

Нейрохимия, 2019, T. 36, № 3, стр. 218-225
Участие серотониновых, глутаматных и ГАМК-рецепторов в проявлении антидепрессивноподобного эффекта циклопролилглицина
А. А. Абдуллина 1, Е. В. Васильева 1, Е. А. Кондрахин 1, Г. И. Ковалёв 1
1 Федеральное государственное бюджетное научное учреждение
“НИИ фармакологии имени В.В. Закусова”
Москва, Россия
Поступила в редакцию 21.12.2018
После доработки 07.02.2019
Принята к публикации 11.02.2019
Аннотация
В работе изучено влияние двухнедельного введения пептида циклопролилглицина (ЦПГ) (1 и 2 мг/кг/сутки, в/б) на характеристики связывания серотониновых 5-HT2A-, NMDA-, метаботропных глутаматных mGluII-, ГАМКА- и ГАМКB-рецепторов в мозге мышей инбредной линии BALB/c в условиях in vitro и ex vivo. Обнаружено, что ЦПГ в дозах 1 и 2 мг/кг/сутки снижает плотность 5‑HT2A-рецепторов в стриатуме мышей на 24 и 28% соответственно. Снижение плотности NMDA-рецепторов в гиппокампе наблюдалось только в дозе 2 мг/кг/сутки и составило 22%. При этом наблюдалось увеличение плотности ГАМКА-рецепторов во фронтальной коре в дозе 1 мг/кг/сутки на 36% по сравнению с контролем. Двухнедельное введение ЦПГ в обеих дозах не сказывалось на величинах Bmax mGluII- и ГАМКB-рецепторов, а также на величинах Kd во всех группах. В радиолигандном анализе in vitro ЦПГ не влиял на специфическое связывание меченых лигандов выбранных рецепторов. Полученные данные впервые указывают на функциональное вовлечение 5‑HT2A-, NMDA- и ГАМКА-рецепторов в формирование антидепрессивноподобного эффекта циклопролилглицина у мышей BALB/c.
ВВЕДЕНИЕ
Разнообразие клинических проявлений депрессии, множественность молекулярных механизмов действия антидепрессантов разных групп свидетельствуют об участии в патогенезе депрессивных расстройств взаимосвязанных нарушений ряда нейрохимических систем. Одной из основных нейромедиаторных систем, задействованных в патогенезе депрессии, является серотонинергическая. Об ее участии в патогенезе депрессии свидетельствуют снижение концентрации серотонина в мозге у больных и ее увеличение при клиническом улучшении, обострение симптомов депрессии при гипотриптофановой диете, снижение плотности транспортеров, осуществляющих обратный захват серотонина, а также нейрорецепторные и генетические исследования. Кроме того, эта система тесно взаимодействуют с глутаматергической и ГАМКергической системами, которые также участвуют в развитии депрессивной симптоматики [1].
Установлено, что на фоне депрессивных расстройств наблюдается гиперпродукция глутамата и чрезмерная активация глутаматергической системы, что приводит к эксайтотоксичности и нарушению нейропластичности. Кроме того, антагонисты NMDA- и mGluII-рецепторов проявляют антидепрессивноподобный эффект в животных моделях депрессии, а также повышают активность серотонинергических нейронов в дорсальном ядре шва и увеличивают концентрацию внеклеточного серотонина в префронтальной коре головного мозга у крыс [2].
ГАМК является функциональным антагонистом глутамата и способна ограничивать его возбуждающее действие. При депрессии наблюдается уменьшение концентрации ГАМК в плазме, спинномозговой жидкости и мозге, снижение уровня ГАМК-активных стероидов, нарушается экспрессия генов, кодирующих субъединицы ГАМКА- и ГАМКB-рецепторов [3, 4].
Известно также, что антагонисты ГАМКB-рецепторов, как и агонисты ГАМКА-рецепторов оказывают антидепрессивноподобный эффект в экспериментальных моделях депрессии на животных и стимулируют нейрогенез [5, 6].
В качестве эндогенного вещества мозга крыс циклопролилглицин был обнаружен в НИИ фармакологии имени В.В. Закусова в 1996 г. [7], позже это было подтверждено другими исследователями [8]. Изучение фармакологической активности ЦПГ показало наличие у пептида психофармакологической активности [9, 10], а также способность увеличивать содержание BDNF в культуре клеток гиппокампа в опытах ex vivo и in vitro [11]. Учитывая эти сведения, а также известные факты о роли снижения продукции BDNF в патогенезе депрессии [12], нами были проведены исследования по выявлению возможной антидепресивноподобной активности ЦПГ. В результате серии экспериментов было обнаружено, что в тесте “Вынужденное плавание” по Порсолту ЦПГ в дозах 1 и 2 мг/кг/сутки демонстрирует на инбредных мышах BALB/c антииммобилизационный эффект, сопоставимый с таковым у антидепрессанта флуоксетина в дозе 10 мг/кг [13]. Однако рецепторные механизмы реализации данного эффекта оставались невыясненными.
Таким образом, целью данной работы стало изучение влияния ЦПГ на характеристики серотониновых 5-HT2A-, NMDA-, метаботропных глутаматных mGluII-, ГАМКА- и ГАМКB-рецепторов в мозге мышей инбредной линий BALB/c после двухнедельного введения в дозах, эквипотенциальных по антидепрессивноподобному эффекту.
МАТЕРИАЛЫ И МЕТОДЫ
Животные
Исследования проводили на самцах мышей линий BALB/c массой 25–30 г, которых содержали в виварии ФГБНУ “НИИ фармакологии имени В.В. Закусова” в стандартных условиях со свободным доступом к воде и корму ad libitum, по 10 особей в клетке в течение 1-й недели до начала эксперимента, на стандартной диете при 12-часовом световом режиме. Содержание животных соответствовало правилам лабораторной практики при проведении доклинических исследований в РФ (ГОСТ 351.000.3-96 и 51000.4-96), Приказу МЗ и СР РФ от 23 августа 2010 г. № 708н “Об утверждении Правил лабораторной практики”. Эксперименты проводили с 10 до 16 ч. Проведение экспериментов одобрено Комиссией по биомедицинской этике ФГБНУ “НИИ фармакологии имени В.В. Закусова”. Животным посредством внутрибрюшинных инъекций в течение 14 дней один раз в сутки вводили физиологический раствор (контрольная группа – NaCl, 0.9%), либо ЦПГ, растворенный в физрастворе (опытные группы) в дозах 1 и 2 мг/кг/сутки.
После тестирования мышей декапитировали, головной мозг извлекали на льду и выделяли фронтальную кору, стриатум и гиппокамп по общепринятой схеме [14] для проведения радиорецепторного анализа.
Вещества
ЦПГ был синтезирован и предоставлен Отделом химии НИИ фармакологии имени В.В. Закусова (зав. – член-корр. РАН Т.А. Гудашева). Для изучения рецепторного связывания использовали следующие лиганды: [3H]Кетансерин с удельной активностью 60 Кюри/ммоль (“Perkin Elmer”), [3H]SR-95531 (49.5 Кюри/ммоль, “Perkin Elmer”), [3H](–)Баклофен (49.7 Кюри/ммоль, “Perkin Elmer”). [3H](+)МК-801 (210 Кюри/ммоль) и [3H]LY354740 (42 Кюри/ммоль) были синтезированы профессором, д. х. н. Ю.А. Золотарёвым в ОХФАВ ИМГ РАН (зав. отделом – академик РАН Н.Ф. Мясоедов). Остальные реактивы были приобретены в коммерческих источниках.
5-HT2A -рецепторы
Выделение плазматических мембран с 5-HT2A-рецепторами стриатума. Изучение радиорецепторного связывания с 5-HT2A-рецепторами серотонина проводили по методу Leysen et al. (1982) с модификациями [15]. Мышей BALB/c декапитировали, извлекали стриатум. Выделенные структуры гомогенизировали в 10 мл ледяного (0–4°C) 50 мМ Tris-HCl буфера (рН 7.4 при 4°C), используя гомогенизатор Potter S “тефлон-стекло”.
Полученную суспензию центрифугировали при 40 000 g в течение 20 мин в ультрацентрифуге “L7-35” (“Beckman Coulter”). После центрифугирования супернатант сливали, осадок ресуспендировали повторной гомогенизацией в том же объеме буфера, затем вновь центрифугировали. Процедуру отмывки проводили трижды, полученный осадок ресуспендировали в 10 мл инкубационного буфера и использовали по 250 мкл в процедуре связывания с [G-3H]-лигандами рецепторов. Инкубационный буфер для связывания с 5-HT2A-подтипом рецепторов стриатума мышей содержал 50 мM Tris-base, 120 мM NaCl, 5 мM KCl, 2 мM CaCl2 ⋅ 2H2O, 1 мM MgCl2 ⋅ 6H2O (рН 7.4 при 37°C).
Радиолигандный анализ 5-HT2A-рецепторов. Инкубационная смесь (конечный объем 0.5 мл) содержала 50 мкл или [3H](+)Ketanserin, 250 мкл буфера и 200 мкл белковой суспензии мембран, для неспецифического связывания добавляли 50 мкл немеченного лиганда (Ketanserin, 1 мМ). Реакционную смесь инкубировали при комнатной температуре в течение 2 часов.
NMDA-рецепторы
Выделение плазматических мембран с NMDA-рецепторами гиппокампа. Выделение плазматических мембран гиппокампа проводили по модифицированным методам [16, 17]. После декапитации ткань немедленно замораживали в жидком азоте и хранили в низкотемпературном холодильнике при –80°С. В день эксперимента гиппокампы размельчали в гомогенизаторе Поттера “тефлон-стекло” в 10 объемах буфера № 1 (5 мМ НЕРЕS, 4.5 мM Tris, 0.32 M сахароза, рН 7.6). Гомогенат разбавляли 50 объемами буфера № 2 (5 мМ НЕРЕS, 4.5 мM Tris, рН 7.6) и центрифугировали при 1000 g 10 мин на ультрацентрифуге “Optima L-70K” (“Beckman Coulter”). Супернатант сливали и вновь центрифугировали при 25 000 g 20 мин. Для увеличения выхода белка эту операцию проводили дважды. Полученный осадок ресуспендировали в 50 объемах буфера № 2 и центрифугировали при 8000 g 20 мин. Супернатант и верхний коричневый осадочный слой сливали и центрифугировали при 25 000 g 20 мин. Осадок ресуспендировали в 50 объемах буфера № 3 (5 мМ НЕРЕS, 4.5 мM Tris, 1 мM Na4EDTA, рН 7.6) и троекратно центрифугировали при 25 000 g 20 мин. Полученный осадок ресуспендировали в 50 объемах буфера № 2 и однократно центрифугировали при 25 000 g 20 мин. Конечный осадок сохраняли в 5 объемах буфера № 2 и замораживали в криопробирках в жидком азоте. В день анализа ткань размораживали, разбавляли в 10 объемах буфера № 2, центрифугировали при 25 000 g 20 мин. Осадок ресуспендировали в необходимом количестве буфера № 2.
Радиолигандный анализ NMDA-рецепторов. Инкубационная смесь (конечный объем 0.5 мл) содержала 50 мкл [3H](+)МК-801, 250 мкл буфера и 200 мкл белковой суспензии мембран, для неспецифического связывания добавляли 50 мкл немеченного лиганда (+)МК-801, 1 мМ). Реакционную смесь инкубировали при комнатной температуре в течение 2 ч.
mGluII-рецепторы
Выделение плазматических мембран с mGluII-рецепторами гиппокампа. Выделение mGluR-рецепторов проводили по методу [18]. После декапитации ткань немедленно замораживали в жидком азоте и хранили в низкотемпературном холодильнике при –80°С. В день эксперимента ткань мозга размельчали в гомогенизаторе Поттера “тефлон-стекло” в 25 объемах буфера (50 мM Tris-HCl, pH 7.1). Гомогенат центрифугировали при 48 000 g 10 мин. Полученный осадок гомогенизировали в буфере (50 мM Tris-HCl, pH 7.1) и инкубировали при 37°С 10 мин, затем центрифугировали при 48 000 g 10 мин. Осадок ресуспендировали в 5 объемах буфера и замораживали в криопробирках при –80°С. В день эксперимента мембраны размораживали и центрифугировали 3 раза в буфере (50 мM Tris-HCl, 2 мM MgCl2, pH 7.4) 48 000 g 10 мин. Осадок ресуспендировали в необходимом количестве буфера (50 mM Tris-HCl, 2 mM MgCl2, pH 7.4). Концентрация белка в образцах мембран составляла 0.25 мг/мл.
Радиолигандный анализ mGluII-рецепторов. Инкубационная смесь (конечный объем 0.5 мл) содержала 50 мкл [3H]LY 354740, 200 или 250 мкл буфера (50 мM Tris-HCl, 2 мM MgCl2, pH 7.4) и 200 мкл белковой суспензии мембран, для неспецифического связывания добавляли 50 мкл немеченого лиганда (глутамат). Реакционную смесь инкубировали при комнатной температуре в течение 1 ч.
ГАМКА-рецепторы
Выделение плазматических мембран с ГАМКА-рецепторами коры мозга. Приготовление мембранных препаратов, содержащих ГАМКА-рецепторы коры мозга мышей BALB/c проводили по модифицированным методам [19, 20]. После декапитации ткань немедленно замораживали в жидком азоте и хранили в низкотемпературном холодильнике при –80°С. В день эксперимента фронтальную кору размельчали в гомогенизаторе Поттера “тефлон-стекло” в 20 объемах ледяного буфера (0.32 M Сахароза; pH 7.1). Гомогенат центрифугировали в ультрацентрифуге “Optima L-70K” (“Beckman Coulter”) в течение 10 мин при 1000 g. Супернатант повторно центрифугировали при 20 000 g в течение 20 мин. Осадок ресуспендировали в 20 мл холодной дистиллированной воды и центрифугировали 8000 g 20 мин. Супернатант и желтый надосадочный слой повторно центрифугировали при 48 000 g 20 мин. Осадок суспендировали в 0.05 М Tris-citrate буфере (pH 7.1) и центрифугировали 48 000 g 20 мин. Полученную мембранную фракцию замораживали и хранили при –80°С. В день эксперимента мембраны суспендировали в 40 объемах 0.05 М Tris-citrate буфера (pH 7.1) и центрифугировали при 48 000 g 20 мин. Полученный осадок суспендировали в 40 объемах 0.05 М Tris-citrate буфера (pH 7.1) и инкубировали при 24°С в течение 30 мин и снова центрифугировали при 48 000 g 20 мин. Конечный осадок ресуспендировали в свежем буфере.
Радиолигандный анализ ГАМКА-рецепторов. Инкубационная смесь (конечный объем 0.5 мл) содержала 50 мкл [3H]SR 95531, 250 мкл буфера и 200 мкл белковой суспензии мембран, для неспецифического связывания добавляли 50 мкл немеченного лиганда SR 95531(габазина), 1 мМ. Реакционную смесь инкубировали при 4°С в течение 1 ч.
ГАМКB-рецепторы
Выделение плазматических мембран с ГАМКB-рецепторами коры мозга. Приготовление мембранных препаратов, содержащих ГАМКB-рецепторы коры мозга мышей BALB/c проводили по модифицированным методам [21, 22]. После декапитации ткань немедленно замораживали в жидком азоте и хранили в низкотемпературном холодильнике при –80°С. В день эксперимента фронтальную кору размельчали в гомогенизаторе Поттера “тефлон-стекло” в 10 объемах ледяного буфера (0.32 M сахароза; pH 7.4). Образовавшийся гомогенат центрифугировали при 1000 g 10 мин. Супернатант повторно центрифугировали при 20 000 g 20 мин. Затем полученный осадок ресуспендировали в дистиллированной воде и вновь центрифугировали 20 мин при 8000 g. Образовавшийся супернатант и верхний надосадочный слой центрифугировали 20 мин при 48 000 g, а осадок суспендировали и вновь центрифугировали в 50 мМ Tris-Сitrate буфере (pH 7.4) 2 раза по 20 мин при 48 000 g, после чего замораживали при –80°С. В день эксперимента мембраны размораживали при комнатной температуре и ресуспендировали в 20 объемах буфера (50 мМ Tris-HCl, 2.5 мM CaCl2, pH 7.4), затем центрифугировали при 8000 g 20 мин. Полученный осадок суспендировали в 20 объемах буфера (50 мМ Tris-HCl, 2.5 мM CaCl2, pH 7.4) и центрифугировали при 20 000 g 20 мин, последнюю процедуру повторяли еще один раз. Конечный осадок ресуспендировали в свежем буфере.
Радиолигандный анализ ГАМКB-рецепторов. Инкубационная смесь (конечный объем 0.5 мл) содержала 50 мкл [3H](–)Баклофена, 250 мкл буфера и 200 мкл белковой суспензии мембран, для неспецифического связывания добавляли 50 мкл немеченного лиганда (–)Баклофена, 1 мМ. Реакционную смесь инкубировали при 4°С в течение 20 мин.
Жидкостно-сцинтилляционная спектрометрия
По окончании инкубации пробы фильтровали через стекловолокнистые фильтры GF/B (“Whatman”), предварительно смоченные в 0.3% полиэтиленимине в течение 2 ч при комнатной температуре. Каждую пробирку промывали два раза холодным буфером, затем фильтры промывали два раза тем же объемом буфера. Фильтры просушивали на воздухе и переносили в сцинтилляционные флаконы. Фильтры заливали 5 мл сцинтилляционной жидкости на основе толуола (4 г PPO, 0.2 г POPOP на 1 л толуола). Радиоактивность проб определяли на счетчике Tri-Carb 2900TR (“Perkin Elmer”) с эффективностью счета 42–46%. Концентрацию белка измеряли по стандартной методике Лоури (1951).
Для обработки результатов радиолигандного связывания использовали программу GraphPad Prism 4 Demo и Statistica 6.0. Результаты представлены в виде “mean ± S.E.M”.
Обработка и представление результатов
В экспериментах in vitro величину IС50 по отношению к связыванию меченых лигандов определяли при добавлении в инкубационную среду 50 мкл исследуемых соединений в конечных концентрациях в диапазоне 10–10–10–4 М. Объем инкубационной смеси составлял 500 мкл. Для построения кривых вытеснения радиоактивных лигандов каждая концентрация исследуемого вещества была взята в 3-х повторностях.
Результаты экспериментов ex vivo оценивали с помощью рассчитанных величин Kd и Bmax, отражающих степень сродства рецептора к лиганду (нМ) и количество мест связывания лиганда (фмоль/мг белка), соответственно. Для анализа насыщения и получения характеристик связывания Bmax и Kdизмеряли специфическое связывание для 5-HT2A-рецепторов – от 0.625 до 20 нМ, для NMDA-рецепторов в диапазоне концентраций от 1.25 до 20 нM, для mGluII-рецепторов – от 12.5 до 200 нМ, для ГАМКА-рецепторов – от 2.5 до 40 нМ, для ГАМКВ-рецепторов – от 2.5 до 20 нМ. Специфическое связывание рассчитывали как разницу между общим и неспецифическим связыванием. Для построения кривых насыщения радиоактивных лигандов каждая концентрация исследуемого вещества была взята в 2-х повторностях.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
В радиолигандном анализе in vitro ЦПГ не влиял на специфическое связывание меченых лигандов выбранных рецепторов во всем интервале используемых концентраций, что говорит об отсутствии прямого конкурентного взаимодействия изучаемого соединения с лигандами 5-HT2A-, NMDA-, mGluII-, ГАМКА- и ГАМКB- рецепторов. Величины IC50 для ЦПГ составили >100 мкмоль/л (табл. 1).
Таблица 1.
Потенциал конкурентного взаимодействия ЦПГ с серотониновыми, глутаматными и ГАМК-рецепторами in vitro (IC50, мкмоль/л) m ± S.E.M. (n = 3)
| Вещества | Рецепторы | ||||
|---|---|---|---|---|---|
| 5-НТ2А (cтриатум) | NMDA (гиппокамп) |
mGluR (гиппокамп) |
ГАМКА (кора мозга) | ГАМКB (кора мозга) | |
| Лиганд | Кетансерин 0.014 ± 0.008 | МК-801 0.007 ± 0.004 | LY354740 0.020 ± 0.001 | SR-95531 1.45 ± 0.002 | Баклофен 1.50 ± 0.002 |
| ЦПГ | >100 | >100 | >100 | >100 | >100 |
В опытах ex vivo различий между контролем и опытными группами в аффинитете (Kd) всех изучаемых типов рецепторов по отношению к их специфическим лигандам выявлено не было. Графики насыщения 5-HT2A-, NMDA-, метаботропных глутаматных mGluII-, ГАМКА- и ГАМКB-рецепторов мозга мышей инбредной линии BALB/c под влиянием двухнедельного введения ЦПГ в дозах 1 и 2 мг/кг/сутки, а также результаты их преобразования по Скетчарду представлены на рис. 1–3. Рассчитанные величины характеристик рецепторного связывания Кd и Bmax для выбранных типов рецепторов – в табл. 2.
Рис. 1.
Влияние циклопролилглицина на связывание [3H]Кетансерина с 5-HT2A-рецепторами стриатума мышей BALB/c (кривая насыщения и график Скетчарда). Примечание: * статистически значимые отличия от контроля (p < 0.05; t-test Стьюдента).
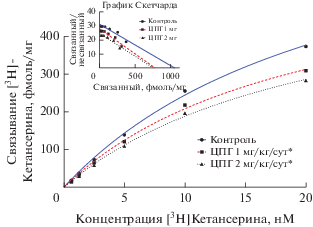
Рис. 2.
Влияние циклопролилглицина на связывание [3H](+)MK-801 с NMDA-рецепторами (а), [3H]-LY354740 с метаботропными глутаматными mGluII-рецепторами (б) гиппокампа мышей BALB/c (кривая насыщения и график Скетчарда). Примечание: * статистически значимые отличия от контроля (p < 0.05; t-test Стьюдента).
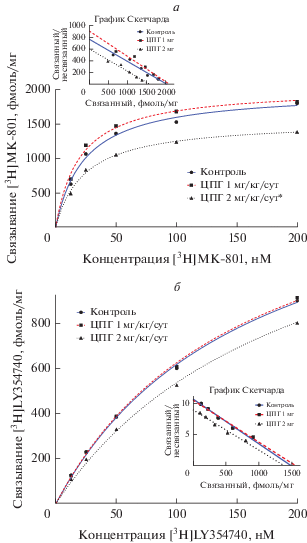
Рис. 3.
Влияние циклопролилглицина на связывание [3H]SR-95531 с ГАМКА-рецепторами (а), [3H](–)Баклофена с ГАМКB-рецепторами (б) коры мышей BALB/c (кривая насыщения и график Скетчарда). Примечание: * статистически значимые отличия от контроля (p < 0.05; t-test Стьюдента).
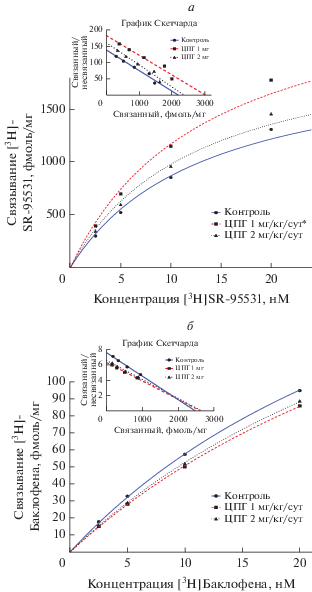
Таблица 2.
Влияние ЦПГ на характеристики связывания с рецепторами мозга мышей BALB/c ex vivo после двухнедельного введения (m ± S.E.M.)
| Рецепторы | Вещества | |||||
|---|---|---|---|---|---|---|
| Контроль | ЦПГ 1 мг/кг/сутки | ЦПГ 2 мг/кг/сутки | ||||
| Bmax, фмоль/мг |
Kd, нМ |
Bmax, фмоль/мг |
Kd, нМ |
Bmax, фмоль/мг |
Kd, нМ |
|
| 5-HT2A | 823 ± 69 | 23.7 ± 3.2 | 626 ± 49* | 20.0 ± 2.6 | 595 ± 45* | 21.6 ± 2.7 |
| NMDA | 1977 ± 79 | 2.4 ± 0.3 | 2024 ± 71 | 2.0 ± 0.3 | 1544 ± 43* | 2.4 ± 0.2 |
| mGluII | 1615 ± 86 | 161.7 ± 15.3 | 1673 ± 78 | 168,3 ± 13.8 | 1518 ± 83 | 181.6 ± 17.0 |
| ГАМКА | 2033 ± 154 | 13.5 ± 2.4 | 2777 ± 222* | 13.8 ± 2.6 | 2213 ± 161 | 12.7 ± 2.3 |
| ГАМКB | 259 ± 9 | 34.9 ± 1.7 | 281 ± 13 | 45.6 ± 2.9 | 285 ± 13 | 44.5 ± 2.8 |
Обнаружено, что под влиянием ЦПГ в дозах 1 и 2 мг/кг/сутки происходило снижение количества мест связывания (Bmax ) 5-HT2A-рецепторов в стриатуме мышей на 24 и 28% соответственно: Bmax = 626 ± 49 и 595 ± 45 фмоль/мг против 823 ± ± 69 фмоль/мг в контроле (p < 0.05, t-тест Стьюдента) (рис. 1).
Двухнедельное введение ЦПГ в дозе 2 мг/кг/ сутки снижало плотность NMDA-рецепторов в гиппокампе мышей BALB/c на 22% Bmax = 1544 ± ± 43 фмоль/мг против 1977 ± 79 фмоль/мг в контроле (p < 0.05, t-тест Стьюдента), в дозе 1 мг/кг/сутки – никаких изменений не происходило (рис. 2а).
В результате двухнедельного введения ЦПГ в дозе 1 мг/кг/сутки увеличивалась плотность ГАМКА-рецепторов в коре мышей BALB/c на 36% Bmax = 2777 ± 222 фмоль/мг против 2033 ± ± 154 фмоль/мг в контроле (p < 0.05, t-тест Стьюдента), в дозе 2 мг/кг/сутки эффектов не обнаруживалось (рис. 3а).
Под воздействием ЦПГ в изучаемых дозах эффектов в отношении Bmax для селективного агониста mGluII-рецепторов [3H]LY354740 и селективного агониста ГАМКB-рецепторов [3H]-Баклофена не было обнаружено (рис. 2б и 3б).
В большинстве доклинических исследований проявление депрессивноподобного фенотипа связывают с активацией 5-HT2A-рецепторов. Показано, что агонист 5-HT2A-рецепторов DOI увеличивает время иммобилизации мышей в тесте “Вынужденное плавание”, а предварительное введение антагониста рецепторов этого подтипа MDL100907 нивелирует этот эффект. Кроме того, снижение экспрессии 5-HT2A-рецепторов, вызванное интрацеребровентрикулярной инъекцией антисмысловых олигонуклеотидов, как и введение антагонистов EMD281014 и MDL100907, уменьшают время иммобилизации грызунов в тесте Порсолта [23].
В ряде клинических исследований было показано, что одновременный прием блокаторов 5-HT2A-рецепторов и селективных ингибиторов обратного захвата серотонина усиливает антидепрессивный эффект последних у пациентов с резистентной депрессией [24]. При посмертных исследованиях у лиц, страдавших депрессией и покончивших жизнь самоубийством, было обнаружено увеличение плотности 5-HT2A-рецепторов в определенных областях префронтальной коры. Известно также, что большинство антидепрессантов при хроническом применении вызывают десенситизацию и снижение плотности 5-HT2A-рецепторов в мозге крыс [25].
Все вышесказанное указывает на то, что антидепрессивный эффект связан со снижением активности 5-НТ2А-рецепторов. Подобная картина наблюдается и в наших результатах: в мозге мышей линии BALB/c, на которых была выявлена антидепрессивноподобная активность ЦПГ, плотность серотониновых рецепторов уменьшалась.
Известно также, что 5-HT2A-рецепторы способны модулировать активность NMDA-рецепторов, а именно активация первых вызывает увеличение активности последних [26], что может приводить к усугублению депрессивной симптоматики.
Большое количество доклинических и клинических исследований показали, что антагонисты NMDA-рецепторов обладают антидепрессивным потенциалом, эффект многих антидепрессантов напрямую связывают с блокадой NMDA-рецепторов [2]. Таким образом, уменьшение количества мест связывания NMDA-рецепторов в мозге мышей BALB/c на фоне снижения 5НТ2А-рецепторов под влиянием ЦПГ в дозе 2 мг/кг/сутки в нашем исследовании не противоречит опубликованным ранее данным.
С помощью ЯМР-спектроскопии было показано, что у пациентов с депрессией концентрация ГАМК в затылочной коре головного мозга была значительно ниже, чем у здоровых людей. Снижение концентрации ГАМК было выражено сильнее у пациентов с меланхолической депрессией. Прием флуоксетина и циталопрама увеличивал концентрацию ГАМК в затылочной области. Кроме того, существуют данные, свидетельствующие о способности ингибиторов обратного захвата норадреналина и серотонина потенцировать ГАМКергическую передачу и синтез нейростероидов [27]. Можно предположить, что эффекты антидепрессантов связаны с активацией ГАМКергической системы. Помимо этого было установлено, что активация 5-HT2А-рецепторов в префронтальной коре запускает внутриклеточный сигнальный каскад, приводящий к усилению фосфорилирования протеинкиназой С γ2-субъединицы ГАМКА-рецептора, что вызывает ослабление ГАМКергического торможения. А дисбаланс возбуждения и торможения, как известно, способен вызвать вторичный дефицит функциональной активности моноаминергических нейронов [28]. Следовательно, эта часть полученных нами данных также не противоречит приведенным выше: у мышей BALB/c под влиянием ЦПГ в дозе 1 мг/кг/сутки происходит увеличение плотности ГАМКА-рецепторов на фоне снижения 5-НТ2А-рецепторов.
Тот факт, что ЦПГ в дозе 2 мг/кг/сутки уменьшает плотность NMDA-рецепторов в гиппокампе также может быть связан и с увеличением продукции ГАМК, так как последняя является функциональным антагонистом глутамата и способна ограничивать его возбуждающее действие [3].
Таким образом, суммируя полученные результаты нашего пилотного исследования, можно сделать заключение, что 5-НТ2А-, NMDA-, ГАМКА-рецепторы принимают участие в реализации антидепрессивноподобного эффекта ЦПГ. При этом наблюдается различная чувствительность к дозам пептида: изменения плотности ГАМКA-рецепторов происходят под влиянием ежесуточной дозы ЦПГ 1 мг/кг, плотности NMDA-рецепторов – при дозе ЦПГ 2 мг/кг, тогда как рецепторы серотонина отзываются на действие обеих доз пептида.
ВЫВОДЫ
1. Двухнедельное введение ЦПГ в дозах 1 и 2 мг/кг/сутки вызывает снижение плотности 5‑HT2A-рецепторов в мозге мышей BALB/c.
2. Двухнедельное введение ЦПГ в дозе 1 мг/кг/сутки приводит к увеличению плотности ГАМКА-рецепторов в коре мозга мышей линии BALB/c, а в дозе 2 мг/кг/сутки – к уменьшению плотности NMDA-рецепторов в гиппокампе мышей этой же линии.
3. 5-НТ2А-, NMDA- и ГАМКА-рецепторы принимают участие в реализации антидепрессивноподобного эффекта ЦПГ.
Список литературы
Левчук Л.А., Шмиголь М.В., Иванова С.А. // СВПН. 2012. № 2. С. 75–79.
Machado-Vieira R., Henter I.D., Zarate C.A. // Prog. Neurobiol. 2015. V. 152. P. 21–37.
Möhler H. // Neuropharmacology. 2012. V. 62. № 1. P. 42–53.
Тюренков И.Н., Перфилова В.Н. // Экспер. клин. фармакол. 2011. Т. 74. № 2. С. 47–52.
Ghose S., Winter M.K., McCarson K.E., Tamminga C.A., Enna S.J. // Br. J. Pharmacol. 2011. V. 162. № 1. P. 1–17.
Smith K.S., Rudolph U. // Neuropharmacology. 2012. V. 62. № 1. P. 54–62.
Gudasheva T.A., Boyko S.S., Akparov V.Kh., Ostrovskaya R.U., Skoldinov S.P., Rozantsev G.G., Voronina T.A., Zherdev V.P., Seredenin S.B. // FEBS Let. 1996. V. 391. № 1–2. P. 149–152.
Tran L.H. US Patent Appl. 2003 / 0109531. 2003.
Середенин С.Б., Гудашева Т.А., Бойко С.С., Ковалев Г.И., Воронин М.В., Яркова М.А. // Бюл. экспер. биол. 2002. Т. 133. № 4. С. 417–419.
Поварнина П.Ю., Колясникова К.Н., Николаев С.В., Антипова Т.А., Гудашева Т.А. // Бюл. экспер. биол. и мед. 2015. Т. 160. № 11. С. 600–603.
Гудашева Т.А., Колясникова К.Н., Антипова Т.А., Середенин С.Б. // ДАН. 2016. Т. 469. № 4. С. 492–495.
Duman R.S., Voleti B. // Trends Neurosci. 2012. V. 35. № 1. P. 47–56.
Ковалёв Г.И., Абдуллина А.А., Васильева Е.В., Гудашева Т.А., Середенин С.Б. // Экспер. клин. фармакол. 2018. Т. 81. № 11. С. 3–6.
Glowinski J., Iversen L.L. // J. Neurochem. 1966. V. 13. № 8. P. 655–669.
Leysen J.E., Niemegeers C.J. // Mol. Pharmacol. 1982. V. 21. № 2. P. 301–314.
Zhou L.M., Gu Z.Q., Costa A.M., Yamada K.A., Manssone P.E. // J. Pharmacol. Exp. Ther. 1997. V. 280. № 1. P. 422–427.
LaPage K.T., Ishmael J.E., Low C.W., Traynelis S.F., Murray T.F. // Neuropharmacology. 2005. V. 49. P. 1–16.
Schaffhauser H., Richards J.G., Cartmell J., Chaboz S., Kemp J.A., Klingelschmidt A., Messer J., Stadler H., Woltering T., Mutel V. // Mol. Pharmacol. 1998. V. 53. P. 228–233.
Ito Y., Koo Lim D., Hayase Y., Murakoshi Y., Ho I. // Neurochem. Res. 1992. V. 4. P. 307–313.
Hawkinson J.E., Acosta-Burruel M., Kimbrought C.L., Goodnough D., Wood P.L. // Eur. J. Pharmacol. 1996. V. 304. P. 141–146.
Bowery N.G., Hill D.R., Hudson A.L. // Neuropharmacology. 1985. V. 24. № 3. P. 207–10.
Szekely A.M., Barbaccia M.L., Costa E. // J. Pharmacol. Exp. Ther. 1987. V. 243. № 1. P. 155–9.
Guiard B., Di Giovanni G. // Front. Pharmacol. 2015. V. 6. № 46.
Celada P., Puig M., Amargós-Bosch M., Adell A., Artigas F. // J. Psychiatry Neurosci. 2004. V. 29. № 4. P. 252–265.
Eison A.S., Mullins U.L. // Behav. Brain Res. 1995. V. 73. № 1–2. P. 177–181.
Abernathy K., Chandler L.J., Woodward J.J. // Int. Rev. Neurobiol. 2010. V. 91. P. 289–320.
Калуев А.В., Натт Д.Дж. // Экспер. клин. фармакол. 2004. Т. 67. № 4. С. 71–76.
Feng J., Cai X., Zhao J., Yan Z. // J. Neurosci. 2001. V. 21. № 17. P. 6502–6511.
Дополнительные материалы отсутствуют.