

Нейрохимия, 2020, T. 37, № 3, стр. 220-227
Превентивная умеренная гипоксия повышает содержание кортикостероидных рецепторов в мозге крыс в экспериментальной модели депрессии
К. А. Баранова
ФГБУН Институт физиологии им. И.П. Павлова РАН
Санкт-Петербург, Россия
Поступила в редакцию 11.10.2019
После доработки 18.11.2019
Принята к публикации 06.12.2019
Аннотация
Вовлечение кортикостероидных рецепторов мозга в антидепрессивные эффекты умеренной гипобарической гипоксии изучали иммуногистохимическим методом в модели депрессии (парадигма “выученной беспомощности”) на крысах. Показано, что формирование депрессивноподобного состояния сопровождается существенной недостаточностью глюкокортикоидных рецепторов мозга, наиболее выраженной в зубчатой извилине гиппокампа, а также значительным сдвигом условного баланса глюко- и минералокортикоидных рецепторов в сторону преобладания последних. Применение гипоксического прекондиционирования перед стрессированием в парадигме “выученной беспомощности”, предотвращавшее развитие экспериментальной депрессии, нивелировало рецепторную недостаточность в гипоталамусе к 10-му постстрессорному дню, а в неокортексе уже в первые сутки наблюдения. В гиппокампе прекондиционированных животных адаптация к стрессу сопровождалась резким и устойчивым повышением иммунореактивности к глюкокортикоидным рецепторам, при этом в области СА1 отмечалось условное равновесие двух типов рецепторов, в то время как в зубчатой извилине – значительный перевес в пользу рецепторов глюкокортикоидов. Можно предположить, что недостаточность и дисбаланс кортикостероидных рецепторов гиппокампа являются одним из важных молекулярных механизмов дисфункции системы глюкокортикоидной регуляции при стресс-индуцированных психопатологиях, в то время как активация экспрессии рецепторов после стресса вносит вклад в проявление антидепрессивных эффектов умеренной гипоксии. Полученные данные расширяют современные представления об эндокринных механизмах формирования и предотвращения депрессивных патологий и повышают трансляционный потенциал умеренной гипоксии в качестве немедикаментозного способа их коррекции.
Основной нейроэндокринной системой, ответственной за поддержание гомеостаза и адаптацию организма к стрессу, считается гипоталамо-гипофизарно-адренокортикальная система (ГАС). Активация ГАС приводит к выбросу надпочечниками глюкокортикоидных гормонов, которые связываются с клеточными рецепторами кортикостероидов, являющимися, в свою очередь, транскрипционными факторами, после чего рецепторы проникают в ядро клетки, где регулируют множество генов. Взаимодействие циркулирующих в крови глюкокортикоидов с клеточными кортикостероидными рецепторами нейроэндокринных ядер гипоталамуса, а также амигдалы, гиппокампа и коры мозга через изменения экспрессии генов-мишеней обеспечивает адекватный эндокринный и поведенческий ответ организма на стресс и играет ключевую роль в саморегуляции ГАС по принципу отрицательной обратной связи, в частности, влияя на гены нейрогормонов-регуляторов ГАС кортиколиберина и вазопрессина [1, 2].
Развитие стресс-индуцированных психопатологий характеризуется нарушениями в активации и регуляции ГАС. При формировании такого распространенного в сегодняшнем мире расстройства как депрессия наблюдается базальная гиперактивность ГАС в сочетании с глюкокортикоидной резистентностью. Нарушения функционирования ГАС отмечаются как при тяжелой депрессии, которая характеризуется длительным снижением настроения, изменениями нейровегетативных и когнитивных функций, апатией, склонностью к суициду, так и в случае моделирования депрессии на животных. В обоих случаях нарушение торможения ГАС, приводящее к ее гиперфункции, может объясняться снижением чувствительности к изменению уровня стероидов крови посредством внутриклеточных глюко- (GR) и минералокортикоидных рецепторов (MR) мозга. Предполагается, что MR обеспечивают тоническое ингибирование ГАС в условиях низкого уровня циркулирующих кортикостероидов, а GR – в условиях стрессорного подъема их уровня. Таким образом, поддержание количества этих рецепторов имеет решающее значение для обеспечения работы механизмов обратной связи. Кортикостероидные рецепторы мозга задействованы в регуляции активности ГАС и адаптации, процессах гибели/выживания нейронов мозга, обучении, памяти, формировании и предотвращении тревожно-депрессивных расстройств [2–4].
В качестве протективного воздействия, предотвращающего формирование депрессивной патологии в моделях на животных, удобно использовать феномен гипоксического прекондиционирования. Гипоксическое прекондиционирование – воздействие краткими эпизодами умеренной гипоксии, приводящее к повышению устойчивости мозга и оказывающее выраженный антидепрессивный эффект, предотвращая формирование депрессивных состояний, вызываемых тяжелыми формами психоэмоционального стресса [5, 6].
Стресспротективное действие умеренной гипоксии, вероятно, включает активацию некоторых универсальных адаптивных механизмов, среди которых ведущая роль принадлежит основной стресс системе организма – ГАС, и можно предположить, что ключевым моментом является коррекция нарушений в ее функционировании, в частности, на уровне кортикостероидных рецепторов мозга. Проверка этого предположения представляет не только теоретический, но и практический интерес, поскольку расшифровка патологических и адаптивных нейроэндокринных механизмов может способствовать разработке и внедрению новых способов повышения устойчивости мозга к стрессам и лечения постстрессорных патологий.
МАТЕРИАЛЫ И МЕТОДЫ
Исследование выполнено на 72 взрослых самцах крыс линии Вистар массой около 230 г из ЦКП “Биоколлекция ИФ РАН”. При проведении экспериментов соблюдались требования Директивы 2010/63/EU Европейского Парламента и Совета ЕС по охране животных, используемых в научных целях. Крыс разделили на контрольную (“Контроль”, n = 6) и 2 экспериментальные группы по 18 животных: “ВБ” – крысы, перенесшие стресс, в классической экспериментальной модели эндогенной депрессии – парадигме “выученная беспомощность”; “гпВБ” – животные, которые перед аверсивным стрессированием подвергались прекондиционированию умеренной гипоксией.
Для выработки депрессивноподобного состояния “выученной беспомощности” (ВБ) крыс подвергали неизбегаемому стрессу – стимулировали 60 асинхронными ударами электрическим током (1 мА, 60 Гц, 15 с) в небольшой клетке в течение часа [7]. В результате у животных значительно снижалась двигательная и исследовательская активность, повышался уровень тревожности, развивалась агедония, устойчиво возрастал уровень кортикостерона в крови и нарушалось глюкокортикоидное торможение гипоталамо-гипофизарно-адренокортикальной системы (ГАС) по механизму отрицательной обратной связи [5, 6].
Гипоксическое прекондиционирование осуществляли в декомпрессионной барокамере проточного типа при давлении 360 мм рт. ст. Животных подвергали 3 сеансам умеренной гипобарической гипоксии (10% O2) по 2 ч с интервалом 24 ч [8], последний сеанс проводили за 24 ч до патогенного стресса в модели ВБ. В этих условиях прекондиционированные крысы успешно адаптировались и экспериментальное депрессивное состояние у них не развивалось, поведенческие и гормональные показатели группы “гпВБ” не отличались от контрольных [5, 6]. Интактных животных из контрольной группы и животных из группы “ВБ” перед стрессированием трижды помещали в барокамеру на 2 часа, не создавая гипоксии.
Спустя 1, 5 и 10 суток после стрессирования (ранний, промежуточный и отсроченный постстрессовый период, соответственно) по 6 крыс из каждой экспериментальной группы декапитировали, затем, после извлечения и фиксации мозга, стандартных процедур обезвоживания и заливки в парафиновую среду, изготавливали срезы во фронтальной плоскости толщиной 7 мкм около –2.8 от брегмы для гиппокампа и неокортекса, и –1.8 для гипоталамуса. Для иммуногистохимической оценки содержания глюкокортикоидных (GR) и минералокортикоидных (MR) рецепторов в нейронах срезы инкубировали с поликлональными антителами к GR или MR (Santa Cruz, USA) в разведении 1 : 100, далее обрабатывали с использованием наборов для детекции фирмы Vector Labs, USA, и проводили оценку иммунореактивности на основании компьютерного анализа микроизображений паравентрикулярного ядра гипоталамуса (крупно- и мелкоклеточной частей), поля СА1 и зубчатой извилины гиппокампа, I–II и V слоев неокортекса – областей мозга, играющих ключевую роль в регуляции стрессорных реакций и обеспечении адаптивного ответа. Используя программу Мастер Морфология (“ВидеоТест”, СПб), в поле зрения производили подсчет числа иммунопозитивных клеток, с их автоматическим разделением на слабо- и интенсивно-иммунопозитивные по величине оптической плотности в условных единицах по сравнению с фоном. Для каждого животного анализировали 2–3 гистопрепарата, использовали усредненные значения по каждой области мозга у данного животного. Эти данные обрабатывали, вычисляя среднюю арифметическую величину и стандартную ошибку среднего в исследуемых подгруппах животных (n = 6 для каждой точки), сравнивали средние значения выборок. Статистическую обработку проводили средствами однофакторного дисперсионного анализа ANOVA (Statistica 7.0), с апостериорным сравнением методом Фишера, если распределение выборки являлось нормальным, а дисперсии групп равны. В противном случае использовали непараметрический тест ANOVA Kruskal–Wallis. Различия между группами считали достоверными и при p ≤ 0.05. Результаты по экспериментальным подгруппам выражены в процентах от среднего значения контрольной группы, принятого за 100%, и представлены в виде средней арифметической величины количества иммунопозитивных клеток, выраженной в процентах от контроля, ± стандартная ошибка среднего, выраженная в %.
РЕЗУЛЬТАТЫ
У контрольных животных выявлен достаточно высокий уровень иммунореактивности к GR во всех исследованных областях мозга, с наибольшими значениями интенсивности экспрессии в гиппокампе.
В паравентрикулярном ядре (ПВЯ) гипоталамуса в течение формирования экспериментальной депрессии общее число иммунопозитивных к GR нейронов достоверно не изменялось (рис. 1а и 1в, черные столбцы). Тем не менее, количество клеток, интенсивно иммунореактивных к GR, выражено (1/2–1/4 от контроля) и устойчиво (до 10 дней) снижалось как в мелко- (F(3.20) = 4.25, p = 0.018), так и в крупноклеточной (F(3.22) = = 3.481, p = 0.033) части ПВЯ (рис. 1б, 1г, 1е). В гипоталамусе крыс из группы “гпВБ” 3 сеанса умеренной гипобарической гипоксии частично нивелировали постстрессорное ослабление экспрессии GR и вызывали ее ап-регуляцию в отсроченный постстрессовый период. Первоначальная редукция числа интенсивно GR-позитивных нейронов в ранний период сменялась постепенным восстановлением до контрольных (в крупноклеточной части ПВЯ) и даже превосходящих контрольные (в мелкоклеточной) значений к 10 суткам (рис. 1, белые столбцы).
Рис. 1.
Модификации иммунореактивности к GR в крупноклеточной (а, б) и мелкоклеточной (в, г) частях ПВЯ гипоталамуса крыс в процессе формирования и коррекции экспериментальной депрессии. д, е – микрофотографии (20×), иллюстрирующие GR-иммунореактивность в ПВЯ контрольных (д) и ВБ (е) животных через 1 сут после стресса. По оси абсцисс – дни после неизбегаемого стресса; по оси ординат – (а, в) общее число иммунореактивных клеток или (б, г) количество интенсивно иммунопозитивных к GR нейронов в % от значений контрольной группы, принятой за 100%. Серые столбцы – интактный контроль; черные столбцы – крысы с экспериментальной депрессией после неизбегаемого ВБ стресса; белые столбцы – прекондиционированные перед стрессированием гпВБ животные, у которых патология не развивалась. * Различия достоверны по отношению к контролю, # достоверные различия относительно группы “ВБ”, р ≤ 0.05.

В экстрагипоталамических областях мозга непрекондиционированных животных воздействие неизбегаемого стресса приводило к заметной редукции GR-иммунореактивности (рис. 2). В поле СА1 гиппокампа через 24 ч оставалось около трети от числа иммунореактивных нейронов с интенсивной окраской по сравнению с контролем, к 10-му постстрессорному дню – около половины (рис. 2а). Наиболее резкие и выраженные изменения экспрессии GR после стрессирования были отмечены в зубчатой извилине (рис. 2б), где практически не выявлялось интенсивно иммунореактивных нейронов (F(3,21) = 4.346, p = 0.016), также в этой области, единственной среди исследованных, существенно и устойчиво понижалась не только интенсивность экспрессии, но и общее число иммунопозитивных клеток (до 18% от контроля). Снижение иммунореактивности наблюдалось и в неокортексе – количество интенсивно-окрашенных клеток здесь достигало минимума к 10 суткам (45% во II слое и 30% в V слое).
Рис. 2.
Постстрессорные изменения числа интенсивно GR-иммунореактивных клеток в мозге непрекондиционированных и прекондиционированных животных. а – область СА1 гиппокампа; б – зубчатая извилина; в – I–II слои неокортекса; г – V слой неокортекса. Обозначения как на рис. 1.

Прекондиционированные умеренной гипоксией животные демонстрировали значительную модификацию постстрессорного паттерна экспрессии GR. В гиппокампе крыс группы “гпВБ” число иммунореактивных клеток резко возрастало, максимально (из всех исследованных структур) – в зубчатой извилине, где увеличение иммунореактивности происходило в основном за счет значительного роста количества интенсивно иммунопозитивных нейронов (F(3,20) = 24.434, p = 0.00006), 8-кратного в 1 день и устойчивого на всем периоде наблюдения (рис. 2б). Пик роста имунореактивности в СА1 также приходился на ранние сроки, достигая 227% (рис. 2а). В неокортексе животных, перенесших трехкратное гипоксическое воздействие, иммунореактивность к GR после стресса не снижалась, как у непрекондиционированных, а оставалась неизменной относительно контрольного уровня (рис. 2г), либо незначительно возрастала на ранних сроках (рис. 2в).
Области неокортекса и гиппокампа контрольных животных демонстрировали умеренное содержание MR-иммунореактивных нейронов, в ПВЯ гипоталамуса MR-содержащие клетки практически не обнаруживались.
В гиппокампе крыс, стрессированных в парадигме ВБ, достоверных изменений в общем числе MR-содержащих нейронов (данные не представлены) и количестве интенсивно иммунопозитивных клеток в области СА1 и зубчатой извилине не обнаружено (рис. 3а и 3б). На раннем этапе формирования экспериментальной депрессии I–II слои неокортекса животных реагировали увеличением числа интенсивно иммунопозитивных к MR нейронов, затем выявлено постепенное снижение, и к 10 суткам наблюдалась значительная редукция их числа относительно контрольной группы (рис. 3в). Нейроны V слоя неокортекса в ответ на стресс продемонстрировали менее выраженные изменения со сходным временным паттерном (рис. 3г).
Рис. 3.
Влияние неизбегаемого стресса и гипоксического прекондиционирования на количество нейронов интенсивно иммунопозитивных к MR в области СА1 (а) и зубчатой извилине (б) гиппокампа, в I–II (в) и V (г) слоях неокортекса. По оси абсцисс – дни после стресса; по оси ординат – число интенсивно MR-иммунореактивных клеток в % от контроля, принятого за 100%. Обозначения как на рис. 1.
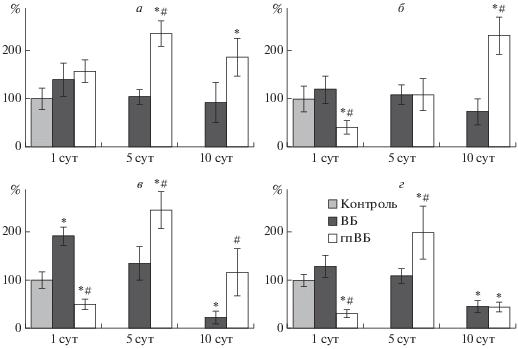
По суммарному количеству MR-иммунореактивных клеток у прекондиционированных перед стрессированием в парадигме ВБ животных достоверных отличий относительно непрекондиционированных или контрольных не наблюдалось. По числу интенсивно иммунореакционноспособных нейронов в группе “гпВБ” отмечено повышение в области СА1 гиппокампа на 5 день после стресса, а в зубчатой извилине на 10-й, в этот же период их число достоверно больше чем в группе “ВБ”. В верхних и нижних слоях неокортекса через 24 ч выявлено снижение количества интенсивно экспрессирующих MR нейронов, к 5 суткам снижение сменилось ростом (246% – I–II слои, 200% – V слой), и в отсроченный постстрессовый период во II слое сравнялось с контролем, а в V слое оказалось вновь ниже контрольного уровня (45%, p = = 0.027), как и в первые сутки (рис. 3в и 3г). Таким образом, интенсивность экспрессии МR нейронами неокортекса гпВБ крыс в первые сутки значительно ниже, а на пятые сутки наоборот достоверно выше, чем у ВБ животных.
ОБСУЖДЕНИЕ
В целом можно заключить, что во всех исследованных областях мозга крыс стресс ВБ вызывал редукцию числа интенсивно иммунопозитивных к глюкокортикоидным рецепторам клеток. Важно отметить, что к интенсивно иммуноокрашенным клеткам, прежде всего, относятся нейроны с ядерной локализацией белка, при том, что общее число GR-содержащих нейронов могло оставаться неизменным, как, например, в кортиколиберин-продуцирующих центрах ПВЯ гипоталамуса. В этом случае снижение интенсивности иммунореактивности может объясняться нарушением связывания глюкокортикоидов с рецепторами и уменьшением транслокации GR из цитоплазмы в ядро, а также усилением их взаимодействия с транскрипционными факторами, что, в свою очередь, негативно влияет на транскрипцию GR-зависимых генов, чем, вероятно, обусловлено ослабление отрицательной обратной связи ГАС, характерное для депрессивной патологии. Вместе с этим в мозге запускаются процессы образования провоспалительных цитокинов, которые препятствуют транслокации комплекса кортизол-глюкокортикоидный рецептор в ядро, а вновь образованные комплексы создают конкуренцию для связывания GR с ДНК [9, 10].
Изменения уровня экспрессии GR, как центрального элемента регуляции ГАС, рассматривались в качестве вероятной причины глюкокортикоидной резистентности при психических расстройствах многими исследователями. В фибробластах пациентов с тяжелыми формами депрессий достоверного снижения GR выявлено не было [11], отдельные постмортальные исследования нескольких областей мозга in situ гибридизацией и ПЦР позволили зафиксировать существенные изменения уровня тотальной мРНК GR лишь в III–VI слоях фронтальной коры [12–14]. Что не позволяет исключить радикального снижения экспрессии этого рецептора в определенных отделах мозга, нарушений на уровне трансляции белка при неизменном уровне тотальной мРНК, а также образования функционально различающихся изоформ этого белка за счет альтернативного сплайсинга мРНК и использования альтернативных стартов трансляции, в том числе изоформы GRβ, которая является доминантным ингибитором нормальной формы GRα [15–17]. Еще одним механизмом, вносящим вклад в формирование резистентности к глюкокортикоидам, могут быть посттрансляционные модификации, в частности, фосфорилирование N-концевого домена белка-рецептора, которое влияет не только на взаимодействия с компонентами транскрипционной машины и кофакторами, но, возможно, и с исследовательскими антителами [18]. В настоящее время не вызывает сомнения тот факт, что при депрессиях имеет место существенная недостаточность глюкокортикоидных рецепторов, предположительно, преимущественно функциональная [9].
Активация ГАС приводит к увеличению содержания глюкокортикоидов в высших отделах мозга, из которых наиболее чувствительным к стрессу, как известно, является гиппокамп, в котором плотность глюкокортикоидных рецепторов максимальна [19]. Интересно отметить, что единственной из исследованных в настоящей работе областей мозга, в которой, наряду с редукцией количества интенсивно иммунореактивных к GR клеток, было выявлено снижение общего числа GR-содержащих нейронов, являлась зубчатая извилина гиппокампа. Таким образом, мы можем сделать вывод о том, что в процессе формирования депрессивной патологии в этой структуре наблюдалось достоверное снижение содержания глюкокортикоидных рецепторов. Поскольку тормозное действие гормонов надпочечников на секрецию кортиколиберина осуществляется на уровне регуляторных областей головного мозга, и прежде всего именно гиппокампа, можно предположить, что обнаруженная редукция глюкокортикоидных рецепторов в зубчатой извилине может вносить существенный вклад в формирование глюкокортикоидной резистентности при моделировании депрессии.
Обратное влияние глюкокортикоидов на биосинтез нейрогормонов гипоталамусом опосредуется не только низкоаффинными глюкокортикоидными, но и намного более чувствительными минералокортикоидными рецепторами, которые экспрессируются преимущественно в нейронах гиппокампа и поддерживают как базовую, так и стрессиндуцированную активность ГАС [20, 21], стимулируя нейрональную активность в гиппокампе [22]. MR считаются главными для первичной, быстрой реакции на кортикостерон [23], предполагают, что их основная функция связана с адаптацией в условиях относительной нормы [24]. Это косвенно подтверждается продемонстрированным в настоящей работе отсутствием существенных изменений их уровня при формировании депрессивной патологии, но при этом условный баланс GR к MR оказывается значительно сдвинут в сторону MR (особенно в зубчатой извилине гиппокампа). Но, несмотря на преобладание MR при ВБ, они не способны связывать гормон в том диапазоне концентраций, который характерен для депрессивных состояний, и ключом к нарушениям в работе ГАС, очевидно, становится нехватка GR.
Считается, что повышенные уровни глюкокортикоидов способны индуцировать изменения в GR и MR гиппокампа, проявляющиеся в функциональной недостаточности, повышении соотношения MR : GR и даже редукции GR [2–4]. Соответственно, с течением времени ингибиторная активность гиппокампа ослабевает, что приводит к нарушению обратной связи и гиперсекреции кортизола. Данное исследование показало, что значительная недостаточность GR мозга выявляется с первых дней развития депрессивного состояния и не является следствием длительного воздействия высоких доз глюкокортикоидов. Скорее, можно предположить обратное – недостаточность GR при ВБ способствует гиперсекреции гормона.
Антидепрессивные эффекты гипоксического прекондиционирования сопровождались резким и устойчивым повышением GR-иммунореактивности в гиппокампе (особенно выраженном в зубчатой извилине) крыс, подвергнутых ВБ стрессу, и при этом наблюдалась их ядерная локализация. Очевидно, что в данном случае GR является активным транскрипционным фактором для генов-мишеней, имеющих в своем промоторе GRE последовательности. Так, прекондиционирование, вероятно, запускает синтез необходимых для включения обратной связи и связывания избыточного при ВБ гормона GR. Таким образом, предварительное воздействие умеренной гипоксией не только предотвращало редукцию GR в ответ на ВБ стресс, но и способствовало постстрессовой оверэкспрессии GR в образованиях мозга, вовлекаемых в нейроэндокринную регуляцию стрессорных реакций. Ранее показано, что длительное введение антидепрессантов, сходным образом может усилить механизм обратной отрицательной связи путем стимуляции экспрессии кортикостероидных рецепторов [25].
Интересно, что если проследить за изменением условного соотношения глюко- и минералокортикоидных рецепторов гиппокампа при развитии и предотвращении депрессивной патологии ВБ, то при ВБ за счет падения числа GR преобладающими оказываются MR, а в результате применения умеренной гипоксии в чувствительной к повреждениям области СА1 наступает условное GR/MR равновесие, в то время как в зубчатой извилине отмечается значительный и устойчивый перевес в пользу GR. Что, вероятно, связано с медленными эффектами глюкокортикоидов, сводящимися к подавлению первичного стрессорного ответа и предотвращению сверхактивации ГАС. В этом отношении, глюкокортикоидные гормоны – это компонент восстановительной фазы, подготавливающей организм к последующим событиям [26].
Таким образом, обнаружено, что при депрессивных состояниях у крыс резко сокращается число глюкокортикоидных рецепторов в гиппокампе, и, вероятно, снижается их функциональная активность в ГАС-регулирующих областях мозга, у прекондиционированных крыс, не развивающих депрессию, эти показатели, напротив, даже возрастают. Можно предположить, что недостаточность глюкортикоидных рецепторов и дисбаланс GR/MR гиппокампа являются одним из молекулярных механизмов дисфункции системы глюкокортикоидной регуляции при стрессиндуцированных психопатологиях, в то время как активация экспрессии рецепторов после стресса вносит вклад в проявление антидепрессивных эффектов умеренной интервальной гипоксии. Полученные данные расширяют современные представления об эндокринных механизмах формирования и предотвращения депрессивных патологий и повышают трансляционный потенциал умеренной гипоксии в качестве немедикаментозного способа их коррекции.
Список литературы
Филаретов А.А. Принципы и механизмы регуляции гипофизарно-адренокортикальной системы. Л.: Наука, 1987. 165 с.
Гуляева Н.В. // Нейрохимия. 2018. Т. 35. № 2. С. 111–114.
de Kloet E.R., Joëls M., Holsboer F. // Nat. Rev. Neurosci. 2005. V. 6. P. 463–475.
de Kloet E.R. // Endocrinology. 2014. V. 155. P. 2754–2769.
Rybnikova E.A., Mironova V.I., Pivina S.G., Ordyan N.E., Tyulkova E.I., Samoilov M.O. // Dokl. Biol. Sci. 2006. V. 411. P. 431–433.
Rybnikova E., Mironova V., Pivina S., Tulkova E., Ordyan N., Nalivaeva N., Turner A., Samoilov M. // Psychoneuroendocrinology. 2007. V. 32. P. 813–823.
Seligman M.E., Beagley G. // J. Comp. Physiol. Psychol. 1975. V. 88. P. 534–541.
Samoilov M.O., Lazarevich E.V., Semenov D.G., Mokrushin A.A. Tiul’kova E.I., Romanovskii D.Iu., Miliakova E.A., Dudkin K.N. // Ross. Fiziol. Zh. Im. I.M. Sechenova. 2001. V. 87. № 6. P. 714–729.
Silverman M.N., Sternberg E.M. // Ann. N.Y. Acad. Sci . 2012. V. 1261. P. 55–63.
Miller A.H., Maletic V., Raison C.L. // Biol. Psychiatry. 2009. V. 65. P. 732–741.
Pariante C.M., Miller A.H. // Biol. Psychiatry. 2001. V. 49. P. 391–404.
Webster M.J., Knable M.B., O’Grady J., Orthmann J., Weickert C.S. // Mol. Psychiatry. 2002. V. 7. P. 985–994, 924.
Lopez J.F., Chalmers D.T., Little K.Y., Watson S.J. // Biol. Psychiatry. 1998. V. 43. P. 547–573.
Alt S.R., Turner J.D., Klok M.D., Meijer O.C., Lakke E.A., Derijk R.H., Muller C.P. // Psychoneuroendocrinology. 2010. V. 35. P. 544–556.
Oakley R.H., Cidlowski J.A. // J. Biol. Chem. 2011. V. 286. P. 3177–3184.
Меркулов В.М., Меркулова Т.И. // Вавиловский журн. генет. селекц. 2011. Т. 4. С. 621–632.
Oakley R.H., Sar M., Cidlowski J.A. // J. Biol. Chem. 1996. V. 271. P. 9550–9559.
Ismaili N., Garabedian M.J. // Ann. N.Y. Acad. Sci. 2004. V. 1024. P. 86–101.
Popoli M., Yan Z., McEwen B.S., Sanacora G. // Nat. Rev. Neurosci. 2011. V. 13. P. 22–37.
Berardelli R., Karamouzis I., Marinazzo E., Prats E., Picu A., Giordano R., Ghigo E., Arvat E. // Eur. J. Endocrinol. 2010. V.162. P. 1067–1074.
Heuser I., Deuschle M., Weber B., Stalla G.K., Holsboer F. // Psychoneuroendocrinology. 2000. V. 25. P. 513–518.
Berardelli R., Karamouzis I., D’Angelo V., Zichi C., Fussotto B., Giordano R., Ghigo E., Arvat E. // Endocrine. 2013. V. 43. P. 51–58.
Joëls M., Karst H., DeRijk R., de Kloet E.R. // Trends Neurosci. 2008. V. 31. P. 1–7.
Joëls M., Krugers H.J., Lucassen P.J., Karst H. // Brain Res. 2009. V. 1293. P. 91–100.
Barden N. // J. Psychiatry Neurosci. 2004. V. 29. P. 185–193.
Munck A., Guyre P.M., Holbrook N.J. // Endocrine Rev. 1984. V. 5. P. 25–44.
Дополнительные материалы отсутствуют.