

Нефтехимия, 2020, T. 60, № 5, стр. 679-685
Медь-содержащие катализаторы в жидкофазном гидрогенолизе глицерина
Г. С. Дмитриев 1, *, В. И. Хаджиев 1, С. А. Николаев 2, Д. И. Эзжеленко 2, И. С. Мельчаков 1, Л. Н. Занавескин 1
1 Институт нефтехимического синтеза им. А.В. Топчиева РАН
119991 Москва, Россия
2 Московский государственный университет имени М.В. Ломоносова, Химический факультет
119234 Москва, Россия
* E-mail: dmitriev.gs@mail.ru
Поступила в редакцию 11.03.2020
После доработки 09.04.2020
Принята к публикации 12.05.2020
Аннотация
Исследованы закономерности жидкофазного гидрогенолиза глицерина в 1,2-пропиленгликоль на катализаторах состава Cu/Al2O3 (Cu = 2–80 мас. %) и индивидуальных частицах меди. Показано, что основным фактором, определяющим активность катализаторов, является удельная площадь поверхности меди. Установлено, что наиболее эффективным является катализатор Cu/Al2O3, содержащий 60 мас. % Cu. На основе полученных данных предложен новый механизм реакции.
1,2-Пропиленгликоль (1,2-ПГ) является сырьем для получения полиэфирных смол, косметических средств, пищевых продуктов и антифризов. В промышленности 1,2-ПГ получают из оксида пропилена [1], который является дефицитным сырьем и обладает высокой стоимостью. Таким образом, в настоящее время существует острая потребность в разработке новых путей синтеза 1,2-ПГ на основе более доступного и недорого сырья.
Одним из перспективных путей синтеза 1,2-ПГ могла бы стать каталитическая конверсия глицерина, который является отходом процессов производства биодизеля и жиро- и маслопереработки. Однако на пути к промышленной реализации синтеза 1,2-ПГ из глицерина стоит препятствие, заключающееся в необходимости разработки новых стабильных, селективных и относительно недорогих катализаторов.
В настоящее время известно, что для гидрогенолиза глицерина в 1,2-ПГ можно использовать недорогие и эффективные, катализаторы состава Cu/Al2O3 [2, 3]. Процесс проводят в жидкой фазе при 180–220°С и давлении водорода 1–100 атм. В описанных выше условиях селективность образования 1,2-ПГ может составлять 90% и более.
Согласно данным [4–12], на катализаторах состава Cu/Al2O3 превращение глицерина в 1,2-ПГ протекает через последовательные стадии (1) и (2):
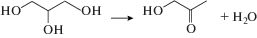

Многие авторы полагают, что дегидратация глицерина в ацетол (1) протекает на оксиде алюминия, а на меди идет гидрирование ацетола в 1,2-ПГ (2). Их предположения основаны на эмпирических наблюдениях о том, что дегидратация спирта протекает на кислотных центрах оксидных носителей, в то время как для диссоциативной адсорбции водорода и гидрирования необходимы редокс-центры на поверхности металла. В то же время имеется ряд экспериментальных данных, которые не согласуются с приведенным выше механизмом. Так, в работах [3, 13] сообщается, что при контакте глицерина и Al2O3 продукты реакции (1) не образуются; в работах [3, 14, 15] сообщается, что в отсутствие кислого носителя реакции (1) и (2) могут протекать как на частицах Cu, так и CuO [8, 16].
Таким образом, в современной литературе отсутствует единое мнение о механизме работы Cu-катализаторов гидрогенолиза глицерина, а так же о влиянии состава Cu-катализатора на его активность и селективность. Настоящая работа призвана по возможности восполнить эти пробелы знаний.
Для решения поставленных в настоящей работе задач методами соосаждения (С) и пропитки (ПР) были получены катализаторы состава Cu/Al2O3 с содержанием меди от 2 до 80 мас. %, а также катализаторы на основе индивидуальных частиц меди. Закономерности каталитического действия модельных систем были изучены в жидкофазном гидрогенолизе глицерина при 200°С в присутствии и в отсутствие водорода. Для идентификации активных центров катализаторов были проведены исследования методами: низкотемпературной адсорбции N2, температурно-программированной десорбции аммиака (ТПД-NH3), сканирующей электронной микроскопии (СЭМ), энергодисперсионным анализом (ЭДА) и рентгенофазовым анализом (РФА).
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Приготовление катализаторов
Катализаторы Cu/Al2O3 (ПР) с содержанием меди 5–40 мас. % получали методом пропитки. Для этого навеску γ-Al2O3 прокаливали при 350°С в течение 3 ч. Прокаленный носитель пропитывали водным раствором Cu(NO3)2, содержащим необходимое количество меди. Влажный катализатор сушили при 25○С в течение 24 ч. Полученную массу прокаливали в муфеле при 120○С в течение 4 ч и при 450○С в течение 2 ч. Прокаленный катализатор измельчали и отбирали фракцию гранул размером 0.1–0.16 мм.
Катализаторы Cu/Al2O3 (С) с содержанием меди 2–80 мас. % готовили методом соосаждения. В типовом синтезе рассчитанные объемы водных растворов Cu(NO3)2 и Al(NO3)3 с концентрациями 1.2 М и раствор NаОН 20 мас. % смешивали при 35–40°С. Полученную суспензию выдерживали в сушильном шкафу при 90–100°С в течение 4 ч. Осадок катализатора отделяли от водного раствора. Для удаления примесей нитрата натрия осадок промывали водой с последующим центрифугированием. Промытый катализатор сушили при 25°С в течение 24 ч, прокаливали при 120°С в течение 4 ч и при 450°С в течение 2 ч, измельчали и отбирали фракцию размером 0.1–0.16 мм.
Катализаторы Cu(ИНД) на основе индивидуальных частиц меди получали тремя способами: восстановлением гранул оксида меди размером 0.1–0.16 мм; кислотным травлением стружки алюминиевой бронзы и латуни размером 0.1–0.16 мм; осаждением из водного раствора Cu(NO3)2 с последующей сушкой, прокалкой и отбором фракции размером 0.1–0.16 мм.
Перед использованием катализаторы восстанавливали в смеси H2 и N2 при плавном повышении температуры от 25 до 300°С и выдерживании при 300°С в течение 2 ч. Восстановление в этих условиях приводит к образованию частиц Cu0.
Каталитическая конверсия глицерина
Конверсию глицерина проводили в стальном автоклаве BuchiGlassUster объемом 300 мл. В стандартном эксперименте в автоклав загружали 150 г водного раствора глицерина (80 мас. % глицерина + 20 мас. % воды) и 7.5 г катализатора. Автоклав нагревали до 200°С, водородом создавали в реакторе давление равное 20 атм (и поддерживали его постоянным в ходе эксперимента) и включали перемешивание (1200 об./мин). Через определенное время перемешивание отключали, реактор остужали, вскрывали, и проводили анализ реакционной смеси методом газовой хроматографии. Анализ реакционной смеси проводили на газовом хроматографе Цвет-800 с детектором по теплопроводности. Условия анализа: набивная колонка диаметром 3 мм и длиной 2 м, заполненная полисорбом-1; температура детектора 200°С; температура испарителя 220°С; программируемый температурный режим колонок 70–180°С.
Проведенные нами ранее исследования [17] показали, что в описанных выше условиях, скорость реакции описывается кинетическим уравнением первого порядка по глицерину и нулевого по водороду. Таким образом, для оценки активности катализаторов можно использовать константу скорости реакции (k), рассчитанную по тангенсу угла наклона прямой, построенной в координатах реакции 1-го порядка: ln(C) = kτ + ln C0 (C – концентрация глицерина [моль/л] в момент времени τ, τ − время реакции [ч], k – константа скорости расхода глицерина [ч–1], C0 – начальная концентрация глицерина [моль/л]).
Для расчета селективности образования продуктов использовали формулу: ${{S}_{i}} = \frac{{{{n}_{{{\text{гл}}~i}}}}}{{\sum {{{n}_{{{\text{гл\;}}}}}} }},$ где nглi – количество глицерина, превратившееся в i продукт реакции, моль; $\sum {{{n}_{{{\text{гл\;}}}}}} $ – количество вещества глицерина, превратившееся во все продукты реакции, моль.
Анализ структуры катализаторов
Структура катализаторов была изучена в их стабильной форме, а именно после проведения каталитических тестов.
Адсорбционные характеристики образцов определяли методом низкотемпературной адсорбции азота на приборе ASAP 2020 (Micromeritics). Стандартная подготовка образцов перед анализом приведена в [18]. Расчет удельной площади поверхности проводили с помощью уравнения БЭТ в интервале относительных давлений P/PS = 0.05–0.3 [18]
Концентрацию кислотных центров на поверхности образцов определяли методом термопрограмированной десорбции аммиака на приборе УСГА-101 (Унисит). В типовом эксперименте навеску образца 0.2 г помещали в проточный кварцевый реактор и нагревали в токе гелия при 500°С в течение 1 ч. После очистки поверхности проводили насыщение образца аммиаком в смеси с азотом при 60°С в течение 15 мин. Удаление физически адсорбированного аммиака проводили при 100°С в токе гелия в течение 1 ч со скоростью продувки гелия 30 мл/мин, затем образец охлаждали. Для получения кривой ТПД образец нагревали в токе гелия (30 см3/мин) до 900°С со скоростью 8 град/мин. Выделяющийся аммиак регистрировали при помощи детектора по теплопроводности. Для расчета числа кислотных центров использовали предварительно построенные калибровочные кривые и программное обеспечение прибора [19].
Микрофотографии образцов получали с помощью сканирующей электронной микроскопии (СЭМ) на приборе JCM-6000 с ускоряющим напряжением электронов 15 кЭВ и максимальной кратностью увеличения ×104 раз. В типовом эксперименте 1–2 гранулы образца помещали на предметный столик и проводили съемку в режиме детектирования вторичных электронов. Элементный состав поверхности определяли с помощью энергодисперсионного анализа (ЭДА) на входящем в комплектацию JCM-6000 рентгеновском спектрометре EX-230. На микрофотографиях СЭМ по данным элементного анализа выделяли индивидуальные частицы оксидов меди и/или алюминия и определяли их размер. Средний размер частиц получали статистической обработкой данных по 200 частицам [20]. Для оценки удельной площади поверхности меди (SCu) использовали формулу: ${{S}_{{{\text{Cu}}}}} = \frac{{\sum\nolimits_{j = 1}^m {\left\{ {{{\left[ {\sum\nolimits_{i = 1}^n {\left( {d_{i}^{2}\frac{\pi }{2}} \right)} } \right]} \mathord{\left/ {\vphantom {{\left[ {\sum\nolimits_{i = 1}^n {\left( {d_{i}^{2}\frac{\pi }{2}} \right)} } \right]} {{{S}_{j}}}}} \right. \kern-0em} {{{S}_{j}}}}} \right\}} }}{m}{{S}_{{{\text{БЭТ}}}}},$ где $\left( {d_{i}^{2}\frac{\pi }{2}} \right)$ – площадь полусферы (мкм2), di – диаметр частиц меди (мкм), n – количество частиц меди, Sj – площадь исследуемого участка образца (длина × × ширина) (мкм2), m – количество микрофотографий, SБЭТ – удельная площадь поверхности образца (м2/г). Погрешность определения SCu данным методом составляет 15–20%.
Фазовый состав катализаторов исследовали с помощью рентгенофазового анализа на приборе с вращающимся медным анодом Rotaflex RU-200 (Rigaku, Япония). Длина волны монохроматизированного излучения составляла 1.542 Å. Пробоподготовка и методика эксперимента приведены в работах [19]. Диапазон измерения углов дифракции составлял от 10° до 100° по шкале 2θ. Измерение вели в режиме непрерывного сканирования со скоростью 2°/мин и шагом 0.04°.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Структура катализаторов и их активность
Согласно данным РФА основное состояние меди в катализаторах настоящего исследования – Cu0. Рассчитанная из изотерм адсорбции N2 удельная площадь поверхности SBET нанесенных катализаторов Cu/Al2O3 приведена на рис. 1. Из приведенных данных видно, что SBET изменяется антибатно массовому содержанию меди. Этот тренд воспроизводится как для образцов, полученных пропиткой, так и для образцов полученных соосаждением. Вероятной причиной наблюдаемой зависимости является заполнение пористой структуры носителя (Al2O3) формируемыми в катализаторе частицами меди. Этот тезис полностью согласуется с данными СЭМ (рис. 2). Так, если низкопроцентные катализаторы содержат морфологически различимые фазы оксида алюминия и частиц меди, то при содержании меди 40 мас. % и выше значительная поверхность гранул оксида алюминия покрыта корочкой из меди (рис. 2).

Рис. 2.
Микрофотографии СЭМ: (a) 5% Cu/Al2O3 (ПР); (б) 40% Cu/Al2O3 (ПР); (в) 10% Cu/Al2O3 (С); (г) 40% Cu/Al2O3 (С); (д–е) СЭМ-ЭДА картирование поверхности 60% Cu/Al2O3 (С).

Очевидно, что уменьшение удельной площади поверхности Al2O3 с ростом содержания меди, должно приводить и к уменьшению числа кислотных центров (Cк) катализаторов. Эта зависимость также наблюдается (рис. 3).

Перед тем как обсуждать активность нанесенных катализаторов необходимо кратко обсудить активность чистого носителя. Оказалось, что нагрев глицерина и Al2O3 до 200°С c последующим перемешиванием в течение 18 ч в среде водорода при давлении 20 атм не приводит к образованию каких-либо новых соединений. Этот результат согласуется с данными по каталитической конверсии глицерина, приводимыми в [3, 14, 15]. Таким образом, в наших условиях тестирования носитель можно рассматривать как относительно инертный компонент катализатора, отвечающий главным образом за диспергирование активной фазы меди.
Данные по активности нанесенных катализаторов приведены на рис. 4. Из рис. 4 видно, что метод пропитки позволяет получать активные катализаторы гидрогенолиза глицерина. Наблюдаемая в присутствии катализатора Cu/Al2O3 (Cu = 5 мас. %) константа скорости реакции составляет 0.0063 ч–1, а селективность образования 1,2-ПГ составляет 98%. Дальнейший рост содержания меди с 5 до 40 мас. % слабо сказывается на активности и селективности полученных пропиткой катализаторов.
Рис. 4.
Зависимость наблюдаемой константы скорости от состава Cu/Al2O3. Условия проведения реакции: 150 г 80% глицерина, 7.5 г катализатора, 200○С, давление H2 20 атм, время 18 ч.

Полученные с помощью соосаждения катализаторы так же обладают высокой селективностью образования 1,2-ПГ (97–98%). При этом зависимость активности таких катализаторов от содержания меди носит экстремальный характер. С ростом содержания меди от 2 до 60 мас. % наблюдается рост константы скорости реакции от 0.0011 до 0.0485 ч–1. Дальнейший рост содержания меди свыше 60 мас. % приводит к уменьшению активности катализаторов.
Анализ данных каталитических экспериментов и определенных экспериментально значений SBET и Cк позволяет сделать вывод об отсутствии корреляции между активностью катализатора и его удельной площадью поверхности или кислотностью. В то же время существует линейная зависимость константы скорости реакции (k) от удельной площади поверхности меди (SCu), причем эта зависимость наблюдается как для образцов, полученных методом соосаждения, так и для образцов, полученных пропиткой (рис. 5).

Наличие прямой корреляции между k и SCu позволяет сделать предположение о том, что основным фактором, определяющим активность катализатора, является именно удельная площадь поверхности меди. Для проверки этой гипотезы были проведены эксперименты с образцами состава 20% Cu/Al2O3 (C) и 60% Cu/Al2O3 (C) таким образом, чтобы удельная площадь поверхности меди (SCu) загруженных образцов катализаторов разного состава совпадала. Это было достигнуто увеличенным количеством загруженного в реактор катализатора состава 20% Cu/Al2O3 по сравнению с массой загрузки катализатора 60% Cu/Al2O3. При прочих равных условиях была получена одна и та же конверсия глицерина 53.8% и селективность по 1,2-ПГ 98%, что свидетельствует в пользу сделанного выше предположения.
В заключение стоит отметить, что, как и катализаторы Cu/Al2O3, катализаторы Cu(ИНД) на основе индивидуальных частиц меди обладают высокой селективностью по 1,2-ПГ (97–98%). В то же время образцы меди Cu(ИНД) показали достаточно низкую активность, что может быть обусловлено интенсивным спеканием активной фазы катализаторов в отсутствие носителя. Действительно, анализ данных СЭМ показал, что индивидуальные частицы меди подвержены агломерации в условиях проведения процесса до массивных агрегатов размером 2–3 мм.
Несмотря на относительно низкую активность катализаторов состава Cu(ИНД), величина самой активности растет с увеличением удельной площади поверхности меди (см. табл. 1). Таким образом, как и в случае с нанесенными образцами, полученными пропиткой и соосаждением, снова видна корреляция между активностью медь-содержащего катализатора и удельной площадью поверхности меди.
Таблица 1.
Структура и активность индивидуальных частиц меди. Условия проведения реакции: 150 г 80% глицерина, 7.5 г катализатора, 200°С, давление 20 атм, 12 ч
| Катализатор | Прекурсор | SCu, м2/г | k, ч–1 |
|---|---|---|---|
| Cu(ИНД) | CuО | 0.2 | 0.17 × 10–3 |
| Cu(ИНД) | Al бронза | 0.7 | 0.59 × 10–3 |
| Cu(ИНД) | Латунь | 0.8 | 0.67 × 10–3 |
| Cu(ИНД) | Cu(NO3)2 | 3.2 | 2.5 × 10–3 |
Вероятный механизм конверсии глицерина
Описанный в литературе механизм образования 1,2-ПГ подразумевает цепочку из реакций: дегидратация глицерина в ацетол (1); гидрирование ацетола до 1,2-ПГ (2). При этом большинство авторов полагают, что дегидратация глицерина протекает на кислотных центрах носителя, а на металле идет гидрирование ацетола в 1,2-ПГ [4–12]. Если бы это было так, то на модельном катализаторе состава CuO/Al2O3 конверсия глицерина должна была бы быть близкой к конверсии, наблюдаемой на катализаторе типа Cu/Al2O3, но с преимущественным образованием ацетола. Проверочный эксперимент показал, что это не выполняется. Так в стандартных условиях тестирования катализатор 65% СuO/Al2O3 (CО) позволил получить конверсию глицерина всего 7.0% при селективности по ацетолу, равной 9.7%, в то время как восстановленная форма того же катализатора дает конверсию глицерина 55% и селективность по 1,2-ПГ 97–98%. Опять же если бы в условиях проведения процесса реакция (1) шла при катализе активными центрами Al2O3, можно было ожидать, что восстановленный катализатор, дающий в среде водорода конверсию 55% с селективностью по 1,2-ПГ 97–98%, должен в инертной среде дать такую же конверсию глицерина, но с образованием ацетола в качестве основного продукта. Проверочный эксперимент показал, что это не выполняется. Так, катализатор 60% Cu/Al2O3 (C) в среде азота при стандартных условиях проведения эксперимента позволил получить конверсию глицерина 6.6%, селективности по 1,2-ПГ и ацетолу 57.3% и 42.7% соответственно. Таким образом, в наших условиях тестирования механизм превращения глицерина в 1,2-ПГ отличается от описанного в [4–12].
Анализ результатов настоящего исследования позволил предложить новый механизм конверсии глицерина в 1,2-ПГ (рис. 6). Принципиальным отличием нового механизма является принятие во внимание факта о том, что активными центрами дегидратации являются частицы меди, а не оксидного носителя.

Из приведенного на рис. 6 механизма видно, что поверхность меди адсорбирует молекулярный H2 с образованием двух атомов водорода [21, 22]. Адсорбция глицерина на меди приводит к формированию координационных связей между α-OH- и β-СH-группами глицерина. Атом водорода, образовавшийся на поверхности катализатора в результате диссоциативной адсорбции Н2, и адсорбированная α-OH-группа глицерина дают молекулу воды. При этом происходит гомолитический разрыв связи С–ОН, который инициирует переход атома водорода β-СH-группы на поверхность меди. В результате из глицерина образуется енол, который быстро изомеризуется в более стабильный ацетол. На финальной стадии ацетол взаимодействует с двумя поверхностными атомами водорода и образует 1,2-пропиленгликоль. Как видно из рис. 6, материальный баланс реакции соответствует уравнению: C3H8O3 + H2 + Cu → → C3H8O2 + H2O + Cu.
ЗАКЛЮЧЕНИЕ
Результаты исследования показывают, что Cu-катализаторы позволяют проводить жидкофазную конверсию глицерина в пропиленгликоль с селективностью не менее 97%. Среди испытанных образцов наибольшую эффективность гидрогенолиза глицерина обеспечивает каталитическая система 60% Cu/Al2O3, полученная методом соосаждения. Основным фактором, определяющим активность Cu катализаторов, является удельная площадь поверхности меди. На основе полученных данных предложен новый механизм реакции.
Список литературы
Тенденции развития мирового производства и рынка пропиленоксида // Евразийский химический рынок. 2018. № 2. С. 30.
Шарада М.Е. // Химия, физика и технология поверхности. 2012. Т. 3. № 1. С. 61.
Akiyama M. // Appl. Catal. A. 2009. V. 371. P. 60.
Dasari M.A., Kiatsimkul P.-P., Sutterlin W.R., Suppes G.J. // Appl. Catal. A. 2005. V. 281. № 1–2. P. 225.
Barbelli M.L., Santori G.F., Nichio N.N. // Bioresource Technology. 2012. V. 111. P. 500.
Wang S., Yin K., Zhang Y., Liu H. // ACS Catal. 2013. V. 3. № 9. P. 2112.
Van Ryneveld E., Mahomed A.S., van Heerden P.S., Green M.J., Friedrich H.B. // Green Chem. 2011. V. 13. № 7. P. 1819.
Wang S., Liu H. // Catal. Lett. 2007. V. 117. № 1–2. P. 62.
Balaraju M., Rekha V., Prasad P.S.S., Prasad R.B.N., Lingaiah N. // Catal. Lett. 2008. V. 126. № 1–2. P. 119.
Xia S., Yuan Z., Wang L., Hou Z. // Appl. Catal., A. 2011. V. 403. № 1–2. P. 173.
Alhanash A., Kozhevnikova E.F., Kozhevnikov I.V. // Catal. Lett. 2008. V. 120. № 3–4. P. 307.
Feng Y., Yin H., Wang A., Shen L., Yu L., Jiang T. // Chem. Eng. J. 2011. V. 168. № 1. P. 403.
Rajkhowa T., Marin G.B., Thybaut J.W. // Appl. Catal. B. 2017. V. 205. P. 469.
Tanielyan S.K., Marin N., Alvez G., Bhagat R., Balaraju M., Augustine R.L., Schmidt S.R. // Org. Process Res. Dev. 2014. V. 18. № 11. P. 1419.
Cui F., Kang H., Chen J., Zhang X., Xia C., Huang Z. // Chem. Mater. 2008. V. 20. № 15. P. 5090.
Mane R.B., Yamaguchi A., Malawadkar A., Shirai M., Rode C.V. // RSC Advances. 2013. V. 3. № 37. P. 16 499.
Khadzhiev V.I., Dmitriev G.S., Melchakov I.S., Shorina T.E., Zanaveskin L.N., Maximov A.L. // Kinet. Catal. 2019. V. 60. № 6. P. 802 [Кинетика и катализ. 2019. № 6. С. 782].
Batova T.I., Obukhova T.K., Kolesnichenko N.V., Nikolaev S.A. // Petrol. Chemistry. 2019. V. 59. № 9. P. 1017 [Heфтexимия. 2019. № 5. C. 569].
Nikolaev S.A., Tsodikov M.V., Chistyakov A.V., Zharova P.A., Ezzgelenko D.I. // J. of Catalysis. 2019. V. 369. P. 501.
Grishin. M.V., Gatin A.K., Dokhlikova N.V., Kirsankin A.A., Kulak A.I., Nikolaev S.A., Shub B.R. // Kinetics and Catalysis. 2015. V. 56. № 4. P. 532. [Кинетика и катализ. 2015. № 4. С. 539].
Álvarez-Falcón L., Viñes F., Notario-Estévez A., Illas F. // Surf. Sci. 2016. V. 646. P. 221.
Clarke L. // J. Am. Chem. Soc. 1937. V. 59. № 7. P. 1389.
Дополнительные материалы отсутствуют.