

Нейрохимия, 2020, T. 37, № 1, стр. 390-45
Синтез дофамина недофаминергическими нейронами в стриатуме у мышей в норме и при деградации нигростриатной дофаминергической системы
В. Е. Блохин 1, Т. С. Пронина 1, М. В. Угрюмов 1, 2
1 Лаборатория нервных и нейроэндокринных регуляций, Федеральное государственное бюджетное
учреждение науки Институт биологии развития им. Н.К. Кольцова РАН
Москва, Россия
2 Отделение психологии, Факультет социальных наук, Национальный исследовательский университет
“Высшая школа экономики”
Москва, Россия
Поступила в редакцию 28.02.2019
После доработки 21.06.2019
Принята к публикации 26.07.2019
Аннотация
В последние пятнадцать лет получены доказательства того, что дофамин (ДА) синтезируется в мозге не только дофаминергическими нейронами, но и недофаминергическими нейронами, экспрессирующими по одному из комплементарных ферментов – тирозингидроксилазу или декарбоксилазу ароматических L-аминокислот (далее – декарбоксилаза). При этом L-ДОФА – промежуточный продукт синтеза ДА, переносится из нейронов, содержащих тирозингидроксилазу в нейроны, содержащие декарбоксилазу. Целью данной работы явилась проверка нашей гипотезы о наличии синтеза ДА недофаминергическими нейронами у мышей в стриатуме – ключевом звене центральной регуляции моторной функции. Для этого использован разработанный нами методический подход, позволяющий определить величину кооперативного синтеза как разницу между суммарным содержанием ДА в срезах мозга и в инкубационной среде после инкубации срезов в растворе Кребса–Рингера и в растворе Кребса–Рингера с добавлением конкурентного ингибитора мембранного переносчика L-ДОФА. В качестве ингибитора впервые использована синтетическая неметаболизируемая 2-амино-2-норборнанкарбоновая кислота, которая, в отличие от ранее использованных ингибиторов (L-тирозин, L-лейцин и др.), не влияет прямо или косвенно на функциональную активность ДА-ергических нейронов. Применение этого ингибитора позволило впервые получить доказательства того, что ДА, содержащийся в стриатуме у мышей в норме, синтезируется не только ДА-ергическими нейронами (в аксонах), но также недофаминергическими нейронами, экспрессирующими по одному из ферментов – тирозингидроксилазу или декарбоксилазу. Показано также, что синтез ДА недофаминергическими нейронами осуществляется у мышей в стриатуме при его частичной ДА-ергической денервации под действием 1-метил-4-фенил-1,2,3,6-тетрагидропиридина – специфического нейротоксина. Предполагается, что синтез ДА недофаминергическими нейронами – важный механизм нейропластичности, способствующий компенсации дефицита ДА при дегенерации нигростриатных ДА-ергических нейронов. Таким образом, впервые получены доказательства синтеза ДА недофаминергическими нейронами в стриатуме у мышей не только при ДА-ергической денервации стриатума, но и в норме.
ВВЕДЕНИЕ
Дофаминергические нейроны (ДА-ергические) нигростриатной системы мозга являются ключевым звеном регуляции моторной функции. Тела нейронов локализованы в черной субстанции, а аксоны проецируются в стриатум, где выделяющийся из них дофамин (ДА) в качестве нейротрансмиттера участвует в регуляции моторной функции [1, 2]. Дегенерация нигростриатных ДА-ергических нейронов и возникающий при этом дефицит ДА в стриатуме приводит к развитию болезни Паркинсона. Однако первые клинические симптомы в виде нарушений моторной функции (тремор, акинетико-ригидность) появляются только через много лет после начала патологического процесса – при дегенерации большей части ДА-ергических нейронов (более 60%) и снижении содержания ДА в стриатуме более, чем на 70% [3].
Считается, что отсутствие моторных симптомов в течение длительного времени, несмотря на прогрессирующую дегенерацию ДА-ергических нейронов, возможно благодаря развивающимся одновременно компенсаторным процессам. К ним относят повышение функциональной активности выживших ДА-ергических нейронов и повышение чувствительности нейронов стриатума к ДА при его дефиците [4]. В наших предыдущих исследованиях был открыт принципиально новый механизм нейропластичности при функциональной недостаточности ДА-ергических нейронов – совместный синтез ДА недофаминергическими нейронами, содержащими по одному из ферментов синтеза ДА (“моноферментные” нейроны) [5]. В процессе такого синтеза L-дигидроксифенилаланин (L-ДОФА), образующийся из L-тирозина в нейронах, содержащих только тирозингидроксилазу, выделяется в межклеточное пространство и захватывается с помощью мембранного переносчика больших нейтральных аминокислот 1-го типа и L-ДОФА (ПБНА-1) в нейроны, содержащие только декарбоксилазу ароматических L-аминокислот (далее – декарбоксилаза), где и происходит синтез ДА. Доказательством кооперативного синтеза ДА недофаминергическими моноферментными нейронами считается снижение синтеза ДА ex vivo в срезах области мозга, содержащей эти нейроны, при конкурентном ингибировании ПБНА-1 [5]. До последнего времени в качестве конкурентного ингибитора использовали большие нейтральные аминокислоты, в основном L-лейцин. При этом были получены доказательства синтеза ДА недофаминергическими моноферментными нейронами в тубероинфундибулярной системе мозга у крыс в норме и в стриатуме у мышей, причем только при дегенерации части ДА-ергических нейронов [6, 7].
Несмотря на то, что L-лейцин не включается в метаболизм ДА в дофаминергических нейронах, нельзя исключить его побочное влияние на ДА-ергические нейроны и нейровоспалительный процесс, спсобствующий нейродегенерации [8]. Поэтому в данной работе для доказательства кооперативного синтеза ДА недофаминергическими моноферментными нейронами использован синтетический неметаболизирующийся конкурентный ингибитор ПБНА-1 – 2-амино-2-норборнанкарбоновая кислота (АНК) [9]. Целью данной работы явилась проверка нашей гипотезы, согласно которой ДА синтезируется в стриатуме недофаминергическими моноферментными нейронами не только при частичной ДА-ергической денервации, но и в норме.
МАТЕРИАЛЫ И МЕТОДЫ
Животные, эксперименты. В данной работе использовали самцов мышей линии C57BL/6J, в возрасте 2–2.5 мес. и массой 23–25 г. Животных содержали в виварии с 12-часовым циклом дня и ночи и свободным (ad libitum) доступом к пище и воде. Все манипуляции с животными были проведены в соответствии с национальными и международными требованиями и правилами, утвержденными комитетом по охране животных ФГБУН Института биологии развития им. Н.К. Кольцова РАН.
Мышам в первой группе (n = 6) подкожно вводили физиологический раствор 4 раза с интервалом в 2 ч. Мышам во второй группе (n = 6) подкожно вводили дважды с интервалом 2 ч 1-метил-4-фенил-1,2,3,6-тетрагидропиридин (МФТП) (разовая доза – 8 мг/кг веса) (Sigma-Aldrich, США) в физиологическом растворе, для моделирования болезни Паркинсона на доклинической стадии. Для моделирования болезни Паркинсона на клинической стадии мышам (n = 7) вводили 4 раза МФТП (разовая доза – 10 мг/кг веса) в физиологическом растворе с интервалом 2 ч между инъекциями.
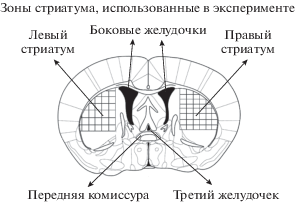
Инкубация срезов ex vivo. Через 2 нед. после инъекций мышей наркотизировали изофлураном (KentScientific, США), декапитировали и выделяли мозг. Далее на вибратоме (Vibratome 1000 plus Sectioning system, Германия) в растворе Кребса–Рингера (NaCl 120 мМ, KCl 4.8 мМ, CaCl2 2 мМ, MgSO4 1.3 мМ, NaHCO3 25 мМ, D-глюкоза 10 мМ, HEPES 20 мМ, аскорбиновая кислота 0.1 мМ, pH 7.4) при 4°C приготавливали фронтальные срезы мозга толщиной 300 мкм на уровне стриатума. Из полученных срезов под контролем бинокулярной лупы при 4°C выделяли стриатум из обоих полушарий в соответствии с атласом мозга мышей [10] (рис. 1). В одну пробу брали 4 среза стриатума из одного полушария мозга от одного животного.
Рис. 1.
Схематическое изображение стриатума. Штриховкой показана вырезаемая область стриатума, использованная в экспериментах.
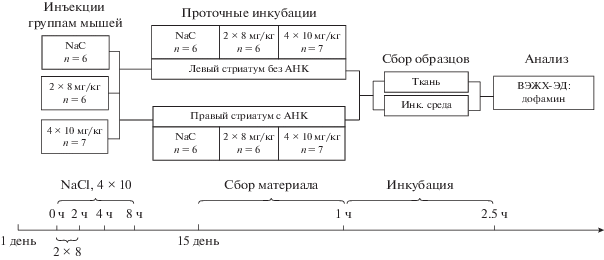
Проточную инкубацию срезов стриатума проводили при 37°C в термостатируемых камерах. Инкубация состояла из двух этапов. Первый этап – стабилизация, когда во все камеры поступал раствор Кребса–Рингера в течение 40 мин. На втором этапе инкубация в течение 60 мин в камеры со срезами стриатума из правых полушарий продолжал поступать раствор Кребса–Рингера, а в камеры со срезами из левых полушарий раствор Кребса–Рингера с АНК в концентрации 0.5 мМ. Инкубационную среду собирали, добавляли к ней 3,4-дигидроксибензиламин (ДГБА), замораживали в жидком азоте и хранили при –70°C. После инкубации срезы замораживали для дальнейшего анализа содержания катехоламинов при помощи высокоэффективной жидкостной хроматографии с электрохимическим детектором (ВЭЖХ-ЭД). Схема эксперимента представлена на рис. 2.
Рис. 2.
Схема эксперимента. NaCl – контрольная группа, получавшая физиологический расствор. 2 × 8 – группа животных получавшая двукратное введение 1-метил-4-фенил-1,2,3,6-тетрагидропиридина (МФТП) в разовой дозе 8 мг/кг. 4 × 10 – группа животных получавшая четырехкратного введение МФТП в разовой дозе 10 мг/кг. Левые стриатумы икубировали в растворе Кребса–Рингера без 2-амино-2-норборнанкарбоновой кислоты (АНК). Правые стриатумы икубировали в растворе Кребса–Рингера с 0.5 мМ АНК. Анализ ткани и инкубационной среды (инк. среда) проводили методом ВЭЖХ-ЭД.
Определение катехоламинов в срезах и среде при помощи высокоэффективной жидкостной хроматографии с электрохимической детекцией. Пробы для измерения катехоламинов в инкубационной среде были приготовлены методом твердофазной экстракции с использованием оксида алюминия. Содержание дофамина в пробах определяли методом высокоэффективной жидкостной хроматографии с электрохимической детекцией (ВЭЖХ-ЭД), по описанному ранее протоколу (Kozina et al, 2017). Для измерения ДА в срезах стриатума замороженную ткань гомогенизировали ультразвуковым гомогенизатором при 4°С в 400 мкл раствора 0.1 М HClO4 и 250 пМ ДГБА. Затем часть гомогената отбирали для измерения концентрации белка с помощью набора “BCA Protein Assay Kit” в соответствии с инструкцией производителя (The Thermo Scientific Pierce, США). Остаток гомогената центрифугировали 20 мин со скоростью 18000 g при 4°C. Супернатант отбирали для дальнейшего анализа.
Определение ДА проводили на обращенно-фазовой колонке (100 × 4 mm ReproSil-Pur C18, 3 μm) (Dr. Maisch, Germany). Подвижной фазой служил 0.1 М цитратно-фосфатный буфер, содержащий 0.25 мМ октансульфоната натрия, 0.1 М ЭДТА и 6% ацетонитрила (pH 2.55). Потенциал веществ выходящих из колонки определяли с помощью электрохимического детектора DECADE II (Antec Leyden, Netherlands). Пики катехоламинов и их метаболитов идентифицировали по времени выхода относительно раствора стандартов. Содержание катехоламинов рассчитывали, используя отношение площадей пиков в образце к стандартам. Площадь пиков измеряли с помощью программного обеспечения LabSolutions (Shimadzu, Япония). Содержание ДА нормировали на концентрацию белка.
Статистическая обработка. Полученные данные были статистически обработаны при помощи программ GraphPad Prism 6.0 (GraphPad Software, США). Для статистического сравнения выбранных групп использовали t-тест Стьюдента. Данные представлены как среднее ± стандартная ошибка среднего (SEM). Значение p < 0.05 считалось статистически значимым.
РЕЗУЛЬТАТЫ
У мышей в контрольной группе (введение 0.9% NaCl) содержание ДА в срезах стриатума при инкубации с 0.5 мМ АНК было меньше (0.037 ± ± 0.009 пмоль), чем при инкубации без АНК на 56% (рис. 3). В среде после инкубации срезов с АНК (18.44 ± 7.3 пмоль/мл) концентрация ДА увеличена в 15 раз по сравнению со средой после инкубации срезов без АНК (рис. 4). Суммарное содержание ДА в среде и ткани внутри группы при инкубации срезов стриатума без АНК (0.085 ± ± 0.02 пмоль) было на 45% больше, чем с АНК (рис. 5).
Рис. 3.
Концентрация дофамина в срезах стриатума, полученных у мышей после введения физиологического раствора (контроль), после двукратного введения 1-метил-4-фенил-1,2,3,6-тетрагидропиридина (МФТП) в разовой дозе 8 мг/кг (2 × 8), после четырехкратного введения МФТП в разовой дозе 10 мг/кг (4 × 10) и инкубированных в растворе Кребса–Рингера без АНК или с АНК (0.5 мМ). Статистическая значимость: * p < 0.05 – относительно контроля, # p < 0.05 – между парами проб, инкубированными без АНК и с 0.5 мМ АНК.
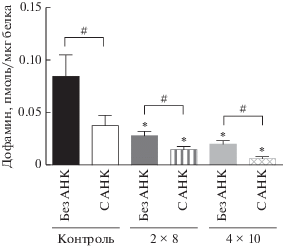
Рис. 4.
Концентрация дофамина в среде после инкубации срезов стриатума, полученных у мышей после введения физиологического раствора (контроль), после двукратного введения МФТП в разовой дозе 8 мг/кг (2 × 8), после четырехкратного введения МФТП в разовой дозе 10 мг/кг (4 × 10) и инкубированных в растворе Кребса–Рингера без АНК или с АНК (0.5 мМ). Статистическая значимость: # p < 0.05 – между парами проб инкубированными без АНК и с 0.5 мМ АНК.
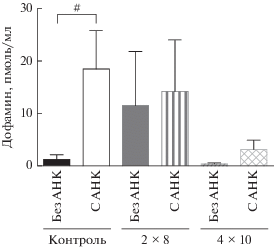
Рис. 5.
Общее содержание дофамина в среде и срезах стриатума, полученных у мышей после введения физиологического раствора (контроль), после двукратного введения МФТП в разовой дозе 8 мг/кг (2 × 8), после четырехкратного введения МФТП в разовой дозе 10 мг/кг (4 × 10) и инкубированных в растворе Кребса–Рингера без АНК или с АНК (0.5 мМ). Статистическая значимость: * p < 0.05 – относительно контроля, # p < 0.05 – между парами проб, инкубированными без АНК и с 0.5 мМ АНК.
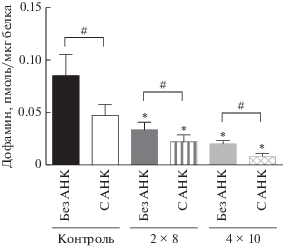
В группе мышей после двукратного введения МФТП (2 × 8 мг/кг) уровень ДА в срезах стриатума без использования АНК (0.027 ± 0.004 пмоль) снизился на 65% относительно контрольной группы, получавшей физиологический раствор (рис. 3). Это свидетельствует об успешном моделировании болезни Паркинсона при помощи МФТП. Внутри группы содержание ДА в срезах стриатума, инкубированных с 0.5 мМ АНК (0.015 ± ± 0.002 пмоль), было меньше на 49% чем при инкубации без АНК (рис. 1). В инкубационной среде срезов стриатума, инкубированных с 0.5 мМ АНК (14.17 ± 9.7 пмоль/мл), концентрация ДА была увеличена на 19.8% по сравнению со средой после инкубации срезов без АНК (рис. 4). Суммарное содержание ДА в среде и ткани внутри группы при инкубации срезов стриатума без АНК (0.033 ± 0.007 пмоль) было больше на 33%, чем с 0.5 мМ АНК (рис. 5).
В группе мышей после четырехкратного введения МФТП (4 × 10 мг/кг) уровень ДА в срезах стриатума без использования АНК (0.0199 ± 0.003 пмоль) снизился на 78% относительно контрольной группы, получавшей физиологический раствор (рис. 3). Внутри группы содержание ДА в срезах стриатума, инкубированных с 0.5 мМ АНК (0.0059 ± 0.002 пмоль), было меньше на 72%, чем при инкубации без АНК (рис. 1, 3). В инкубационной среде срезов стриатума, инкубированных с 0.5 мМ АНК (3.11 ± 1.76 пмоль/мл), концентрация ДА была в 10 раз выше по сравнению со средой после инкубации срезов без АНК (рис. 4). Суммарное содержание ДА в среде и ткани внутри группы при инкубации срезов стриатума без АНК (0.02 ± 0.003 пмоль) было больше на 60%, чем с АНК (рис. 5).
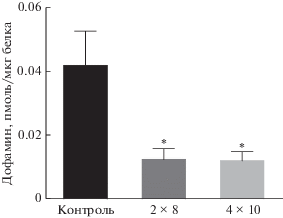
Проведен сравнительный анализ разницы в общем содержании дофамина в среде и срезах стриатума после инкубации без АНК и с АНК (0.5 мМ) срезов, полученных у мышей после введения физиологического раствора (контроль), после двукратного введения МФТП в разовой дозе 8 мг/кг (2 × 8), после четырехкратного введения МФТП в разовой дозе 10 мг/кг (4 × 10) (рис. 6). Показано, что эта разница у животных в контроле превосходит этот показатель в обеих группах животных, которым вводили МФТП. Напротив, по этому показателю обе группы животных, которым вводили МФТП, не различаются (рис. 6).
Рис. 6.
Разница в общем содержании дофамина в среде и срезах стриатума после инкубации без АНК и с АНК (0.5 мМ) срезов, полученных у мышей после введения физиологического раствора (контроль), после двукратного введения МФТП в разовой дозе 8 мг/кг (2 × 8), после четырехкратного введения МФТП в разовой дозе 10 мг/кг (4 × 10). Статистическая значимость: * p < 0.05 – относительно контроля.
ОБСУЖДЕНИЕ
В данной работе для доказательства наличия кооперативного синтеза ДА недофаминергическими моноферментными нейронами в стриатуме и его количественной оценки в норме и при различном уровне деградации нигростриатной ДА-ергической системы был использован методический подход, предложенный нами первоначально для доказательства наличия такого синтеза ДА в тубероинфундибулярной ДА-ергической системе гипоталамуса [11, 6 ]. При этом уровень кооперативного синтеза ДА определяли как разницу между суммарным содержанием ДА в срезах мозга и в инкубационной среде после инкубации срезов в течение часа в растворе Кребса–Рингера без добавления или с добавлением конкурентного ингибитора ПБНА-1 [12].
В предыдущих работах в качестве ингибитора ПБНА-1 использовали одну из больших нейтральных L-аминокислот – L-лейцин или L-тирозин [6, 7], хотя обе аминокислоты прямо или косвенно влияют на функциональную активность ДА-ергических нейронов. Так, L-тирозин является субстратом для синтеза катехоламинов и ДА, в частности, а L-лейцин является предшественником глютамата, который участвует в регуляции секреции ДА ДА-ергическими нейронами [13]. Для того чтобы при оценке синтеза ДА недофаминергическими нейронами избежать побочного действия больших нейтральных L‑аминокислот, в данной работе для предотвращения поступления L‑ДОФА из межклеточной среды в моноферментные нейроны, содержащие декарбоксилазу, нами впервые при изучении стриатума использован АНК – синтетический неметаболизируемый ингибитор ПБНА-1 [12]. Важно подчеркнуть, что, помимо того, что АНК неметаболизируемый ингибитор, а следовательно не должен прямо или опосредовано влиять на ДА-ергические нейроны, он обладает не меньшим сродством к ПБНА-1 по сравнению с ранее использованными большими нейтральными аминокислотами – L-лейцином и L-тирозином [9].
Следует отметить, что L-ДОФА, секретируемый в значительном количестве только моноферментными нейронами, содержащими тирозингидроксилазу, может превращаться в ДА не только в моноферментных нейронах, содержащих декарбоксилазу, но и в любой клетке, содержащей этот фермент [6, 14]. Так, в стриатуме в кооперативном синтезе ДА могут участвовать аксоны дофаминергических, серотонинергических и норадренергических нейронов, содержащих декарбоксилазу. Однако, учитывая то, что число норадренергических аксонов в стриатуме крайне мало [15], их участием в кооперативном синтезе ДА можно пренебречь.
В данной работе впервые удалось получить прямые доказательства синтеза ДА недофаминергическими моноферментными нейронами в стриатуме у мышей в норме, используя разработанный нами ранее подход к оценке синтеза ДА в слайсах мозга в присутствии и в отсутствии АНК в качестве ингибитора ПБНА-1. Наши предыдущие попытки доказать наличие синтеза ДА моноферментными нейронами в стриатуме у интактных животных не увенчались успехом [7]. Это, возможно, объясняется использованием в качестве ингибитора ПБНА-1 L-лейцина, обладающего побочными эффектами на функциональную активность ДА-ергических нейронов [16].
Полученные нами данные о кооперативном синтезе ДА моноферментными нейронами в стриатуме у мышей в норме хорошо согласуются с результатами нашего предыдущего иммуногистохимического исследования с использованием двойного мечения – тирозингидроксилазы и декарбоксилазы. В этой работе в стриатуме у мышей в норме выявлены единичные тела и многочисленные отростки моноферментных нейронов, содержащих тирозингидроксилазу [7]. В таком же количестве были выявлены тела и отростки нейронов, содержащих только декарбоксилазу. В совокупности, результаты, полученные в данной работе и в предыдущем иммуногистохимическом исследовании, свидетельствуют о том, что в стриатуме у мышей в норме осуществляется кооперативный синтез ДА, причем не столько в телах моноферментных нейронов, сколько в их отростках.
Предполагается, что кооперативный синтез ДА недофаминергическими моноферментными нейронами приобретает особенно большое значение при дегенерации нигростриатных ДА-ергических нейронов и соответствующей деафферентации стриатума, что характерно для болезни Паркинсона. Для проверки этого предположения оценивали количественно кооперативный синтез ДА в стриатуме у мышей при различном уровне деградации нигростриатной ДА-ергической системы на моделях доклинической и клинической стадий болезни Паркинсона. Моделирование этого заболевания осуществляли путем системного введения мышам протоксина – МФТП, превращающегося в организме в 1-метил-4-фенилпиридиний (МФП+) – токсин ДА-ергических нейронов [17, 18]. Как было показано в нашей предыдущей работе, содержание ДА в стриатуме при моделировании доклинической стадии болезни Паркинсона снижалось на 46%, а при моделировании ранней клинической стадии на 73% [7]. В данной работе при использовании в качестве ингибитора ПБНА-1 АНК показано, что величина кооперативного синтеза ДА не отличается у мышей с различным уровнем деградации нигростриатной ДА-ергической системы, вызванной МФТП. Однако кооперативный синтез в стриатуме у мышей с паркинсонизмом оказался меньше, чем в стриатуме у мышей в контроле. Снижение уровня кооперативного синтеза ДА в стриатуме у мышей на моделях болезни Паркинсона по сравнению с нормой, вероятно, объясняется частичной деградацией под действием МФТП ДА-ергических, норадренергических и серотонинергических афферентов, которые содержат декарбоксилазу, обеспечивающую синтез ДА из L-ДОФА, образующегося в моноферментных тирозингидроксилаза-содержащих нейронах.
Таким образом, впервые получены доказательства кооперативного синтеза ДА недофаминергическими нейронами в стриатуме у мышей не только при ДА-ергической денервации стриатума, но и в норме.
Список литературы
Crocker A.D. // Reviews in the neurosciences. 1997. V. 8. P. 55–76.
Joshua M., Adler A., and Bergman H. // Curr. Opin. Neurobiol. 2009. V. 19. P. 615–620.
Cheng H-C., Ulane C.M., Burke R.E. // Ann. Neurol. 2010. V. 67. P. 715–725.
Blesa J., Trigo-Damas I., Dileone M, Lopez-Gonzalez del Rey N., Hernandez L. F., Obeso J. A. // Exp. Neurol. 2017. V. 298. P. 148–161.
Ugrumov M.V. // Adv. Pharmacol. 2013. V. 68. P. 37–91.
Ugrumov M.V., Taxi J., Pronina T., Kurina A., Sorokin A., Sapronova A., Calas A. // Neurosci. 2014. V. 277. P. 45–54.
Kozina E.A., Kim A.R., Kurina A.Y., Ugrumov M.V. // Neurobiol. Dis. 2017. V. 98. P. 108–121.
Sperringer J.E., Addington A., Hutson S.M. // Neurochem. Res. 2017. V. 6. P. 1697–1709.
Yanagida O., Kanai Y., Chairoungdua A., Kim D.K., Segawa H., Nii T., Cha S.H., Matsuo H., Fukushima J., Fukasawa Y., Tani Y., Taketani Y., Uchino H., Kim J.Y., Inatomi J., Okayasu I., Miyamoto K., Takeda E., Goya T., Endou H. // BBA. 2001. V. 1514. P. 291–302.
Franklin K.B.J. и Paxinos G. // The Mouse Brain in Stereotaxic Coordinates, Compact 3rd Edition London: Academic press, 2008.
Ugrumov M.V., Melnikova V.I., Lavrentyeva A.V., Kudrin V.S., Rayevsky K.S. // Neurosci. 2004. V. 124. P. 629–635.
Kageyama T., Nakamura M., Matsuo A., Yamasaki Y., Takakura Y., Hashida M., Kanai Y., Naito M., Tsuruo T., Minato N., Shimohama S. // Brain Res. 2000. V. 879. P. 115–121.
Shimizu N., Duan S., Hori T., Oomura Y. // Brain Res. 1990. V. 25. P. 99–102.
Arai R., Karasawa N., Geffard M., Nagatsu I. // Neurosci. Lett. 1995. V. 195. P. 195–198.
Swanson L.W., Hartman B.K. // J. Comp. Neur. 1975. V. 163. P. 467–506.
Cachope R., Cheer J.F. // Front. Behav. Neurosci. 2014. V. 8. P. 1–7.
Javitch J.A., D’Amato R.J., Strittmater S.M., Snyder S.H. // Proc. Natl. Acad. Sci. 1985. V. 82. P. 2173–2177.
Ransom B.R., Kunis D.M., Irwin I., Langston J.W. // Neurosci. Lett. 1987. V. 75. P. 323–328.
Дополнительные материалы отсутствуют.