

Нейрохимия, 2020, T. 37, № 1, стр. 640-74
Особенности метилирования ДНК и гистона Н3 в гиппокампе и неокортексе крыс, переживших патологические воздействия в пренатальном периоде развития
Е. И. Тюлькова 1, Л. А. Ватаева 2, В. А. Стратилов 1, В. С. Барышева 1, О. В. Ветровой 1, 3
1 ФГБУН Институт физиологии им. И.П. Павлова РАН
Санкт-Петербург, Россия
2 Российский государственный педагогический университет им. А.И. Герцена
Санкт-Петербург, Россия
3 ФГБОУ ВО Санкт-Петербургский государственный университет,
биологический факультет, кафедра биохимии
Санкт-Петербург, Россия
Поступила в редакцию 03.04.2019
После доработки 13.05.2019
Принята к публикации 29.07.2019
Аннотация
Стресс во время беременности может быть причиной структурных и функциональных изменений головного мозга плода, которые в дальнейшем приводят к формированию различных нервно-психических заболеваний, включая синдром дефицита внимания и гиперактивности, депрессию, шизофрению, аутизм и др. Среди стрессовых факторов, способных привести к нарушениям развития плода, особое место занимает пренатальный гипоксический стресс. Эпигенетические механизмы могут играть ключевую роль в развитии нарушений функциональной активности мозга, вызываемых стрессовыми воздействиями различной природы. Целью настоящей работы было изучение возрастных особенностей метилирования ДНК (meDNA) и гистона Н3 по лизинам 4 (meН3К4) и 9 (meН3К9) в клетках гиппокампа и неокортекса крыс – наиболее чувствительных к гипоксии структур мозга, вследствие воздействия тяжелой гипобарической гипоксии (180 мм рт. ст., 3 ч) или введения синтетического глюкокортикоида дексаметазона (0.8 мг/кг), моделирующего стрессорный ответ материнского организма, на 14–16-е сут пренатального онтогенеза. С использованием иммуногистохимического метода были обнаружены возрастные модификации степени meDNA и meН3К9 (но не meН3К4) у контрольных животных, а именно снижение уровня meDNA и meH3K9 у старых 18-месячных крыс по сравнению с 3-месячными. Предъявление тяжелой гипобарической гипоксии, так же, как и введение дексаметазона, на 14–16-е сут пренатального онтогенеза приводят к длительному (до 18 мес.) увеличению уровня meDNA во всех исследованных структурах мозга. При этом наблюдается прогрессирующее с возрастом снижение уровня meН3К4 – активационной модификации хроматина, в гиппокампе и неокортексе крыс. В то же время уровень meH3K9 – тормозной модификации хроматина, у животных, переживших неблагоприятные воздействия в пренатальном периоде, с возрастом снижается в меньшей степени, чем у контрольных крыс. Эти данные косвенно указывают на снижение транскрипционной активности генома пренатально стрессированных животных, что может лежать в основе длительных изменений их поведения и способности к обучению.
ВВЕДЕНИЕ
Эпидемиологические и клинические данные свидетельствуют о том, что стресс во время беременности может быть причиной структурных и функциональных изменений головного мозга плода, которые в дальнейшем приводят к формированию различных нервно-психических заболеваний, включая синдром дефицита внимания и гиперактивности, депрессию, шизофрению, аутизм и др. [1–7]. Многочисленные клинико-доклинические исследования позволили сформулировать теорию “эмбрионального происхождения болезни взрослых” [8–10], показав существенную корреляцию между неблагоприятными воздействиями на организм матери и развитием различных заболеваний в дальнейшей жизни, включая сердечно-сосудистые заболевания, диабет и неврологические заболевания [11–14]. Среди стрессовых факторов, способных привести к нарушениям развития плода, особое место занимает пренатальный гипоксический стресс [15, 16].
Механизмы влияния стрессовых воздействий на развитие мозга плода до конца не изучены, однако считается, что ключевым негативным фактором риска формирования патологии является избыточный уровень глюкокортикоидов (ГК) в организме беременной женщины. При тяжелом хроническом стрессе и длительном повышении уровня ГК в крови матери, несмотря на высокоэффективную конверсию ГК в кортизон с помощью 11-бета-гидроксистероид-дегидрогеназы 1 типа в плаценте, значительное количество глюкокортикоидов попадают в кровоток плода и напрямую воздействуют на его мозг [17–19]. Воздействие ГК на плод также может быть индуцировано во время терапевтического введения синтетических ГК для ускорения созревания легких плода. Применение ГК в акушерской практике продолжается, хотя многие клинические исследования показывают, что введение ГК в пренатальный период оказывает неблагоприятное влияние на познавательные способности и вызывает долгосрочные изменения в поведении детей [20–22]. В настоящее время, в связи с накоплением данных о долгосрочном влиянии введения глюкокортикоидов при беременности, в особенности на нервную систему, оправданность их применения в этот период вызывает сомнения [23, 24]. Недостаток глюкокортикоидов, также как и их избыток, может играть фатальную роль в развитии организма. Таким образом, необходимо проведение дальнейших исследований в этой области [25].
Недостаточное понимание механизмов влияния стрессовых воздействий на развивающийся организм требует изучения в экспериментальных моделях на животных. Результаты модельных экспериментов подтверждают клинические наблюдения, касающиеся влияния стрессовых факторов на формирование мозга и поведения. Так, в наших предыдущих исследованиях были выявлены нарушения формирования оборонительного поведения у крыс, подвергшихся воздействию дексаметазона или гипоксии в пренатальный период. Было установлено, что характер нарушений зависит от сроков гестации, на которые приходилось воздействие стресса, и вида стресса [26].
Одним из общих механизмов, с помощью которого материнский стресс может быть связан с фенотипическими изменениями в дальнейшей жизни, является эпигенетическое программирование генов, которое играет центральную роль в определении функционального выхода информации, хранящейся в геноме. Патологические воздействия во время беременности влияют как на развитие матери, так и плода посредством изменения генетических признаков индивида, приобретенных в течение нескольких поколений естественным отбором, и изменения экспрессии генов путем эпигенетических модификаций. Эпигенетические модификации регулируют экспрессию генов без изменений последовательности ДНК в основном путем метилирования собственно ДНК и ковалентными посттрансляционными модификациями гистонов, такими как ацетилирование, фосфорилирование, метилирование, убиквитинилирование, сумоилирование. Метилирование/деметилирование ДНК является основным механизмом в регуляции изменения экспрессии генов [27]. Модификация гистонов изменяет структуру хроматина, которая определяет доступность регуляторных факторов к основной ДНК [28].
Эпигенетические модификации играют значительную роль в пре- и постнатальные периоды развития, внося вклад в пластичность организма к изменяющимся условиям среды обитания, регулируя процессы пролиферации и дифференциации клеток путем активации/репрессии транскрипционных факторов и изменения экспрессии генов.
Достаточно доказательств участия эпигенетической регуляции в реакции плода на внутриутробный стресс, приводящий к долгосрочным изменениям профилей экспрессии генов, которые потенциально приводят к заболеваниям в более позднем возрасте [13, 29, 30]. Важно отметить, что как эндогенные, так и синтетические глюкокортикоиды могут вызывать эпигенетические изменения, которые оказывают влияние на развитие плода [31]. Кроме того, глюкокортикоиды могут оказывать быстрые негеномные эффекты, влияющие, например, на возбудимость нейронов или вызывающие ингибирование пролиферации нейронов, сопровождающееся неврологическими дефектами, наблюдаемыми у младенцев после хронического стресса у матерей или после лечения синтетическими глюкокортикоидами [32].
Молекулярные механизмы влияния гипоксии или других стрессорных воздействий при программировании нарушений головного мозга в течение длительного постнатального периода развития остаются в значительной степени малоизученными.
В последние годы интенсивно развивающейся областью исследований механизмов развития мозга стало изучение роли метилирования ДНК в периоде нейрогенеза. Показано, что патологические изменения внешних условий во время эмбрионального развития приводят к значительным модификациям уровня метилирования ДНК [33]. Основная функция метилирования ДНК заключается в том, чтобы транскрипционно ингибировать экспрессию генов [34, 35]. Как известно, посттрансяционные модификации гистонов также вовлечены в регуляцию экспрессии генов. Так метилирование гистона Н3 по лизину 4 (meH3K4), также как ацетилирование, стимулируют транскрипцию, а метилирование по лизину 9 (meH3K9) – тормозит [36].
Ранее нами были показаны изменения уровня ацетилирования гистона Н3 по лизину 24 в гиппокампе и неокортексе взрослых крыс после предъявления им на 14-16-е сут пренатального развития тяжелой гипобарической гипоксии [37] или введения дексаметазона [38]. Представляет большой интерес проведение исследований по изучению эпигенетического статуса мозга крыс, подвергавшихся воздействиям повреждающих факторов в период пренатального онтогенеза, на разных этапах дальнейшего постнатального развития – от раннего постнатального онтогенеза до старости.
Целью настоящей работы было изучение особенностей метилирования ДНК и гистона Н3 в гиппокампе и сенсомоторной коре мозга 2-недельных, 3- и 18-месячных крыс, переживших воздействие тяжелой гипоксии или введение синтетического гормона дексаметазона в течение третьей недели пренатального онтогенеза – периода формирования исследуемых структур мозга.
МАТЕРИАЛЫ И МЕТОДЫ
Работа с животными. Использовали крыс линии Вистар из “Коллекции лабораторных млекопитающих разной таксономической принадлежности” Института физиологии им. И.П. Павлова РАН, поддержанной программой биоресурсных коллекций ФАНО России. При проведении экспериментов соблюдали требования, сформулированные в Директивах Совета Европейского сообщества (86/609/ЕЕС) об использовании животных для экспериментальных исследований. Протоколы экспериментов были утверждены Комиссией по гуманному обращению с животными Института Физиологии им. И.П. Павлова РАН.
Животные были рождены интактными самками и самками, которых на 14–16-е сут беременности подвергали действию тяжелой гипобарической гипоксии или введению синтетического гормона дексаметазона. Для создания тяжелой гипоксии беременных самок помещали в барокамеру проточного типа при температуре от 20 до 25°С и ступенчато понижали давление до 180 мм рт. ст. (продолжительность воздействия – по 3 ч в течение 3-х сут с интервалом 24 ч между сеансами). Смертность животных в барокамере составляла 15% [39]. Синтетический гормон дексаметазон вводили внутрибрюшинно трехкратно с интервалом в сутки в течение 14–16-х сут беременности в дозе 0.8 мг/кг. При выборе дозы дексаметазона мы учитывали данные других авторов и наши более ранние исследования [40, 41].
Крысят отлучали от кормившей их матери в возрасте 30 суток. После отлучения крысы находились в клетках размером 60 × 30 × 20 см по 6 животных в каждой. В течение всего периода проведения экспериментов крысы содержались при режиме свет : темнота 12 : 12 ч, температуре 20–23°С и при постоянном доступе к воде и пище. В настоящей работе эксперименты были поставлены на 2-недельных ювенильных животных, 3-месячных молодых половозрелых самцах с активным сперматогенезом, и 18-месячных стареющих крысах. Для каждой временной точки были использованы животные из одного помета. Воспроизводимость результатов была оценена в двух независимых экспериментах.
Иммуногистохимические исследования проведены на животных всех возрастных групп. Декапитацию крыс каждой из экспериментальных групп (по 6 крыс) для взятия головного мозга осуществляли гильотиной. После декапитации вскрывали череп, извлекали мозг, отрезали мозжечок и помещали мозг в фиксатор. Далее образцы ткани мозга обрабатывали согласно стандартному гистологическому протоколу: фиксировали в молекулярном фиксаторе FineFix (разведение: 28 мл фиксатора + 72 мл 96% этанола; Milestone, Italy) в течение 24 ч при температуре 4°C. Затем образцы промывали в проточной воде в течение 2 ч и обезвоживали, проводя через этанол возрастающих концентраций (50 → 70 → 80 → 96 → 96% по 1 ч в каждом). На ночь оставляли в бутаноле. Затем материал проводили через 2 порции ксилола (по 30–40 мин), помещали в парафин (2 смены парафина, по 1 ч в каждой) в термостате при температуре 56°C и изготавливали парафиновые блоки. На ротационном микротоме (Reichert, Austria) изготавливали серийные срезы мозга во фронтальной плоскости толщиной 7 мкм на уровне 2.8–3.6 мм от брегмы. Полученные срезы монтировали на предметные стекла, обработанные полилизином.
Далее срезы депарафинизировали в ксилоле (2 смены по 5 мин) и подвергали регидратации в спиртах (96 → 96 → 96 → 70% по 5 мин в каждом). Для оценки степени ацетилирования гистона Н3 по лизину 24 использовали иммуногистохимический метод. Основные этапы метода: 1) инкубация с поликлональными кроличьими антителами к гистону Н3, метилированному по Lys4 (meН3К4, активационная модификация хроматина, антитела ab8580, 1 : 500) и Lys9 (meН3К9, тормозная модификация хроматина, антитела ab8898, 1 : 500), а также с моноклональными мышиными антителами к метилированному цитидину ДНК (meDNA, антитела sc-56615, 1 : 200) 2) инкубация с вторичными биотинилированными противокроличьими либо противомышиными антителами (Vectastein ABC kit, Vector Laboratories, Inc., США, 1 : 200); 3) инкубация с комплексом авидина и биотинилированной пероксидазы (ABC, Vector Laboratories, Inc., США, разведение реагентов А и В 1 : 100); 4) визуализация реакции с помощью диаминобензидинового набора (DAB substrate kit for peroxidase, Vector Laboratories, Inc., США).
Анализ препаратов проводили с помощью морфометрической установки, состоящей из светового микроскопа Jenaval (Carl Zeiss, Германия), цифровой камеры Baumer CX05c (Baumer Optronic, Германия) и компьютера IBM PC с программным обеспечением ВидеоТест Мастер Морфология (разработка ООО Видео Тест, Санкт-Петербург). Клетки подсчитывали в поле зрения площадью 460 × 340 мкм при увеличении объектива 40×). Определяли величину средней оптической плотности каждой клетки в усл. ед. уровня серого, обратно пропорциональных единицам яркости. Для анализа проводили иммуногистохимическую реакцию на 4-х гистологических препаратах от каждого животного, усредняя значения для каждой области мозга c одного поля зрения конкретной области мозга на срезе. Результаты обрабатывали с помощью пакетов анализа данных STATISTICA 7.0 Stat Soft, Inc и Microsoft Excel’2003, использовали непараметрический критерий Манн–Уитни (Mann–Whitney U-test). Изменения считали достоверными при P ≤ 0.05. Все результаты представлены в виде среднего арифметического и его ошибки. Результаты выражены в процентах от контроля, принятого за 100%.
РЕЗУЛЬТАТЫ
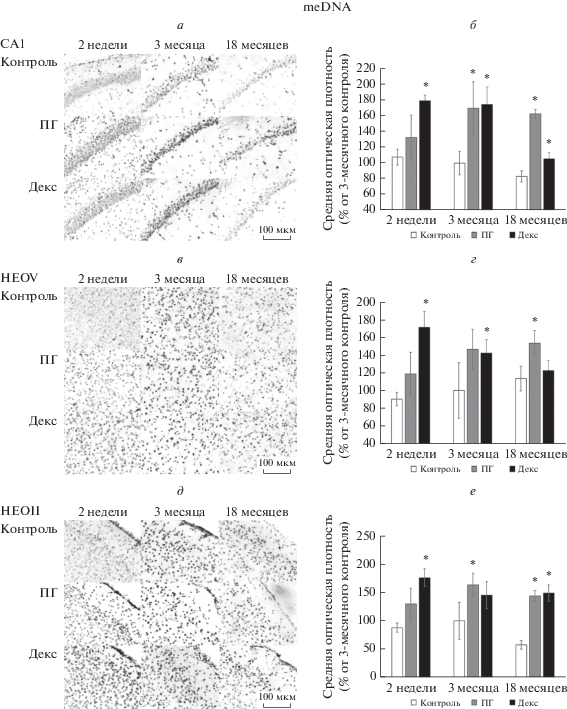
Метилирование ДНК (meDNA). На рис. 1 представлены репрезентативные микрофотографии иммунопозитивных к метилированной ДНК клеток в поле СА1 гиппокампа и неокортексе (II и V слои) контрольных крыс и крыс, подвергавшихся воздействию тяжелой гипобарической гипоксии или введению дексаметазона на 14–16-е сут пренатального онтогенеза. На графиках представлены изменения содержания meDNA в клетках СА1 области гиппокампа и во II и V слоях неокортекса 14-суточных, 3-месячных и 18-месячных крыс после воздействия тяжелой гипобарической гипоксии или введения дексаметазона на 14–16-е сут пренатального онтогенеза по отношению к контролю (за 100% приняли оптическую плотность клеток, иммунопозитивных к meDNA в срезах мозга 3-месячных контрольных животных). У 18-месячных контрольных животных снижается степень метилирования ДНК в области СА1 гиппокампа и во II слое неокортекса по сравнению с 3-месячными (на 17 и 42% соответственно). Увеличение уровня метилирования ДНК при предъявлении гипобарической гипоксии или введении дексаметазона на 14–16-е сут пренатального онтогенеза наблюдаются во всех исследованных структурах мозга крыс вне зависимости от их возраста.
Рис. 1.
Микрофотографии (40×) (а, в, д) СА1 поля гиппокампа (а), 5-го слоя неокортекса (НЕОV) (в) и 2-го слоя неокортекса (НЕОII) (д) 2-недельных, 3-месячных и 18-месячных контрольных крыс (Контроль) и крыс, переживших тяжелую гипобарическую гипоксию (пренатальная гипоксия, ПГ) либо инъекции дексаметазона (Декс) на 14–16-е сут пренатального развития. Иммуногистохимическая реакция на метилирование ДНК (meDNA). Маркер, 100 мкм. Средняя оптическая плотность иммунопозитивных к meDNA клеток (б, г, е) СА1 поля гиппокампа (б), 5го слоя неокортекса (НЕОV) (г) и 2го слоя неокортекса (НЕОII) (е) 2-недельных, 3-месячных и 18-месячных контрольных крыс (Контроль) и крыс, переживших тяжелую гипобарическую гипоксию (пренатальная гипоксия, ПГ) либо инъекции дексаметазона (Декс) на 14–16-е сут пренатального развития. По оси абсцисс – наименование экспериментальных групп; по оси ординат – средняя оптическая плотность иммунопозитивных клеток и временные точки, выраженная в % от 3-месячного контроля. * – различия с контролем статистически достоверны, р ≤ 0.05.
Метилирование Н3 по лизину 4 (meH3K4). На рис. 2 представлены репрезентативные микрофотографии иммунопозитивных к метилированному гистону Н3 по лизина 4 клеток в поле СА1 гиппокампа и неокортексе (II и V слои) контрольных крыс и крыс, подвергавшихся воздействию тяжелой гипобарической гипоксии или введению дексаметазона на 14–16-е сут пренатального онтогенеза. На графиках представлены изменения содержания meН3К4 в клетках СА1 области гиппокампа и во II и V слоях неокортекса 14-суточных, 3-месячных и 18-месячных крыс после воздействия тяжелой гипобарической гипоксии или введения дексаметазона на 14–16-е сут пренатального онтогенеза по отношению к контролю (за 100% приняли оптическую плотность клеток, иммунопозитивных к meH3K4 в срезах мозга 3‑месячных контрольных животных).
Рис. 2.
Микрофотографии (40×) (а, в, д) СА1 поля гиппокампа (а), 5-го слоя неокортекса (НЕОV) (в) и 2-го слоя неокортекса (НЕОII) (д) 2-недельных, 3-месячных и 18-месячных контрольных крыс (Контроль) и крыс, переживших тяжелую гипобарическую гипоксию (пренатальная гипоксия, ПГ) либо инъекции дексаметазона (Декс) на 14–16-е сут пренатального развития. Иммуногистохимическая реакция на гистон Н3, метилированного по Lys4 (meН3К4). Маркер, 100 мкм. Средняя оптическая плотность иммунопозитивных к meН3К4 клеток (б, г, е) СА1 поля гиппокампа (б), 5-го слоя неокортекса (НЕОV) (г) и 2го слоя неокортекса (НЕОII) (е) 2-недельных, 3-месячных и 18-месячных контрольных крыс (Контроль) и крыс, переживших тяжелую гипобарическую гипоксию (пренатальная гипоксия, ПГ) либо инъекции дексаметазона (Декс) на 14–16-е сут пренатального развития. По оси абсцисс – наименование экспериментальных групп и временные точки; по оси ординат – средняя оптическая плотность иммунопозитивных клеток, выраженная в % от 3-месячного контроля. * – различия с контролем статистически достоверны, р ≤ 0.05.
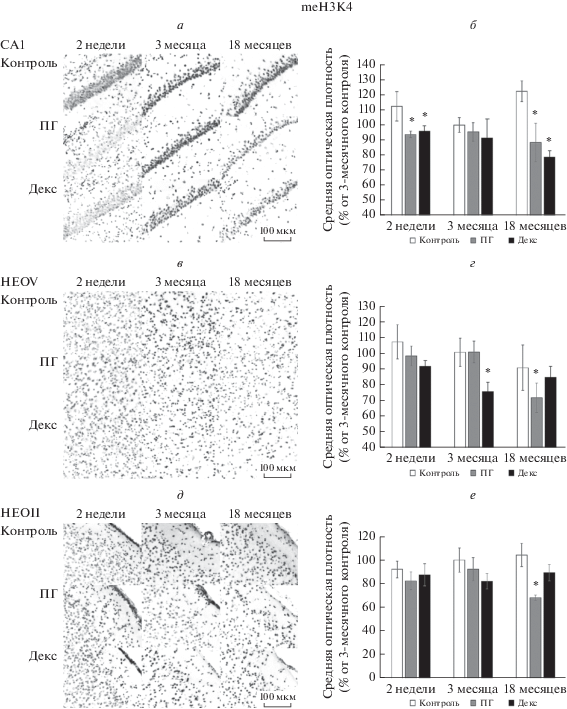
Уровень метилирования гистона Н3 по лизину 4 не зависел от возраста контрольных животных. Пренатальная гипоксия, так же, как и введение дексаметазона, приводила к снижению средней оптической плотности иммунопозитивных к meH3K4 клеток в области СА1 гиппокампа 14-суточных (до 83 и 85% соответственно, по отношению к своему контролю) и 18-месячных (до 72 и 64% соответственно) крыс. У взрослых 3-месячных экспериментальных крыс количество meH3K4 не отличалось от контроля.
В неокортексе наибольшие изменения уровня meH3K4 вследствие пренатальной гипоксии наблюдались у старых полуторогодовалых животных (снижение до 31 и 34% по сравнению с контролем в V и II слоях соответственно). Введение дексаметазона не приводило к снижению уровня meH3K4 в неокортексе крыс ювенильных и старых экспериментальных крыс. Однако, наблюдалось достоверное снижение средней оптической плотности иммунопозитивных к meH3K4 клеток в V слое неокортекса после введения дексаметазона на 14–16-е сут пренатального онтогенеза.
Метилирование Н3 по лизину 9 (meН3К9). На рис. 3 представлены репрезентативные микрофотографии иммунопозитивных к метилированному гистону Н3 по лизина 9 клеток в поле СА1 гиппокампа и неокортексе (II и V слои) контрольных крыс и крыс, подвергавшихся воздействию тяжелой гипобарической гипоксии или введению дексаметазона на 14–16-е сут пренатального онтогенеза. На графиках представлены изменения содержания meН3К9 в клетках СА1 области гиппокампа и во II и V слоях неокортекса 14-суточных, 3-месячных и 18-месячных крыс после воздействия тяжелой гипобарической гипоксии или введения дексаметазона на 14–16-е сут пренатального онтогенеза по отношению к контролю (за 100% приняли оптическую плотность клеток, иммунопозитивных к meH3K9 в срезах мозга 3‑месячных контрольных животных).
Рис. 3.
Микрофотографии (40×) (а, в, д) СА1 поля гиппокампа (а), 5-го слоя неокортекса (НЕОV) (в) и 2-го слоя неокортекса (НЕОII) (д) 2-недельных, 3-месячных и 18-месячных контрольных крыс (Контроль) и крыс, переживших тяжелую гипобарическую гипоксию (пренатальная гипоксия, ПГ) либо инъекции дексаметазона (Декс) на 14–16-е сут пренатального развития. Иммуногистохимическая реакция на гистон Н3, метилированный по Lys9 (meН3К9). Маркер, 100 мкм. Средняя оптическая плотность иммунопозитивных к meН3К9 клеток (б, г, е) СА1 поля гиппокампа (б), 5-го слоя неокортекса (НЕОV) (г) и 2-го слоя неокортекса (НЕОII) (е) 2-недельных, 3-месячных и 18-месячных контрольных крыс (Контроль) и крыс, переживших тяжелую гипобарическую гипоксию (пренатальная гипоксия, ПГ) либо инъекции дексаметазона (Декс) на 14–16-е сут пренатального развития. По оси абсцисс – наименование экспериментальных групп и временные точки; по оси ординат – средняя оптическая плотность иммунопозитивных клеток, выраженная в % от 3-месячного контроля. * – различия с контролем статистически достоверны, р ≤ 0.05. # – различия с 3-месячным контролем статистически достоверны, р ≤ 0.05.
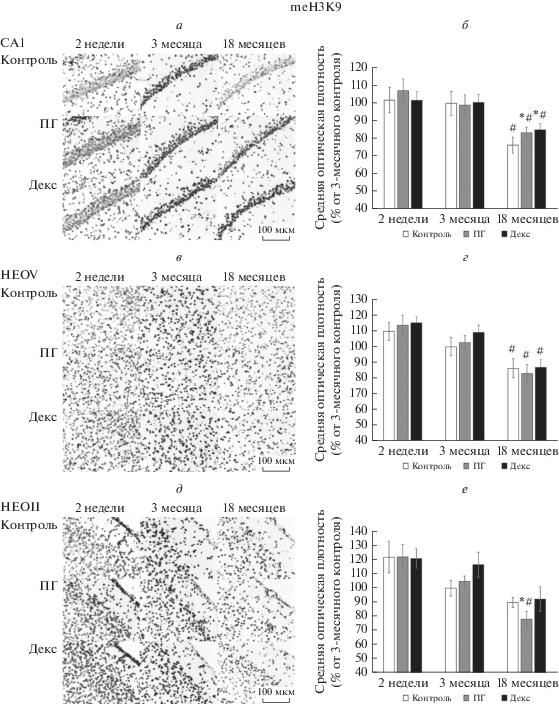
У контрольных животных обнаружено достоверное снижение средней оптической плотности иммунопозитивных к meH3K9 клеток в области СА1 гиппокампа (до 75%) и пятом слое неокортекса (до 85%) стареющих (18-месячных) крыс по сравнению с взрослыми (3-месячными).
У ювенильных 2-недельных и взрослых 3-месячных животных экспериментальной группы средняя оптическая плотность клеток, иммунопозитивных к meH3K9, не отличался от контроля. То есть, пренатальное воздействие гипобарической гипоксии или введение дексаметазона в большей степени затрагивает активационные сайты гистонов Н3. Уровень meH3K9 в области СА1 гиппокампа старых экспериментальных животных снижался в меньшей степени, чем у контрольных.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Основной целью проведенных исследований было изучения характера вовлечения эпигенетических механизмов в формирование показанных нами ранее [42, 43] нарушений функциональной активности мозга на молекулярно-клеточном и поведенческом уровнях вследствие воздействия гипоксии или введения дексаметазона в течение третьей недели пренатального онтогенеза крыс. Особое внимание в настоящей работе уделяется сопоставлению результатов, полученных на животных разных возрастных групп, включая период геронтогенеза (период старения).
В нашей работе в период постнатального онтогенеза у контрольных животных были обнаружены возрастные модификации степени метилирования ДНК и гистона Н3 по лизину 9 (но не по лизину 4), а именно снижение уровня meDNA и meH3K9 у старых 18-месячных крыс по сравнению с 3-месячными. Возрастные изменения уровня meDNA и meH3K9, модификаций, способствующих торможению экспрессии генов, могут вовлекаться в обнаруженные нами нарушения способности к обучению крыс в лабиринте Морриса в процессе естественного старения [44].
Воздействие стресса в пренатальный период является примером “программирования развития мозга на ранней стадии” – явления, которое обусловлено действием стрессовых фактор в критические периоды развития, вызывающих стойкие изменения в физиологических функциях и поведении в последующей жизни [45, 46]. Перепрограммирование эпигенома в течение раннего развития организма является очень сложным и хорошо организованный процессом, включающим взаимодействие молекулярных изменений ДНК и гистоновых белков [47, 48], определяющим баланс экспрессии генов, участвующих в поддержании пластичности клеток при адаптации к изменяющимся условиям среды. Кроме того, изменение баланса метилирования/деметилирования ДНК, модификации гистонов в период эмбрионального развития играет существенную роль в регуляции дифференцировки клеток-предшественников между нейрогенезом и астроглиогенезом [49, 50].
Пренатальная гипоксия на 14–16-е сут пренатального онтогенеза приводит к увеличению уровня meDNA во всех исследованных структурах мозга вне зависимости от возраста крыс. Введение в те же сроки беременности дексаметазона так же повышает степень метилирования ДНК, причем в наибольшей степени это проявляется у ювенильных животных. Метилирование ДНК – фундаментальный эпигенетический механизм контроля экспрессии генов у млекопитающих, связанный, в основном, с репрессией транскрипции, который заключается в присоединении метильной группы к углероду в 5-м положении молекулы цитозина с образованием 5-метилцитозина. Усиление метилирования ДНК подавляет экспрессию гипоксия-индуцибельного фактора HIF-1α [51, 52], который является основным медиатором транскрипционных ответов на гипоксию, способствующим формированию гипоксически-индуцируемого фенотипа [53]. В то же время показана обратная зависимость между базовым уровнем HIF-1α в нейронах неокортекса и генетически запрограммированной толерантностью организма к гипоксии [54]. Увеличение степени метилирования ДНК согласуются со снижением базовой экспрессии HIF-1α в неокортексе этих животных [44].
При анализе результатов по модификации метилирования гистона Н3 по лизину 4 и 9 оказалось, что основные изменения проявляются, в основном, в отдаленные сроки после воздействия, а именно в структурах мозга 18-месячных старых крыс. При этом увеличивается уровень meH3K4, способствующего активации транскрипции, и снижается meH3K9, связанный, как и meDNA, с репрессией генов.
Обнаруженное уменьшение степени метилирования гистона Н3 по лизину 4 в исследованных структурах мозга старых крыс, подвергавшихся действию тяжелой гипобарической гипоксии на 14–16-е сут пренатального онтогенеза, согласуются с показанными нами ранее [37] модификациями ацетилирования гистона Н3 по лизину 24 в гиппокампе и неокортексе взрослых крыс после таких же воздействий, acH3K24 также способствует активации генов и коррелирует с показанными нами ранее нарушениями экспрессии глюко- и минералокортикоидных рецепторов, регулирующих экспрессию генов-мишеней, в том числе транскрипционных факторов HIF-1α, нейрогормонов кортиколиберина и вазопрессина, антиоксидантов [55]. Отсроченные изменения степени метилирования гистона Н3 могут обуславливать усиливающиеся с возрастом нарушения функциональной активности мозга, приводящие к ослаблению памяти и способности к обучению животных, подвергавшихся действию тяжелой гипобарической гипоксии.
Можно предположить, что модификации эпигенетического статуса лежат в основе ранее показанных нами длительных изменений поведения и способности к обучению, обусловленных изменениями активности основных внутриклеточных регуляторных систем (кальциевой и фосфоинозитидной), нарушений глутаматергической сигнальной трансдукции, соотношения про- и антиоксидантных систем в мозге крыс, подвергавшихся тяжелой гипобарической гипоксии в период пренатального развития [26, 42, 43]. Профиль модификаций метилирования гистонов и ДНК, вызванных пренатальным введением повышенных доз глюкокортикоидных гормонов (дексаметазона) отличается по степени выраженности от действия пренатальной гипоксии, что может лежать в основе неоднозначных проявлений этих воздействий на молекулярно-клеточном и поведенческом уровне, показанными нами ранее [26, 43].
ЗАКЛЮЧЕНИЕ
Полученные данные могут иметь важное значение для клинической практики, способствуя раскрытию механизмов нарушений функциональной активности мозга, связанных с гипоксией и другими неблагоприятными воздействиями в раннем онтогенезе. Следует отметить, что представленные сведения о механизмах повреждения мозга после перенесенных повреждающих воздействий в период пренатального онтогенеза создают теоретическую основу для перспективного поиска медикаментозных и немедикаментозных способов коррекции возникающих в более поздние периоды жизни неврологических и психических болезней. Кроме того, полученные данные могут иметь важное значение для клинической практики, способствуя выявлению механизмов нарушения формирования когнитивных расстройств, связанных с применением глюкокортикоидной терапии в период беременности, в частности, как предотвращение ранних родов.
Список литературы
van Os J., Selten J.P. // Br. J. Psychiatry. 1998. V. 172. P. 324–326.
Van den Bergh B.R., Marcoen A. // Child Dev. 2004. V. 75. № 4. P. 1085–1097.
Beversdorf D.Q., Manning S.E., Hillier A, Anderson S.L., Nordgren R.E., Walters S.E., Nagaraja H.N., Cooley W.C., Gaelic S.E., Bauman M.L. // J. Autism. Dev. Disord. 2005. V. 35. № 4. P. 471–478.
Khashan A.S., Abel K.M., McNamee R, Pedersen M.G., Webb R.T., Baker P.N., Kenny L.C., Mortensen P.B. // Arch. Gen. Psychiatry. 2008. V. 65. № 2. P. 146–52.
Van den Bergh B.R., Van Calster B., Smits T., Van Huffel S., Lagae L. // Neuropsychopharmacology. 2008. V. 33. № 3. P. 536–545.
Grizenko N., Fortier M.E., Zadorozny C., Thakur G., Schmitz N., Duval R., Joober R. // J. Can. Acad. Child. Adolesc. Psychiatry. 2012. V. 21. № 1. P. 9–15.
Graignic-Philippe R., Dayan J., Chokron S., Jacquet A.Y., Tordjman S. // Neurosci. Biobehav. Rev. 2014. V. 43. P. 137–162.
de Boo H.A., Harding J.E. // Aust. N. Z. J. Obstet. Gynaecol. 2006. V. 46. P. 4–14.
Warner M.J., Ozanne S.E. // Biochem. J. 2010. V. 427. P. 333–347.
Langley-Evans S.C., McMullen S. // Med. Princ. Pract. 2010. V. 19. P. 87–98.
Barker D.J., Osmond C., Kajantie E., Eriksson J.G. // Ann. Hum. Biol. 2009. V. 36. P. 445–458.
Harris A., Seckl J. // Horm. Behav. 2011. V. 59. P. 279–289.
Gluckman P.D., Hanson M.A., Cooper C., Thornburg K.L. // N. Engl. J. Med. 2008. V. 359. P. 61–73.
Gluckman P.D., Hanson M.A. // Science. 2004. V. 305. P. 1733–1736.
Gonzalez-Rodriguez P.J., Xiong F., Li Y., Zhou J., Zhang L. // Neurobiol. Dis. 2014. V. № 65. P. 172–179.
Li Y., Gonzalez P., Zhang L. // Prog. Neurobiol. 2012. V. 98. P. 145–165.
Wilcoxon J.S., Redei E.E. // Horm. Behav. 2007. V. 51. № 3. P. 321–327.
Salomon S., Bejar C., Schorer-Apelbaum D., Weinstock M. // J. Neuroendocrinol. 2011. V. 23. № 2. P. 118–128.
Reynolds R.M. // Clin. Obstet. Gynecol. 2013. V. 56. № 3. P. 602–609.
Crowther C.A., Doyle L.W., Haslam R.R., Hiller J.E., Harding J.E., Robinson J.S. // N. Engl. J. Med. 2007. V. 357. P. 1179–1189.
French N. P., Hagan R., Evans S. F., Mullan A., Newnham J.P. // Am. J. Obstet. Gynecol. 2004. V. 190. P. 588–595.
Braun T., Challis J.R., Newnham J.P., Sloboda D.M. // Endocr. Rev. 2013. V. 34. P. 885–916.
Moors M., Bose R., Johansson-Haque K., Edoff K., Okret S., Ceccatelli S. // Toxicol. Sci. 2012. V. 125. P. 488–495.
Sorrells S.F., Munhoz C.D., Manley N.C., Yen S., Sapolsky R.M. // Neuroendocrinology. 2014. V. 100. № 2. P. 129–140.
Stutchfield P.R., Whitaker R., Gliddon A.E., Hobson L., Kotecha S., Doull I.J. // Arch. Dis. Child. Fetal. Neonatal. Ed. 2013. V. 98. № 3. P. 195–200.
Ватаева Л.А., Тюлькова Е.И., Алёхин А.Н., Стратилов В.А. // Журн. эволюционной физиологии и биохимии. Т. 54. № 6. 2018. С. 404–410.
Bhutani N., Burns D.M., Blau H.M. // Cell. 2011. V. 146. P. 866–872.
Suganuma T., Workman J.L. // Annu. Rev. Biochem. 2011. P. 80. P. 473–499.
Egger G., Liang G., Aparicio A., Jones P.A. // Nature. 2004. V. 429. P. 457–463.
Chen M., Zhang L. // Drug. Discov. Today. 2011. V. 16. P. 1007–1018.
Concepcion K.R., Zhang L. // Drug. Discov. Today. 2018. V. 23. № 10. P. 1718–1732.
Oakley R., Busada T., Cidlowski J. // FASEB J. 2018. V. 32. № 10.
Jobe E.M., Zhao X. // Brain. Plasticity. 2017. V. 3. № 1. P. 5–26.
Freitag M., Selker E.U. // Curr. Opin. Genet. Dev. 2005. V. 15. P. 191–199.
Weber M., Hellmann I., Stadler M.B., Ramos L., Pääbo S., Rebhan M., Schübeler D. // Nat. Genet. 2007. V. 39. P. 457–466.
Perez-Perri J.I., Acevedo J.M., Wappner P. // Int. J. Mol. Sci. 2011. V. 12. P. 4705–4721.
Тюлькова Е.И., Ветровой О.В., Сариева К.В., Ватаева Л.А., Глущенко Т.С. // Нейрохимия. 2017. Т. 34. № 4. С. 310–316.
Тюлькова Е.И., Ватаева Л.А., Ветровой О.В., Сариева К.В., Стратилов В.А. // Цитология. 2019. Т. 61. № 2. С. 98–105.
Строев С.А., Тюлькова Е.И., Ватаева Л.А., Самойлов М.О., Пельто–Хуикко М.Т. // Нейрохимия. 2011. Т. 28. С. 226–231.
Slotkin T.A., Kreider M.L., Tate C.A., Seidler F.J. // Neuropsychopharmacology. 2006. 31. № 5. P. 904–911.
Vilaca Junior P.E.A., Teixeira A.A.C., Wanderley-Teixeira V., Moraes E.F., Araujo A.C.C., Maia C.S. // Int. J. Morphol. 2008. V. 26. № 3. P. 523–527.
Тюлькова Е.И., Ватаева Л.А., Самойлов М.О., Отеллин В.А. // Журн. акушерства и женских болезней. 2010. Т. 59(4). С. 99–110.
Тюлькова Е.И., Ватаева Л.А., Ветровой О.В., Романовский Д.Ю. // Журн. эволюц. биохим. и физиол. им. И.М. Сеченова. 2015. Т. 51. № 2. С. 115–121.
Тюлькова Е.И., Ватаева Л.А., Ветровой О.В. // Детская медицина Северо-Запада. 2018. Т. 7. № 1. С. 319–320.
Barker D.J. // Mol. Med. Today. 1995. V. 1. № 9. P. 418–423.
Nyirenda M.J., Seckl J.R. // Int. J. Mol. Med. 1998. V. 2 № 5. P. 607–614.
Dasgupta C., Chen M., Zhang H., Yang S., Zhang L. // Hypertension. 2012. V. 60. P. 697–704.
Liu X., Wang C., Liu W., Li J., Li C., Kou X., Chen J., Zhao Y., Gao H., Wang H., Zhang Y., Gao Y., Gao S. // Nature. 2016. V. 537. P. 558–562.
Sauvageot C.M., Stiles C.D. // Curr. Opin. Neurobiol. 2002. V. 12. P. 244–249.
Takizawa T., Nakashima K., Namihira M., Ochiai W., Uemura A., Yanagisawa M., Fujita N., Nakao M., Taga T. // Dev. Cell. 2001. V. 1. P. 749–758.
Koslowski M., Luxemburger U., Tureci O., Sahin U. // Oncogene. 2011. V. 30. P. 876–882.
Walczak-Drzewiecka A., Ratajewski M., Pulaski L., Dastych J. // Biochem. Biophys. Res. Commun. 2010. V. 391. P. 1028–1032.
Watson J.A., Watson C.J., McCann A., Baugh J. // Epigenetics. 2010. V. 5. № 1. P. 293–296.
Кирова Ю.И. // Патол. физиология и эксперим. терапия. 2012. Т. 56. № 3. С. 51–55.
Kodama T., Shimizu N., Yoshikawa N., Makino Y., Ouchida R., Okamoto K., Hisada T., Nakamura H., Morimoto C., Tanaka H. // J. Biol. Chem. 2003. V. 278. № 35. P. 33384–33391.
Дополнительные материалы отсутствуют.