимо проводить при повышенной температуре с подбором соответствующего углеводородного растворителя в зависимости от типа полимера. Результаты исследования показывают, что процесс гомогенизации полиэтилена в тяжелых нефтяных остатках можно успешно проводить в температурном интервале 380–420°С, в котором наблюдается частичная деструкция как ПЭВП, так и компонентов тяжелого нефтяного остатка с образованием низкомолекулярных алканов и алкенов, являющихся растворителями ПЭВП [61].
С учетом современных тенденций в добыче жидких УВ, проявляющихся в постепенном утяжелении нефти, направление совместной переработки полимерных отходов с тяжелыми нефтяными остатками представляется весьма перспективным. Это направление изучено пока еще в недостаточной мере и от соответствующих исследований, в частности, с применением метода каталитической гидроконверсии с применением новейших наноразмерных катализаторов, можно ожидать достижения эффективных результатов.
МЕТАТЕЗИСНАЯ ДЕПОЛИМЕРИЗАЦИЯ
Реакция метатезиса олефинов, открытая всего лишь в середине 20-го века, быстро превратилась в эффективный инструмент органической и полимерной химии и нашла промышленное воплощение в процессах получения ряда низко- и высокомолекулярных продуктов [62]. Известны промышленные полимеры, полученные с помощью олефинового метатезиса: полинорборнен (торговая марка Norsorex), полидициклопентадиен (марки Telene®, Metton®, Pentam® и Proxima™), полиоктенамер (торговая марка Vestenamer) [62]. Необычность этой реакции заключается в разрыве двойных связей исходных соединений и образовании новых непредельных продуктов в мягких условиях под действием металлокарбеновых активных центров:
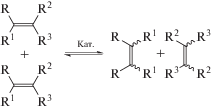
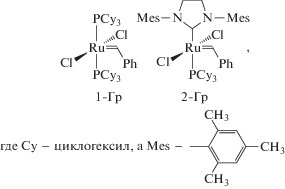
Метатезисная полимеризация циклоолефинов с раскрытием цикла (ROMP) и метатезисная полимеризация α,ώ-диенов (ADMET) приводит к полимерным продуктам. Полученные полимеры также могут претерпевать метатезис, который в зависимости от условий протекает по меж- или внутримолекулярной схеме. В результате, из полимеров под действием катализатора образуются циклоолигомеры по внутримолекулярной схеме. Последние могут быть отделены, а полимер снова подвергнут воздействию катализатора вплоть до полного превращения [62]. В качестве объектов деполимеризации, естественно, могут выступать лишь непредельные полимеры, такие как полиоктенамер, полибутадиен, полиизопрен, полинорборнен, блок-сополимеры стирола и бутадиена. В качестве катализаторов используются “классические” системы на базе соединений WCl6–SnBu4 [63], Mo- и W-содержащие системы Шрока – M(NAr)(CHC(CH3)2PH)(OCCH3(CF3)2)2 [64–66] и Ru-содержащие системы Граббса 1-Гр и 2-Гр [67–74]:
При использовании межмолекулярной схемы в качестве атакующего агента – обрывателя цепи – хорошо зарекомендовал себя этилен. В случае применения катализаторов Шрока этенолиз проводится в среде толуола при комнатной температуре [75, 76]. Показано, что деполимеризация осуществляется при необходимом для эффективной реакции давлении этилена или барботированием этилена через реакционную смесь ‒ раствор полимера в толуоле:

При этом в случае деполимеризации полибутадиена с Mn около 100 000 г/моль, образуются олигомеры с Mn порядка 1000–2000 г/моль. Дальнейшие попытки деполимеризации с помощью этилена не привели к дополнительной конверсии, т.е. по-видимому, избыток этилена каким-то образом реагирует с катализатором, дезактивируя его [75]. Процесс с участием этилена экономически оправдан, поскольку последний является самым дешевым агентом метатезисной деполимеризации.
Для сравнения, двойные связи некоторых непредельных полимеров, таких как полинорборнен, стерически затруднены и лишь в малой степени вступают в обменные реакции [75].
В качестве деполимеризующих агентов могут быть использованы 1-алкены или функциональные производные этилена, приводящие к ценным реакционноспособным олигомерам с химически активными концевыми группами. Следует отметить, что Mo-системы Шрока более толерантны к функциональным группам, чем W-системы:
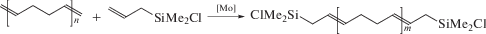
Механизм, приводящий к низкомолекулярным структурам с функциональными концевыми группами (так называемым, телехеликам), реализуется через первичное формирование циклических олигомеров. Интересно, что коммерческий полибутадиен может деполимеризоваться даже в отсутствие этилена и растворителя, в том числе, внутримолекулярно, на катализаторе Граббса первого поколения:

Деполимеризацию следует проводить при концентрациях ниже равновесной для того, чтобы способствовать более полному превращению в низкомолекулярные продукты. Ранее это было показано при использовании “классических” каталитических систем типа WCl6–EtAlCl2–EtOH. Если же исходная концентрация полимера выше равновесной, происходит лишь частичная деполимеризация, причем свежая доза катализатора инициирует дальнейшую деградацию [77, 78].
Попытки осуществить деполимеризацию бутилкаучука (сополимера изобутилена и изопрена) не привели к однозначным результатам. Процесс деполимеризации наблюдается, но осложнен и замедлен, возможно, из-за стерических затруднений, создаваемых изопреновыми звеньями [76, 79]. В то же время катализатор Граббса второго поколения оказался способным инициировать деполимеризацию зашитого серой цис-1,4-полиизопрена до жидких олигомеров [71]. Это представляет несомненный практический интерес.
К переработке ненасыщенных коммерческих полимеров с помощью реакции метатезиса следует отнести реакции межцепного кросс-метатезиса в смесях полимеров, исследование которых ведется в последнее время в разных научных группах, в том числе в ИНХС РАН [80–91]. Реакция включает взаимодействие катализатора (1-Гр, 2-Гр) с макромолекулами гомополимеров и образованием фрагмента макроцепи с металлокарбеном на конце. Образующийся полимерный металлокарбен вступает в межцепные обменные реакции, в результате которых сначала образуются ди-, а затем мультиблок-сополимеры.

Указанные исследования направлены на получение статистических мультиблок-сополимеров – ценных и малодоступных продуктов, сочетающих свойства исходных гомополимеров. Показано, что в сополимеры, обладающими улучшенными механическими свойствами, могут быть переработаны полимеры, синтезированные по различным механизмам, например, 1,4-полибутадиен или полиоктенамер и ненасыщенные полиэфиры, поликарбонаты или полиуретаны [82–84]. Кросс-метатезисом между несовместимыми коммерческими полибутадиеном и полиизопреном можно получить однофазные блок-сополимеры [85]. Указанный способ обеспечивает получение сополимеров, которые зачастую не могут быть синтезированы другим путем. Например, промышленные полинорборнен, полибутадиен, полиоктенамер и его замещенные могут быть переработаны кросс-метатезисом в сополимеры норборнена, малодоступные из-за высокой полимеризационной активности последнего, и обладающие регулируемыми термическими и кристаллическими свойствами [86–90].
Список литературы
Plastics – The Facts 2010–2018 / Plastics Europe Market Research Group (PEM RG). https://www.plasticseurope.org/en/resources/market-data. Дата обращения: 25.06.2019.
Plastics – the Facts 2016: An analysis of European plastics production, demand and waste data. Association of Plastics Manufactures. https://www.plasticseurope.org/ application/files/4315/1310/4805/plastic-the-fact-2016.pdf. Дата обращения: 09.12.2019.
Lettieri P., Al-Salem S. Thermochemical treatment of plastic solid waste. In: A Handbook for Management. Ed. by Letcher T.M., Vallero D., 2011. P. 233.
Scott D.S., Czernik S.R., Piskorz J., Radlein D.S.A.G. // Energy Fuels. 1990. V. 4. № 44. P. 407.
Zhou C., Fang W., Xu W., Cao A., Wang R. // J. of Cleaner Production. 2014. № 80. P. 80.
Achilias D.S., Antonakou E., Roupakias C., Megalokonomos P., Lappas A. // Global NEST J. 2008. V. 1. № 10. P. 114.
Polystyrene (PS). https://www.plasticseurope.org/en/ about-plastics/what-are-plastics/large-family/polystyrene. Дата обращения: 09.12.2019.
Waste: The Challenges Facing Developing Countries. 2012. https://www.proparco.fr/sites/proparco/files/ 2017-10/RevueSPD15_Waste_UK.pdf. Дата обращения: 09.12.2019.
Соловьянов А.А. // Твердые бытовые отходы. 2010. № 8. Р. 38.
Егорова М.В. // Вестник Казанского технологического университета. 2011. № 5. Р. 67.
Hopewell J., Dvorak R., Kosior E. // Phil. Trans. R. Soc. B. 2009. № 364. P. 2115.
Panda A.K., Singh R.K., Mishra D.K. // Renew. Sustain. Energy Rev. 2010. № 14. P. 233.
Williams P.T., Slaney E. // Resources, Conservation and Recycling. 2007. V. 51. № 4. P. 754.
Zhou J., Gui B., Qiao Y., Zhang J., Wang W., Yao H., Yu Y., Xu M. // Fuel. 2016. № 166. P. 526.
Montaudo G., Puglisi C., Samperi F. // Polymer Degradation and Stability. 1993. V. 42. № 1. P. 13.
Nonobe T., Ohtani H., Usami T., Mori T., Fukumori H., Hirata Y., Tsuge S. // J. Anal. Appl. Pyrolysis. 1995. V. 33(C). P. 121.
Shapi M.M., Hesso A. // J. Anal. Appl. Pyrolysis. 1990. V. 18. № 2. P. 143.
Dimitrov N., Krehula L.K., Siročić A.P., Hrnjak-Murgić Z. // Polymer Degradation and Stability. 2013. V. 98. № 5. P. 972.
Buekens A.G., Huang H. // Resources, Conservation and Recycling. 1998. V. 23. № 3. P. 163.
Zeaiter J. // Fuel. 2014. № 133. P. 276.
Seo Y.-H., Lee K.-H., Shin D.-H. // J. Anal. Appl. Pyrolysis. 2003. V. 70. P. 383.
Renzini M.S., Sedran U., Pierella L.B. // J. Anal. Appl. Pyrolysis. 2009. V. 86. P. 215.
Jin S., Cui K., Guan H., Yang M., Liu L., Lan Ch. // Applied Clay Science. 2012. V. 56. P. 1.
Nishino J., Itoh M., Ishinomori T., Kubota N., Uemichi Y. // J. Mater. Cycles Waste Manag. 2003. V. 5. P. 89.
Shah J., Jan M.R., Adnan // J. of Industrial and Engineering Chemistry. 2014. V. 20. P. 3604.
Adnan, Shah J., Jan M.R. // J. of the Taiwan Institute of Chemical Engineers. 2014. V. 45. P. 2494.
Adnan, Shah J., Jan M.R. // J. Anal. Appl. Pyrolysis. 2014. V. 109. P. 196.
Shah J., Jan M.R., Adnan // J. Anal. Appl. Pyrolysis. 2015. V. 114. P. 163.
Chauhan R.S., Gopinath S., Razdan P. Delattre C., Nirmala G.S., Natarajan R. // Waste Manag. 2008. V. 28. P. 2140.
Kim Y.S., Hwang G.C., Bae S.Y., Yi S.C., Moon S.K., Kumazawa H. // Korean J. Chem. Eng. 1999. V. 16. P. 161.
Onwudili J.A., Insura N., Williams P.T. // J. Anal. Appl. Pyrolysis. 2009. V. 86. P. 293.
Jung S.-H., Kim S.-J., Kim J.-S. // Fuel Process. Technol. 2013. V. 116. P. 123.
Kang S.H., Son S.M., Song P.S., Kang Y., Choi M.J. // Stud. Surf. Sci. Catal. 2006. V. 159. P. 529.
Liu Y., Qian J., Wang J. // Fuel Process. Technol. 2000. V. 63. P. 45.
Artetxe M., Lopez G., Amutio M., Barbarias I., Arregi A., Aguado R., Bilbao J., Olazar M. // Waste Manag. V. 45. P. 126.
Ng S.H. // Energy Fuels. 1995. V. 9. P. 216.
Bartoli M., Rosi L., Frediani M., Undri A., Frediani P. // J. Anal. Appl. Pyrolysis. 2015. V. 113. P. 281.
Undri A., Frediani M., Rosi L. Frediani P. // J. Anal. A-ppl. Pyrolysis. 2014. V. 105. P. 35.
de la Puente G., Arandes J.M., Sedran U.A. // Ind. Eng. Chem. Res. 1997. V. 36. P. 4530.
de la Puente G., Klocker C., Sedran U. // Appl. Catal. B Environ. 2002. V. 36. P. 279.
Arandes J.M., Erena J., Azkoiti M.J., Olazar M., Bilbao J. // J. Anal. Appl. Pyrolysis. 2003. V. 70. P. 747.
Passamonti F.J., Sedran U. // Appl. Catal. B Environ. 2012. V. 125. P. 499.
Dement’ev K.I., Palankoev T.A., Alekseeva O.A., Babkin I.A., Maksimov A.L. // J. Anal. Appl. Pyrolysis. 2019. V. 142. P. 104612.
de la Puente G., Sedran U. // Appl. Catal. B: Environ. 1998. V. 19. P. 305.
Arandes J.M., Erena J., Bilbao J., López-Valerio D., de la Puente G. // Ind. Eng. Chem. Res. 2003. V. 42. P. 3952.
Arandes J.M., Abajo I., Lopez-Valerio D., Fernández I., Azkoiti M.J., Olazar M., Bilbao J. // Ind. Eng. Chem. Res. 1007. V. 36. P. 4523.
Karagoz S., Karayildirim T., Ucar S., Yuksel M., Yanik J. // Fuel. 2003. Vol. 82. P. 415.
Pakhmanova O.A., Antonov S.V., Dement’ev K.I., Gerzeliev I.M., Khadzhiev S.N. // Petrol. Chemistry. 2012. V. 52. P. 401.
Siddiqui M.N., Redhwi H.H. // J. Anal. Appl. Pyrolysis. 2009. V. 86. № 1. P. 141.
Ali M.F., Siddiqui M.N., Redhwi H.H. // J. Mater. Cycles Waste Manag. 2004. V. 6. № 1. P. 27.
Ali M.F., Qureshi M.S. // Petrol. Sci. and Technol. 2013. V. 31. № 16. P. 1665.
Ali M.F., Siddiqui M.D. // J. Anal. Appl. Pyrolysis. 2005. V. 74. № 1–2. P. 282.
Ahmaruzzaman M., Sharma D.K. // Energy Fuels. 2006. V. 20. № 6. P. 2498.
Yanik J., Uddin M.A., Sakata Y. // Energy Fuels. 2001. V. 15. № 1. P. 163.
Lovett S., Berruti F., Behie L.A. // Ind. Eng. Chem. Res. 1997. V. 36. № 11. P. 4436.
Khadzhiev S.N. // Petr. Chem. 2011. V. 51. № 1. P. 1.
Kadiev Kh.M., Khadzhiev S.N., Kadieva M.Kh. // Petrol. Chemistry. 2013. V. 53. № 5. P. 298.
Khadzhiev S.N., Kadiev Kh.M., Kadieva M.Kh. // Petrol. Chemistry. 2014. V. 54. № 5. P. 323.
Kadiev Kh.M., Khadzhiev S.N., Kadieva M.K., Dogova E.S. // Petrol. Chemistry. 2017. V. 57. № 7. P. 608.
Кадиев Х.М., Дандаев А.У., Гюльмалиев А.М., Батов А.Е., Хаджиев С.Н. // Химия твердого топлива. 2013. № 2. С. 65.
Гюльмалиев А.М., Зекель Л.А., Батов А.Е., Дандаев А.У., Кадиев Х.М. // Журнал прикладной химии. 2018. Т. 91. № 8. С. 1203.
Ivin K.J., Mol J.C. Olefin Metathesis and Metathesis Polymerization. Academic Press: San Diego. London, 1997, 472 p.
Seyferth K., Taube R., Dahlke, M. // German Patent. № 146 053. 1981.
Shrock R.R., De Pue R.T., Feldman J., Schaverian C.J., Dewan J.C., Liu A.H. // J. Am. Chem. Soc. 1988. V. 110. P. 1423.
Shrock R.R., Murdzek J.S., Bazan G.C., Robbins J., DiMare M.O, Regan M. // J. Am. Chem. Soc. 1990. V. 112. P. 3875
Wagener K.B., Puts R.D., Smith D.W. // J. Makromol. Chem. Rapid Commun. 1991. V. 24. P. 6073.
Fu G.C., Nguyen S.T., Grubbs R.H. // J. Am. Chem. Soc. 1993. V. 115. P. 9856.
Nguyen S.T., Grubbs R.H. // J. Am. Chem. Soc. 1993. V. 115. P. 9858.
Watson M.D., Wagener K.B. // J. Polym. Sci. Part A: Polym. Chem. 1999. V. 37. P. 1857.
Marmo J.C., Wagener K.B. // Macromolecules. 1993. V. 26. P. 2137.
Craig S.W., Manzer J.A., Coughlin E.B. // Macromolecules. 2001. V. 34. P. 7929.
Thorn-Csanyi E., Ruhland K. // Macromol. Chem. Phys. 1999. V. 200. P. 1662.
Schulz M.D., Ford R.R., Wagener K.B. // Polym. Chem. 2013. V. 4. P. 3656.
Watson M.D., Wagener K.B. // Macromolecules. 2000. V. 33. P. 1494.
Wagener K.B., Puts R.D., Smith D.W., Jr. // Makromol. Chem. Rapid Commun. 1991. V. 12. P. 419.
Wagener R.B., Wolfe P.S., Watson M.D. In: Metathesis Polymerization of Olefins and Polymerization of Alkynes. Ed. by Yavus Imamoglu, 1998. P. 277.
Коршак Ю.В., Тленкопачев М.А., Тимофеева Г.Л., Павлова С.А., Долгоплоск Б.А. // ДАН СССР. 1976. Т. 226. С. 1344.
Korshak Y.V., Dolgoplosk B.A., Tlenkopatchev M.A. // Rec. Trav. Chim. Pays-Bas. 1977. V. 96. P. M64.
Konzelman J. Acyclic diene metathesis (ADMET) polymerization: a structure reactivity study. Diss. University of Florida, 1993.
Gringolts M.L., Denisova Yu.I., Finkelshtein E.Sh., Kudryavtsev Y.V. // Beilstein J. Org. Chem. 2019. V. 15. P. 218.
Wagner N.L., Timmers F.J., Arriola D.J., Jueptner G., Landes B.G. // Macromol. Rapid Commun. 2008. V. 29. P. 1438.
Otsuka H., Muta T., Sakada M., Maeda T., Takahara A. // Chem. Commun. 2009. P. 1073.
Maeda T., Kamimura S., Ohishi T., Takahara A., Otsuka H. // Polymer. 2014. V. 55. P. 6245.
Ohishi T., Suyama K.; Kamimura S., Sakada M., Imato K., Kawahara S., Takahara A., Otsuka H. // Polymer. 2015. V. 78. P. 145.
Daniele S., Mariconda A., Guerra G., Longo P., Giannino L. // Polymer. 2017. V. 130. P. 143.
Gringolts M.L., Denisova Yu.I., Shandryuk G.A., Krentsel L.B., Litmanovich A.D., Finkelshtein E.Sh., Kud-ryavtsev Y.V. // RSC Adv. 2015. V. 5. P. 316.
Denisova Yu.I., Gringolts M.L., Krentsel’ L.B., Shandryuk G.A., Litmanovich A.D., Finkelshtein E.Sh., Kudryavtsev Ya.V. // Polymer Science. Series B. 2016. V. 58. P. 292.
Denisova Yu.I., Gringolts M.L., Peregudov A.S., Krentsel L.B., Litmanovich E.A., Litmanovich A.D., Finkelshtein E.Sh., Kudryavtsev Y.V. // Beilstein J. Org. Chem. 2015. V. 11. P. 1796.
Denisova Y.I., Gringolts M.L., Roenko A.V., Shandryuk G.A., Finkelshtein E.Sh., Kudryavtsev Y.V. // Mendeleev Commun. 2017. V. 27. P. 416.
Shandryuk G.A., Denisova Y.I., Gringolts M.L., Krentsel L.B., Litmanovich A.D., Finkelshtein E.Sh., Kud-ryavtsev Y.V. // Eur. Polym. J. 2017. V. 86. P. 143.
Denisova Yu.I., Gringolts M.L., Krentsel L.B., Shandryuk G.A., Peregudov A.S., Finkelshtein E.Sh., Kudryavtsev Ya.V. // Роlymtr Sciens. Series B. 2017. V. 59. P. 412.
Дополнительные материалы отсутствуют.