

Нейрохимия, 2021, T. 38, № 2, стр. 111-126
Роль транскрипционного фактора NF-κB в нейровоспалении
И. И. Бабкина 1, 2, С. П. Сергеева 3, Л. Р. Горбачева 1, 2
1 ФГАОУ ВО РНИМУ им. Н.И. Пирогова Минздрава России
Москва, Россия
2 Московский государственный университет им. М.В. Ломоносова
Москва, Россия
3 ФГАОУ ВО Первый МГМУ им. И.М. Сеченова Минздрава России (Сеченовский университет)
Москва, Россия
Поступила в редакцию 10.01.2021
После доработки 11.01.2021
Принята к публикации 12.01.2021
Аннотация
Транскрипционный ядерный фактор NF-κB представляет семейство транскрипционных факторов, играющих ведущую роль в патогенезе многих хронических воспалительных процессов и в регуляции иммунных реакций. В конце прошлого столетия NF-κВ преимущественно рассматривали в качестве медиатора апоптоза в клетках иммунной системы. Последующие исследования продемонстрировали его участие в процессах развития ЦНС, пластичности, дифференцировки нейронов, нейродегенерации и при травмах мозга. Однако, его роль в регуляции нейровоспаления и в выживании зрелых нейронов ЦНС оказалась противоречивой. В представленном обзоре предпринята попытка обобщить имеющие в литературе данные о вовлечении NF-κB в формирование ответа разных типов клеток мозга на действие провоспалительных факторов. Разные субъединицы семейства NF-κB формируют димеры, вовлечение которых в регуляцию экспрессии генов определяется типом стимула. Интересно, что NF-κB вовлекается в провоспалительную активацию микроглии и астроцитов, а в нейронах опосредованные им протекторные или дегенеративные эффекты зависят не только от типа стимула, но и от времени отсроченного ответа клетки. Стимул-зависимый характер активации NF-κB определяется широким спектром рецепторов, с которых запускаются целый ряд сигнальных каскадов клетки, завершающихся активацией транскрипционного фактора. Рецепторы фактора некроза опухоли (TNFR), или рецепторы смерти, толл-подобные рецепторы (TLR), рецепторы, активируемые протеазами (PAR) – вот только часть рецепторов плазматической мембраны, способных инициировать внутриклеточные каскады, ведущие к активации NF-κB. Важным фактом является возможность модулирования активности NF-κB и его эффектов другими факторами транскрипции. STAT1, STAT3, Sirt1 – вот те молекулы, которые, как показывают последние исследования, способны изменять характер NF-κB-опосредованных клеточных ответов. Итак, NF-κB – фактор транскрипции, один из ключевых факторов, определяющий исход негативных влияний на нервную систему, в связи с чем представляющий определенный интерес для терапии нейродегенеративных процессов, связанных с воспалением, в качестве фармакологической мишени.
ВВЕДЕНИЕ
Транскрипционный ядерный фактор NF-κB представляет семейство транскрипционных факторов, играющих ведущую роль в патогенезе воспалительных процессов и иммунных реакций, через регуляцию экспрессии генов, кодирующих цитокины, хемокины, белки острой фазы, молекулы адгезии, индуцибельные ферменты [1, 2]. Более того, тяжелые заболевания человека, такие как рак, аутоиммунные заболевания, диабет обоих типов и др., сопровождаются нарушением работы сигнальных механизмов, сопряженных с NF-κB [3]. Примером тому является способность NF-κB запускать процесс фосфорилирования IRS-1 по остаткам серина, что ингибирует передачу инсулинового сигнала [4]. Со времени открытия NF-κВ [5] и его роли как медиатора апоптоза в клетках иммунной системы [6] ученые активно изучали его функции, в том числе и в нервной системе.
Экспрессия NF-κB показана во всех типах клеток головного мозга. Наибольшее количество представителей семейства NF-κB обнаружено в церебральных кровеносных сосудах и глиальных клетках. В последние годы наблюдается стремительный рост числа работ, посвященных участию NF-κB в процессах развития, пластичности, нейрональной дифференцировки, нейродегенерации и травмах мозга [7–11]. Однако, данные о его роли в регуляции нейровоспаления и в выживании зрелых нейронов ЦНС оказались противоречивыми. Показано, что базальный уровень NF-κB в нейронах необходим для поддержания их нормального функционирования, роста синапсов и синаптической пластичности, а его повышение обеспечивает нейропротекцию при отдельных патологиях [12, 13]. Так, было обнаружено, что ингибирование NF-κВ в нейронах переднего мозга приводит к их апоптозу в результате нейротоксического поражения [14], а в кортикальных нейронах этот фактор может предотвратить апоптоз при экспериментальной ишемии мозга [15]. Активация же NF-κB необходима для поддержания гомеостаза и целостности нейронов [16–18].
В то же время для глии характерна низкая базальная активность NF-κB, а экспрессия генов, индуцированная NF-κB, защищает нейроны, но вместе с тем, хроническая или чрезмерная активация NF-κB демонстрирует токсический эффект [12, 13, 19–22].
В связи с этим, особенно важны четкая систематизация и обобщение уже имеющихся экспериментальных данных о вовлечение NF-κB в воспалительные процессы ЦНС для дальнейшего изучения роли фактора NF-κB в процессах нейровоспаления разной этиологии и локализации.
СЕМЕЙСТВО БЕЛКОВ NF-κB
Транскрипционный фактор NF-κB был впервые идентифицирован Дэвидом Балтимором в 1986 году в клетках B лимфоцитов, как плейотропный транскрипционный фактор, связанный с κВ в энхансере гена легкой каппа-цепи иммуноглобулина [5].
NF-κB является гетеродимерным комплексом и включает белки двух подсемейств: NF-κB и гомологи вирусного онкогена ретикулоэндотелиоза (Rel). Семейство факторов транскрипции NF-κB у млекопитающих состоит из пяти белков: p65 (RelA), RelB, c-Rel, p105/p50 (NF-κB1) и p100/p52 (NF-κB2), которые связываются друг с другом с образованием отдельных гомо- и гетеродимерных комплексов [23]. Все эти белки имеют в своем составе Rel домен – высоко консервативную N‑концевую последовательность из 300 аминокислот – который необходим для димеризации, связывания с ДНК и ассоциации с белками семейства IκB – цитоплазматическим ингибитором NF-κB [24]. Структура белков подсемейства Rel включают трансактивирующий домен в С-концевой области. Синтезируются эти белки из белков-предшественников p105 и p100, которые содержат длинные С-концевые домены с несколькими копиями анкириновых повторов, обладающих ингибиторной активностью. Р105 и р100 подвергаются селективной убиквитин-зависимой протеасомной деградации в С-концевой области, содержащей анкириновые повторы, что приводит к образованию активных форм р52 из р100 и р50 из р105 (рис. 1). Гомодимеры р50 и р52 играют роль репрессоров генной экспрессии, тогда как р65, с-Rel и RelB в любых сочетаниях, в том числе и с р50 и р52, играют роль активаторов транскрипции. Помимо Rel домена данные белки имеют специальные NLS (nuclear localizing signal) последовательности, которые необходимы для транспорта данных белков в ядро из цитоплазмы. Члены семейства Rel содержат активационные домены, необходимые для индукции генов, и отличаются друг от друга по своим ДНК-связывающим свойствам, которые обеспечивают дополнительный уровень контроля генов [25].
Рис. 1.
Пути активации NF-κB. Канонический, неканонический и атипичный пути активации NF-κB. В условиях покоя димеры NF-κB связаны с ингибирующими IκB белками в цитоплазме. Индуцированная стимулом деградация белков IκB инициируется путем фосфорилирования комплексом киназы IκB (IKK), который состоит из двух каталитически активных киназ, IKKα и IKKβ, и регуляторной субъединицы IKKγ (NEMO). Фосфорилированные белки IκB нацелены на убиквитинирование и протеасомную деградацию, которая, таким образом, высвобождает связанные димеры NF-κB с последующим их перемещением в ядро. Канонический путь (в центре) активации NF-κB индуцируется большинством физиологических стимулов, например, цитокинами через TNFR1. Стимуляция TNFR1 приводит к связыванию TRADD, который обеспечивает платформу сборки для рекрутирования FADD и TRAF2. TRAF2 связывается с RIP1 для активации IKK. В каноническом пути IκBα фосфорилируется IKKβ- и NEMO-зависимым образом, что приводит к ядерной транслокации в основном р65-содержащих гетеродимеров. Неканонический путь (справа), индуцируемый некоторыми цитокинами семейства TNF, такими как CD40L, BAFF и лимфотоксин-β (LT-β, LT-βR, рецептор лимфотоксина-β), включает IKKα-опосредованное фосфорилирование p100, связанного с RelB, что приводит к частичному процессингу p100 и генерации активных гетеродимеров p52-RelB. Активация IKKα и фосфорилирование p100 зависит от NIK, который подвергается сложной регуляции с помощью TRAF3, TRAF2 и дополнительныхубиквитинлигаз. Атипичный путь (слева) активации NF-κB запускается в ответ на гипоксию, ультрафиолетовое излучение, перекиси и др., не зависит от IKKβ и NEMO и требует казеинкиназы 2 (СК2) вместо IKK.
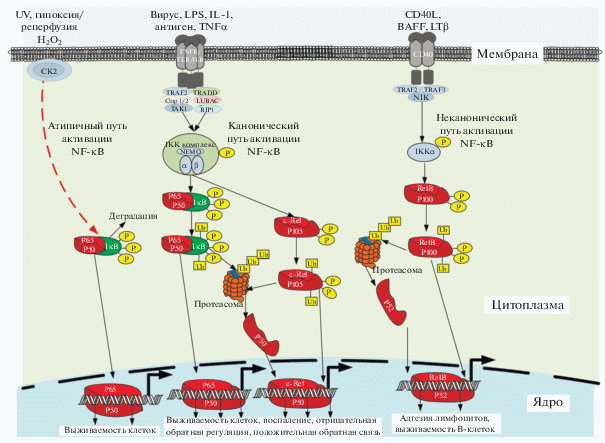
В цитоплазме NF-κB находится в виде комплекса с одним из членов семейства ингибиторных белков IκB (рис. 1). Определены пять членов семейства белков IκB: IκBα, IκBβ, IκBγ, IκBε и BCL-3. Высокая аффинность белков IκB к NF-κB обеспечивает жесткий контроль процесса активации этого транскрипционного фактора. Все члены семейства IκB содержат в своей структуре анкириновый повтор, который позволяет им связываться с NF-κB и обеспечивать его цитоплазматическую локализацию [26, 27]. IκB являются мишенями для разнообразных сигнальных путей, активируемых внутри- и внеклеточными стимулами (цитокины, вирусы, оксиданты, факторы роста) [24, 28–31]. Белки-предшественники p105 и p100 функционируют подобно белкам IκB, подавляя и удерживая NF-κB в цитоплазме [8] (рис. 1).
ПУТИ АКТИВАЦИИ NF-κB
Провоспалительные цитокины (TNF-α, IL1-β и др), LPS, повреждение ДНК, ионизирующее излучение [23, 32] и другие провоспалительные, токсические или повреждающие воздействия способны индуцировать фосфорилирование IKK комплекса, в состав которого входят субъединицы IKKα, IKKβ и регуляторная субъединица IKKγ (NEMO), с его последующей диссоциацией из комплекса с NF-κB и деградацией. NF-κB, высвободившийся из IKK комплекса, транслоцируется в ядро, где запускает индукцию фактор-зависимых генов-мишеней (рис. 1). Убиквитин участвует по крайней мере в трех стадиях пути активации NF-κB, которые включают деградацию IκB ингибитора NF-κB, процессинг предшественников NF-κB и активацию IKK. Убиквитинирование – это обратимая ковалентная модификация, катализируемая тремя ферментативными стадиями [33].
Активация NF-κВ может осуществляться по каноническому, неканоническому или альтернативному (атипичному) пути [3, 8, 34] (рис. 1). Первый путь сопровождается образованием димеров p50/p65 или p50/c-Rel и запускается в ответ на инфекцию, провоспалительные цитокины и ростовые факторы. В результате активируется IKKβ (IκB kinase β) [8, 35, 36], которая фосфорилирует IκB, с высвобождением димеров NF-κB и перемещением последних в ядро. Канонические сигнальные пути NF-κВ являются временными, а транскрипционные реакции ограничены ауторегуляторными механизмами, которые вовлекают NF-κВ-зависимую индукцию негативных регуляторов, таких как IκBα или убиквитин-редактирующий фермент A20 (TNFAIP3), препятствующих активации IKKβ [37].
Неканонический путь активации запускается специфическими членами TNF семейства цитокинов, такими как CD40L, лимфотоксин β (LTβ) и активирующий фактор B-клеток (BAFF) [36]. Данный путь зависит от IKKα-опосредованного фосфорилирования p100, связанного с RelB, что приводит к образованию p52/RelB комплекса [38] (рис. 1). Атипичный путь активации NF-κB запускается в ответ на гипоксию, ультрафиолетовое излучение, перекиси и др., не зависит от IKKβ и NEMO и требует казеинкиназы 2 (СК2) вместо IKK. Деградация ингибитора NF-κBIκB реализуется без участия протеасом, с использованием калпаин-зависимых механизмов. Это событие приводит к разрушению цитоплазматических ингибиторов после чего димеры NF-κB, способны транслоцироваться в ядро [38] (рис. 1).
NF-κB-зависимая транскрипция гена включает рекрутинг субъединицы RelA/p65 для узнавания геномных сайтов κB и требует транскрипционных коактиваторов, которые индуцируют специфичные для последовательности активаторы и приводят к изменению структуры хроматина и индукции транскрипции. Одним из известных коактиваторов субъединицы RelA/p65 является CBP-связывающий белок и его структурный гомолог p300. Протеинкиназа A (PKA) фосфорилирует субъединицу p65 и усиливает ее связь с PCAF. PCAF является членом семейства гистонацетилтрансфераз и индуцирует ацетилирование, изменяет хроматин и приводит к индукции транскрипции. Обнаружено обогащенное ацетилирование гистонов на промоторах провоспалительных цитокинов (таких как IL-1, IL-2, IL-8 и IL-12) с повышенной активацией транскрипции этого гена в результате воспаления.
KLF – семейство Krüppel-подобных ДНК-связывающих факторов транскрипции с консервативными доменами, известными как цинковые пальцы – играют разнообразную роль в биологических процессах, в том числе дифференциации, развитии, росте и воспалении. Известна роль KLF2 в NF-κB-опосредованной регуляции воспаления. А именно, NF-κB ингибирует экспрессию KLF2 посредством прерывания связывания факторов MADS-бокса фактора энхансера транскрипции 2 (MEF2) и доступа молекул гистондеацетилазы (HDAC) к промотору KLF2 [39]. При этом, KLF2 ингибирует транскрипционную активность NF-κB, напрямую взаимодействуя с p300 и PCAF, которые являются критическими ко-активаторами для NF-κB-опосредованной транскрипционной активности [40]. KLF2 уменьшает рекрутирование коактиваторов в NF-κB, а также ослабляет ацетилирование гистона 3 по лизину 9 (H3K9) и гистону 4 по лизину 8 (H4K8) [39].
Обнаружено, что эффект активации NF-κB является временным, при этом ядерная форма остается активной менее часа. Активированный NF-κB также индуцирует транскрипцию генов, кодирующих белки IκB, и приводит к петле обратной связи по ингибированию белками IκB. Вновь синтезированный IκB перемещается в ядро и образует комплексы с гетеродимером NF-κB (p65-p50), прерывает связывание с сайтами κB, а комплекс p65-p50-IκB перемещается в цитоплазму.
Множество генов, регулируемых NF-κB, содержат одну или несколько NF-κB связывающих последовательностей. Видимо, в регуляции разных генов, индуцируемых в ответ на разные стимулы, участвуют разные комбинации субъединиц NF-κB [41, 42]. Например, в ответ на действие TNFα в ядро быстро транслоцируются димерные комплексы p50-p65, p50-c-Rel, p65-c-Rel, p52-c-Rel и p52-p65. Это связывают с необходимостью разных димеров для регуляции разных генов [41–43].
МОДУЛЯТОРЫ ПРОЦЕССА АКТИВАЦИИ NF-κB
NF-κB уникален тем, что может активироваться широким спектром внутриклеточных сигнальных каскадов, индуцируемых воспалительными цитокинами, продуктами окисления липидов, липосахаридами, гипоксией, ROS, некоторыми микроорганизмами (сальмонеллы, шигеллы, листерии, Helicobacter, E. coli) и вирусами [44], при этом может происходит запуск порочного круга и обострение воспалительного процесса [45]. В большинстве случаев данный процесс опосредован разными типами рецепторов: рецепторами антигенов и членами суперсемейства TNFR (Tumour necrosis factor receptor), TLR (Toll-like receptors), IL-1R (Interleukin1 receptor) [3, 46] (рис. 2).
Рис. 2.
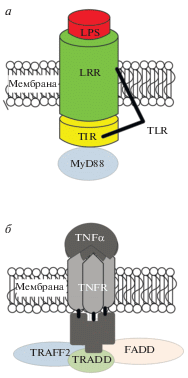
Рецепторы цитокинов. Структура рецептора TLR. MyD88 – критический домен рецепторного комплекса TLR и важный адаптерный белок сигнального пути NF-κB, способствующий экспрессии воспалительных генов (а). Структура рецептора TNFR1. TNF-α связывается со своим рецептором (TNFR1), а белок домена смерти (TRADD) обеспечивает связывание нейровоспаления и апоптоза.
Такая особенность активации обеспечивает NF-κB способность контролировать многие процессы, включая воспаление, апоптоз, иммунитет, выживаемость клеток и рак, при этом общепризнанна его ключевая роль в регуляции индуцируемой экспрессии генов иммунного и воспалительного ответа [47, 48]. NF-κB-зависимая транскрипция не только строго контролируется положительными и отрицательными регуляторными механизмами, но также модулируется другими сигнальными путями, что обеспечивает преобразование разнообразных биологических функций NF-κB в специфические, адаптивные клеточные ответы.
PI3K/Akt, MAPK и UPR как модуляторы активации NF-κB
Известно, что сигнальные пути, такие как PI3K/Akt и MAPK, взаимодействуют с путем NF-κB. Показано, что PI3K/Akt-опосредованное фосфорилирование и активация RelA/p65 субъединицы NF-κB реализуется с участием как IKKα, так и IKKβ [49]. G. Haegeman [50] предложил модель индукции провоспалительных генов, включающую несколько сигнальных путей, сопряженных с модулированием эффектов NF-κB. С одной стороны, транскрипционный фактор NF-κB освобождается от ингибирующего его IκB комплекса в цитоплазме, направляется в ядро, где связывается с различными промоторами провоспалительных генов. С другой стороны, происходит активация MAPK, p38 и ERK, которые также могут запускаться через TNFR с последующей активацией их общего субстрата, MSK1. В свою очередь MSK1 фосфорилирует NF-κB p65 по ключевому аминокислотному остатку (например, Ser 276), что и обеспечивает транскрипционную активность NF-κB p65, формируя способность транскрипционного фактора к взаимодействию с ядерными коактиваторами (CREB, СBP, p300 и др.). Кроме этого, MSK1, будучи рекрутированной в регионы воспалительных промоторов, фосфорилирует хроматиновые хвосты, способствует релаксации местного хроматина и облегчает генную транскрипцию [45, 50].
Guha и Mackman показали, что ингибирование пути PI3K/Akt увеличивает LPS-индуцированную продукцию TNF-α через MAPK (ERK, p38 и JNK) и ядерную транслокацию NF-κB, индуцируя активацию GSK-3β [51].
UPR (Unfolded protein response) является интегрированным внутриклеточным сигнальным путем, который обеспечивает адаптивные реакции клетки при ER-стрессе (стресс эндоплазматического ретикулума). Исследования показали, что UPR, стимулируя активацию провоспалительных стресс-киназ, таких как IKK, JNK, активирует транскрипционные факторы NF-κB, AP-1 (Activator protein 1), и генерацию ROS, что вызывает индукцию секреции IL-1β, IL-18, TNFα, IL-6 и MCP-1, в том числе и при разных формах нарушений метаболизма глюкозы [52–54].
Сопряжение транскрипционных факторов Nrf2 и NF-κB
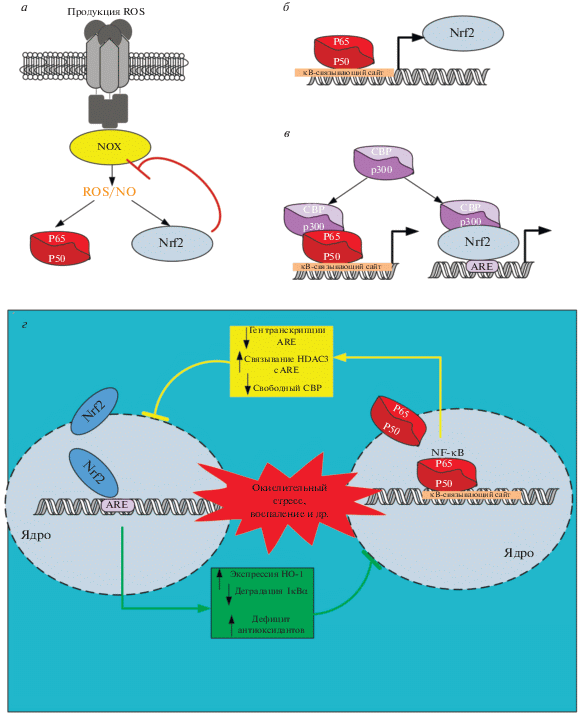
Другим модулятором активности NF-κB через IKK или напрямую через субъединицу р65 является фактор транскрипции Nrf2 (nuclear E2-related factor 2). В клетках Nrf2 находится под постоянным контролем репрессорного белка Keap1 (Kelch-like ECH associating protein 1), являющегося своеобразным молекулярным “сенсором” изменения внутриклеточного гомеостаза. Активация пути Nrf2 приводит к диссоциации комплекса Nrf2-Keap1 и увеличение свободного Keap1 в цитозоле. Установлено, что Keap1, связываясь с IKKβ, предотвращает фосфорилирование ингибиторного комплекса NF-κB и препятствует транслокации p65 в ядро, блокируя передачу сигналов через NF-κB [55, 56]. С другой стороны, Keap1 способен напрямую связываться с субъединицей р65 NF-κB [57]. Взаимодействие между двумя путями опосредуется также через CREB-связывающий белок (CBP), который является ко-активатором транскрипции, способным связываться как с Nrf2, так и с субъединицей р65 NF-κB [58] (рис. 3). Таким образом, Nrf2 способен регулировать NF-κB-сигнализацию, конкурируя с ним за белок CBP (рис. 3). Активация Nrf2 в ответ на острые травмы, воспаление, ксенобиотики и многие другие стимулы обеспечивает протекцию в основном через защиту клеток от окислительного повреждения. Индукция NF-κB происходит в ответ на аналогичные стимулы, что позволяет рассматривать работу сигнальных каскадов Nrf2-NF-κB как целостного механизма, направленного на уравновешивание активности друг друга [59, 60] (рис. 3).
Рис. 3.
Взаимная модуляция активности транскрипционных факторов NF-κB и Nrf2, происходящая на разных уровнях. Активные формы кислорода (ROS), продуцируемые во время воспаления, активируют NF-κB и Nrf2. Nrf2, снижая уровень активных форм кислорода, вызывает снижение активности NF-κB (а). Сайты связывания NF-κB идентифицированы в промоторной области NFE2L2 (б). Факторы транскрипции Nrf2 и NF-κB конкурируют за связывание с коактиватором транскрипции CREB-связывающим белком (CBP/p300). Схема взаимной модуляции транскрипционной активности NF-κB и Nrf2 (Г). Сокращения: HO-1 – гемоксигеназа-1, HDAC3 – гистондиацетилаза 3, NOX – NADPH-оксидазы, CBP – CREB связывающий белок.
Участие сигнального пути JAK2/STAT3 в модуляции активации NF-κB
Известно, что цитокины и окислительный стресс индуцируют фосфорилирование JAK2 и активацию фактора транскрипции STAT3, тем самым способствуя ассоциированной с воспалением экспрессии генов [61]. В связи с этим, можно предположить сопряжение этого сигнального каскада с активацией NF-κB. Guzzo с соавторами было продемонстрировано [62] участие JAK2/STAT3 в регуляции транскрипционной активности NF-κB, о чем свидетельствует снижение ДНК-связывающей активности субъединиц p50 и p65 NF-κB на фоне предварительного ингибирования JAK2/STAT3. Другие исследования подтвердили, что транскрипционная активность NF-κB и STAT тесно связана с их ацетилированием по лизину, которое регулируются балансом между гистоновыми ацетилтрансферазами (HATs) и гистоновыми деацетилазами (HDAC) [63].
Особый интерес в контексте регуляции транскрипционной активности NF-κB представляют сиртуины (Silent Information Regulator 2 proteins, SIR2, у млекопитающих это гомологичный белок SIRT1), третий класс НАД-зависимых гистоновых деацетилаз. Показано, что SIRT1 и SIRT6 снижают транскрипционную активность NF-κB. SIRT1 деацетилирует NF-κB p65 по остатку лизина 310, что способствует его эффективному связыванию с IκB, снижая связывание с промотором целевого гена и экспрессию соответствующих цитокинов [64–66]. Кроме того, было показано, что активация SIRT1 подавляет передачу сигналов JAK2/ STAT3/NF-κB при β-амилоидной нейротоксичности [67]. SIRT6, связывается с субъединицей RELA NF-κB, приближается к промоторам генов, экспрессию которых регулирует NF-κB, деацетилирует гистон H3 по девятому остатку лизина, что способствует конденсации хроматина и, следовательно, ослабляет действие NF-κB.
НЕЙРОВОСПАЛЕНИЕ И NF-κB
Нейровоспаление как особый тип воспалительного процесса
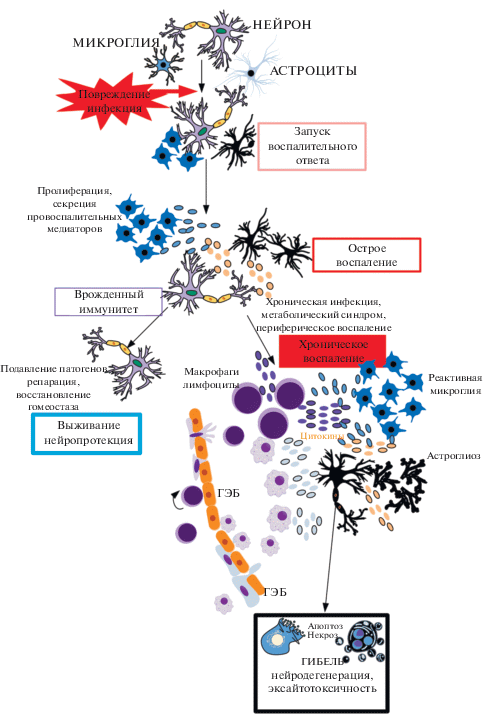
Воспаление, как биологический процесс, инициируемый в ответ на повреждение, инфекцию и травму, нанесенную клеткам или тканям в пределах нервной системы, имеет ряд особенностей. В частности, это связано со спецификой врожденного и адаптивного иммунитета мозга по отношению к периферии. Нейровоспаление может быть отрицательным фактором. Так хроническое воспаление с участием активированной микроглии и астроглии становится отличительной чертой многих заболеваний человека, включая нейродегенерацию [68, 69]. Иммунная активация в ЦНС также включает микроглию (резидентные макрофаги) и астроциты (потенциально иммунокомпетентные клетки), которые в условиях нормы вносят свой вклад в регуляцию гомеостаза ткани мозга. Наряду с нейроглией клетки эндотелия и периваскулярные макрофаги также важны для интерпретации и распространения воспалительных сигналов в ЦНС [70] (рис. 4). Микроглия сканирует микросреду, продуцируя факторы, которые влияют на соседние астроциты и нейроны, особенно в ответ на инфекцию или повреждение. Это приводит к активации воспалительного ответа, который в дальнейшем включает временный, самоограничивающийся ответ через иммунную систему и инициирует восстановление тканей. В патологических условиях, когда нормальные механизмы разрешения не работают, происходит аномальная активация и повышенная продукция воспалительных факторов, что приводит к хроническому нейровоспалению и прогрессированию нейродегенеративных изменений (рис. 4). Нейродегенеративные факторы влияют на функцию глии за счет сверхактивации как микроглии, так и астроцитов, вызывая производство и высвобождение большого количества провоспалительных цитокинов и активных форм кислорода и азота (ROS, RNS). Хроническая активация микроглии связана с деградацией белка, дисфункцией митохондрий и дефектами аксонального транспорта и апоптоза, которые пагубно влияют на функцию нейронов и приводят к гибели клеток. Кроме того, нейровоспаление приводит к последующей инфильтрации клеток иммунной системы от периферии к ЦНС через гематоэнцефалический барьер (ГЭБ), что ускоряет нейровоспаление и нейродегенерацию [71] (рис. 4). Практически во всех этих процессах активно участвует NF-κB.
Рис. 4.
Две стороны нейровоспаления. Нейровоспалительные реакции могут быть полезными или вредными, в зависимости от их продолжительности и силы активации. Хроническое воспаление обычно ведет к прогрессивной нейродегенерации.
Интересно, что результат активации NF-κB имеет определенную клеточную специфичность, так в нейронах он способствует преимущественно выживанию и пластичности. С другой стороны, активация NF-κB в глиальных клетках играет важную роль в воспалительных процессах, которые в большинстве своем ведут к нейродегенерации [19].
Роль TLR/NF-κB сигнального пути в регуляции нейровоспаления
Как видно, из предыдущего раздела данного обзора, во всех эукариотических клетках большинство внеклеточных сигналов, включая инфекции, воспалительные цитокины и множественные стрессовые факторы, могут активировать NF-κB. Одними из важных структур мембраны, обеспечивающих передачу сигналов в системе врожденного иммунитета и воспалительной реакции, являются Toll-подобные рецепторы (TLR) (рис. 2). Микроглия экспрессирует ряд TLR, которые активируют эти клетки и инициируют нейровоспалительную реакцию [72]. Миелоидный дифференцирующий фактор 88 (MyD88), являясь адаптерным белком, связывается с TLR через их TIR-домены, что активирует несколько путей передачи сигнала и, в конце концов, приводит к активации NF-κB и воспалению (рис. 2). MyD88 играет роль в инфицировании ЦНС и последующей активации астроцитов. Он может также участвовать в повреждениях зрительного нерва, болезни Паркинсона и болезни Альцгеймера [73, 74]. Пути передачи сигналов TLR4/MyD88/NF-κB могут быть полезной терапевтической мишенью для фармакологического лечения нейровоспалительных повреждений [75]. Функционирование некоторых TLR может зависеть от ко-рецепторов. Например, TLR4 для распознавания бактериального липополисахарида (LPS) требует наличия MD-2, CD14 и липополисахарид-связывающего белка (LBP). LPS связывается с LBP для индуцированной активации микроглии M1. Этот комплекс взаимодействует с TLR4 и ко-рецепторами CD14, MD2 стимулируя трансмембранные сигнальные пути. Передача сигналов через TLR4 разделена на два разных сигнальных пути, MyD88-зависимый, и MyD88-независимые. В MyD88-зависимых путях, происходит активация NF-κB [76].
Имеются данные о том, что воспалительный процесс может запускаться TLR4-NF-κB-зависимым путем при его активации с помощью HMGB1 [77–79]. HMGB1 это высококонсервативный, негистоновый ДНК-связывающий белк, который признан ключевым медиатором воспаления и нейровоспаления, секретируемый активированными макрофагами и моноцитами. Будучи ядерным белком, HMGB1 может высвобождаться при некрозе клеток, что в условиях повреждения нервной ткани может потенцировать развитие воспаления. Внеклеточный HMGB1 запускает воспалительные реакции посредством активации таких рецепторов как TLR4, TLR2 и RAGE (receptor for advanced glycation endproducts) в иммунокомпетентных клетках, нейронах и астроцитах [80, 81]. Передача сигналов HMGB1 через эти рецепторы способствует активации NF-κB, через деградацию IκB запускаемую фосфорилированием IκBα, что инициирует экспрессию медиаторов воспаления, таких как TNF-α, IL-1β и IL-6, и выживания клеток [82, 83].
Интересные факты были получены об участии HMGB1 в развитии астроцитарного отека спинного мозга через повышение экспрессии AQP4 после кислородно-глюкозной депривации и реперфузии. Установлено, что этот процесс опосредуется через передачу сигналов HMGB1/TLR4/ MyD88/NF-κB и зависит от IL-6 [84].
Участие TNF-α/TNFR/NF-κB сигнального пути в регуляции нейровоспаления
Микроглия играет ведущую роль в инициации процессов как воспаления, так и гибели клеток в нервной системе. Эти функции микроглии связаны с врожденным иммунным ответом и преимущественной передачей сигналов, вызванных цитокином TNF-α. Известно, что степень апоптоза нейронов гиппокампа определяется уровнем TNF-α (мРНК), что указывает на существование критической пороговой концентрации TNF-α необходимой для инициации апоптотических путей [72, 85]. Повышение TNF-α и IL-1β наблюдалось в тканях мозга ещё до гибели нейронов. IL-1β и TNF-α играют важную роль в патологическом воспалении и ускорении болезни [72]. Они могут вызывать разрушение ГЭБ, активировать экспрессию молекул адгезии и стимулировать диффузию реактивных веществ, таких как оксид азота (NO) [86]. IL-1β играет решающую роль в прогрессировании хронических нейродегенеративных заболеваний, таких как болезни Альцгeймера и Паркинсона, а также острых нейровоспалительных состояний, включая инсульт, ишемию и повреждение головного мозга [87]. Эффекты TNF-α могут определятся областью мозга. В черной субстанции и стриатуме он способствует нейродегенерации, тогда как в гиппокампе – нейропротекции [88]. Запуск воспаления обычно определяется по высвобождению провоспалительных цитокинов, таких как TNF-α, IL-1β, а также молекул адгезии.
Апоптотическое действие TNF-α реализует через рецепторы фактора некроза опухоли (TNFR), или рецепторы смерти, – надсемейство тримерных белков, рецепторов цитокинов, которые распознают и связывают цитокины семейства TNF и содержат гомологичную цитоплазматическую последовательность, идентифицирующую внутриклеточный домен гибели (рис. 2). Один из подтипов этих рецепторов – TNFR2 способен инициировать активацию NF-κB через PI3K/Akt путь или через рекрутирование NIK, что обеспечивает канонический и неканонический тип активации NF-κB соответсвенно [89, 90]. Тип активации NF-κB определяет и особенности димеризации его субъединиц, что, вероятно, может определять и направленность конечного эффекта [91]. Так, Grosh с соавторами обнаружили, что TNF-α играет защитную роль в гиппокампе мыши, активируя TNFR2-опосредованный сигнальный путь и рекрутируя гетеродимер p65-p52. Этот же гетеродимер участвует в GDNF-опосредованной нейропротекции дофаминергических нейронов черной субстанции у крыс в модели болезни Паркинсона [92]. Эти данные подтверждают ранее полученные результаты о том, что TNF-α обеспечивает нейропротекцию первичных нейронов коры при вызванной глутаматом эксайтотоксичности, активируя NF-κB через TNFR2 [93]. Активация же другого рецептора TNF-α – TNFR1 может вызывать быстрый апоптоз нейронов через включение сигнальных комплексов смерти, таких как AP-1, NF‑κB и каспаза-3 [69, 94], и потенцировать прогрессирующее воспаление, индуцируя димеризацию p65/p50 гетеродимера [91].
Другим членом суперсемейства рецепторов TNF-α является рецептор-активатор ядерного траскрипционного фактора NF-κB (RANK), который является центральным активатором NF-κB [95]. Внутриклеточная сигнальная трансдукция RANK опосредуется прямым взаимодействием с факторами TRAF (рис. 2). TRAF6 является критической сигнальной молекулой, запускающей активацию MAPKs, p38 и JNK, а также канонического пути NF-κB в ответ на передачу сигналов через RANK. Недавно было показано, что TRAF3 вовлекается в RANK-опосредованную негативную регуляцию неканонического пути NF-κB [96]. В связи с этим, есть основание считать, что RANK, обладая способностью инициировать оба – канонический и неканонический – пути активации NF-κB, функционально обособлен от некоторых других членов суперсемейства TNFR.
Таким образом, тип рецептора смерти, его локализация и тип формирующегося в результате его активации гетеродимера NF-κB будет, вероятно, определять провоспалительное или протекторное действие цитокина.
Вовлечение NF-κB в ROS-зависимое нейровоспаление
Воспаление вызывает окислительный стресс и повреждение ДНК, что приводит к перепроизводству ROS макрофагами и микроглией. Помимо прямого токсического воздействия ROS на биологические макромолекулы, они могут запускать воспалительную реакцию, стимулируя ряд генов, регулирующих каскады воспалительных сигналов. В ткани мозга есть уникальные источники окислительного стресса, такие как возбуждающие аминокислоты и нейротрансмиттеры. Метаболизм этих аминокислот и нейротрансмиттеров приводит к образованию ROS [97]. Митохондрии являются важным источником утечки ROS из цепи переноса электронов, поскольку они восприимчивы к окислительному повреждению, которое приводит к дисфункции митохондрий и повреждению тканей. NF-κB может быть активирован митохондриальной дисфункцией через окислительный стресс. На линии клеток нейробластомы человека показано, что токсическое воздействие марганца опосредовано активацией NF-κB и приводит к высвобождению провоспалительных цитокинов (TNF-α, IL-1β и IL-6) и индукции ферментов (iNOS и COX-2) [98]. Генерация ROS, приводящая к NF-κB-вызванной индукции iNOS, усиливает продукцию оксида азота глиальными и эндотелиальными клетками. Избыточное количество NO приводит к воспалительным нейродегенеративным заболеваниям [99]. Сигнальные молекулы NO и ROS в низких концентрациях регулируют пролиферацию клеток. С другой стороны, при высоких концентрациях они являются ключевыми цитотоксическими молекулами. При этом NF-κB, выступая мишенью для ROS и индуктором экспрессии iNOS, выступает звеном патологического процесса нарастания нейровоспаления при окислительном стрессе.
ОСОБЕННОСТИ ТРАНСКРИПЦИОННОЙ АКТИВНОСТИ NF-κB В НЕЙРОНАХ
Нейроны, в отличие от других клеток характеризуются наличием конститутивной экспрессии NF-κB p65, в частности, это относится к нейронам гиппокампа и коры головного мозга. Предполагают, что конститутивная экспрессия NF-κB является следствием синаптической активности. Конститутивная активность NF-κB достигается за счет глутаматергических рецепторов, Са2+ каналов L-типа, а также за счет генерации потенциала действия. Активация NF-κB в основном запускается проникновением внеклеточного Ca2+ [100–103] (рис. 5). Таким образом, NF-κB действует как преобразователь сигнала, передавая информацию в ядро из активных синапсов, участвуя в поддержании синаптической пластичности. При этом, подавление нейровоспаления в пожилом возрасте путем ингибирования патологической гиперактивации NF-κB может стимулировать обучение, хотя физиологическое присутствие NF-κB в нейронах необходимо для процесса обучения.
Рис. 5.
Схема последствий конститутивной и индуцибельной активации NF-κB в нервной ткани. BIM, NOXA – проапоптотические белки; Bcl-2, Bcl-XL – антиапоптотические белки; DMT1, переносчик двухвалентных металлов 1; UCP4, митохондриальные разобщающие белки; BIRC3 – белок 3, содержащий бакуловирусный повтор IAP; и MnSOD, марганцевая супероксиддисмутаза. Двойными стрелками обозначено повышение/снижение регулируемых параметров.
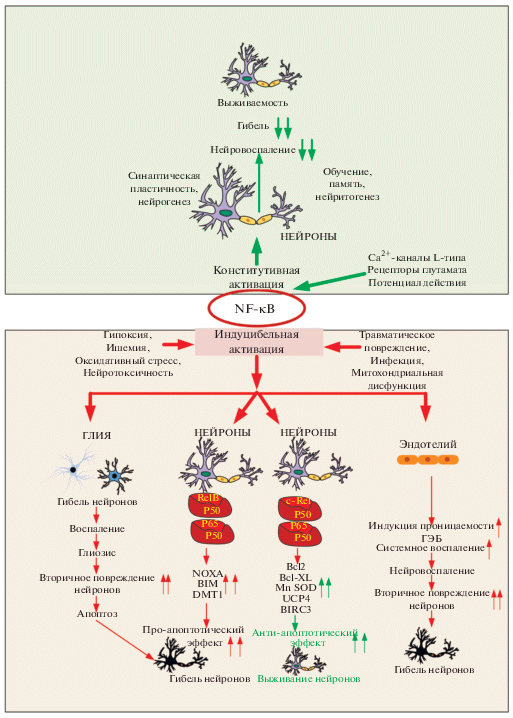
У пациентов с болезнью Альцгеймера активированный NF-κB обнаруживается главным образом в нейронах и глиальных клетках в областях, окружающих бляшки Ab [104]. Активированные клетки генерируют воспалительные цитокины, такие как TNF-α, IL-1β, а также iNOS, что ведет к продукции свободных радикалов, таких как оксид азота [105, 106]. Эти провоспалительные молекулы влияют на прогноз болезни Альцгеймера.
В целом функции NF-κB, которые связаны с нейрональной передачей сигналов, подразумевают, что роль NF-κB в нейронах заметно отличается от других клеток [107–110]. Факторы транскрипции NF-κB являются ключевыми регуляторами воспаления и апоптоза при старении и во время повреждения мозга. Компоненты димеров NF-κB, а также эпигенетические модификации, способствующие ацетилированию гистонов, тонко уравновешивают ответ нейронов на поражение мозга (рис. 5). Активация NF-κB/c-Rel стимулирует нейропротекторную активность, способствуя транскрипции ключевых антиапоптотических генов в моделях инсульта. Напротив, есть данные, указывающие на то, что аберрантная активация NF-κB/RelA, демонстрирующая снижение ацетилирования, а скорее сайт-специфическое ацетилирование Lys 310, запускает экспрессию проапоптотических генов [111, 112] (рис. 5).
В ЦНС различные стимулы, такие как молекулярные структуры, связанные с повреждениями и патогенами, синаптическая активность, окислительный стресс, цитокины, хемокины, нейротрансмиттеры, нейротрофические факторы и нейротоксины, а также экспериментальные повреждения и невропатологические расстройства индуцируют активацию NF-κB [8, 11, 16, 113–117]. При этом активация NF-κB в глиальных клетках в основном связана с воспалением [118, 119], а в нейронах NF-κB, напротив – преимущественно участвует в дифференцировке, синаптической пластичности и способствует выживанию клеток [113, 120]. Различие эффектов предположительно связано с рекрутированием различных субъединиц NF-κB (рис. 5). Так повышенная экспрессия субъединицы р50 в гиппокампальных нейронах крыс, при моделировании закрытой травмы головы (CHI), способствует восстановлению и регенерации мозговой ткани [121]. Исследования показывают, что NF-κB может оказывать положительное или отрицательное влияние в зависимости от типа клеток и характера повреждения [7]. Например, травматическое повреждение мозга (TBI), эксайтотоксичность и ишемия характеризуются активацией NF-κB [15, 122, 123]. Однако точная роль этого фактора транскрипции в поврежденных нейронах до конца не изучена. Известно, что супрессия NF-κB в нейронах усиливает острую посттравматическую смертность и ухудшает неврологический исход выживших в различные интервалы посттравматического периода, в том числе усиливая реактивный астроглиоз и апоптоз, и способствует усилению неврологического и моторного дефицита после черепно-мозговой травмы (ЧМТ) [20].
Эти результаты свидетельствуют о том, что базальная активность NF-κB в нейронах необходима для защиты этих клеток от травматического повреждения (рис. 5). Интересно, что подобные протекторные эффекты NF-κB возможны только в определенный момент времени после ЧМТ. Возможно, эндогенная активация NF-κB защищает нейроны в условиях стресса, вызванного повреждением, путем подавления опосредованного р53 апоптоза через внутренний митохондриальный путь [124]. Кроме того, защитная роль NF-κB в нейронах включает антиапоптотические эффекты, опосредованные индукцией эндогенного ингибитора каспазы 8 или путем запуска экспрессии антиоксидантных генов, таких как марганцевая супероксиддисмутаза или кальций-связывающий белок кальбиндин D28k [125–127]. При этом ингибирование NF-κB в микроглии и астроцитах наоборот способствовало благоприятному исходу после травм ЦНС [128–130].
В других работах, было показано, что противоположность эффектов NF-κB может быть опосредована двухфазным характером активации NF-κB в нейронах. Повышение уровней p65 и c-Rel в разные периоды времени позволяет предположить, что в пределах одного нейрона NF-κB может участвовать в различных патофизиологических процессах. Известно, что NF-κB способствует повреждению нейронов, индуцируя провоспалительные цитокины, но, в то же время способствует выживанию клеток при гипоксии-реперфузии посредством усиления экспрессии антиапоптотических медиаторов, таких как Bcl-2 и Bcl-XL [131] (рис. 5).
Это указывает на двойную роль NF-κB в регуляции выживания нейронов при повреждении головного мозга.
Травматическое повреждение нейронов в модели “царапины” in vitro вызывало посттравматическую двухфазную активацию NF-κB, через 1 и через 24 ч. При этом, первичное возрастание активации NF-κB способствовало гибели нейронов, тогда как второй пик активации фактора не влиял на данный показатель [132]. Двухфазная активация NF-κB описана Nijboer с соавторами [133] в модели ишемического повреждения головного мозга у новорожденных крысят, где активация фактора во время раннего постишемического периода (0–3 ч) проявляла негативное влияние, а в поздний период (до 24 ч – нейропротекторное). Pizzi и соавторы [134] при анализе выживаемости нейронов при токсическом действии глутамата и протекторном действии IL-1β продемонстрировали противоположные эффекты разных представителей семейства NF-κB. Субъединицы р65 опосредуют гибель клеток, вызванную глутаматом, а c-Rel вовлечены в протекторное действие IL-1β в нейронах мозжечка крысы и на гиппокампальных переживающих срезах мыши [134]. Эксперименты на первичных культурах нейронов при моделировании травмы in vitro также демонстрируют корреляцию выживаемости клеток с отсроченной активацией субъединицы c-Rel, в отличие от р65 [132]. Таким образом, анти- и проапоптотических эффекты NF-κB могут определяться типом субъединиц фактора, вовлекаемых в клеточную сигнализацию при повреждении, и временным интервалом посттравматической активации фактора.
Роль PAR-зависимой регуляции активности NF-κB в нейровоспалении
Механизмы повреждения мозга, вызванные геморрагией, а также нарушение проницаемости ГЭБ, при воспалении, несомненно, затрагивают вопрос сопряжения системы гемостаза и процессов воспаления. В этой связи особый интерес представляют данные о вовлечении специфических рецепторов, активируемых протеазами (PAR) в регуляцию активности транскрипционного фактора NF-κB. Тромбин – ключевая протеаза гемостаза является высокоспецифичным агонистом PAR1 (одного из подтипов PAR) и мощным модулятором эндотелиальной функции. Эта PAR1-зависимая функция тромбина реализуется, посредством стимуляции NF-κB, который индуцирует экспрессию адгезивных молекул ICAM-1 и VCAM-1 эндотелием. Известно, что передача сигналов PAR-1/NF-κB зависит от активации PKC. Было показано, что сигнальный комплекс белков, содержащих каркасный белок (CARMA3), линкерный белок (Bcl10) и эффекторный белок (MALT1) связывают активацию PAR-1 со стимуляцией IκB киназного комплекса. IκB киназа в свою очередь фосфорилирует IκB, что приводит к его деградации и высвобождению активного NF-κB [135]. Все три белка, CARMA3, Bcl10 и MALT1 необходимы для эффективной стимуляции тромбином канонический путь NF-κB в эндотелиальных клетках. Delecta с соавторами показали, что запуск NF-κB пути тромбином включает несколько ключевых участников: РКС (фосфорилирует CARMA3), PI3K (рекрутирует PDK1 через фосфорилирование), СВМ сигналосома (активирует IKKβ) и Akt (фосфорилирует субъединицу р65, увеличивая ее транскрипционную активность) [135]. Активация фактора транскрипции NF-κB представляет собой один из ключевых механизмов, ответственных за индуцированную тромбином эндотелиальную дисфункцию, которая на уровне мозга проявляется в нарушении ГЭБ и инфильтрации мозговой ткани макрофагами, потенцируя нейровоспаление (рис. 5).
Также необходимо отметить, что увеличение внутриклеточной концентрации кальция, вызываемое активацией PAR, может влиять на ряд внутриклеточных факторов как в ядре, так и в цитоплазме (транскрипционные факторы – NF-κB, AP-1 и др.) и запускать или тормозить экспрессию различных генов, в том числе тех белков (Вах/Bad семейства Bcl, AIF, MAP-киназ, каспаз), активность которых определяет развитие апоптоза при токсическом действии глутамата. Было показано, что длительное токсическое действие глутамата на культивируемые гиппокампальные и кортикальные нейроны вызывает активацию NF-κB и транслокацию субъединицы р65 в ядро [13].
ЗАКЛЮЧЕНИЕ
Нейровоспаление это сложный комплексный ответ нервной ткани на травму, инфекцию, эксайтотоксичность и другие повреждающие факторы. Исход этого процесса определяется совместной работой разных типов клеток: нейронов, микроглии, астроцитов, эндотелиоцитов, макрофагов и перицитов. Принимая хроническую форму, нейровоспаление ведет к нейродегенерации, что позволяет рассматривать его не только как патологический фактор при травме и инсульте, но и как прогностический фактор при нейродегенеративных заболеваниях. В связи с этим, изучение механизмов нейровоспаления с целью выявления терапевтических мишеней для эффективной коррекции данного процесса является актуальным. В качестве такой мишени может выступать транскрипционный ядерный фактор NF-κB, который экспрессируется во всех клетках-участниках нейровоспаления. Многочисленные данные экспериментальных исследований свидетельствуют, что эффекты опосредованные активацией NF-κB определяются как уровнем его базальной экспрессии, так и типом клетки, типом формирующегося димера, временны́м интервалом активации в посттравматический период (рис. 5). Более того, тонкий контроль активности NF-κB обеспечивается многочисленными модулирующими влияниями со стороны других транскрипционных факторов и сигнальных каскадов. Таким образом, баланс и переключение с провоспалительной на протекторную функцию NF-κB регулируются сложной многокомпонентной системой связей с учетом локально-временны́х параметров. При всей привлекательности NF-κB в качестве терапевтической мишени, необходимо дальнейшее исследование механизмов его действия с использованием избирательной активации/ингибирования отдельных субъединиц фактора, дальнейшего изучения его клеточной специфичности и приуроченности его активации к определенным повреждающим факторам.
Список литературы
Hayden M.S., Ghosh S. // Genes. Dev. 2004. V. 18. P. 2195–2224.
Lawrence T., Fong C. // Int. J. Biochem. Cell Biol. 2010. V. 42. P. 519–523.
Ghosh S. // Handbook of Transcription Factor NF-κB / Boca Raton: CRC Press by Taylor & Francis Group, 2006.
Gao Z., Hwang D., Bataille F., Lefevre M., York D., Quon M.J., Ye J. // J. Biol. Chem. 2002. V. 277. P. 48115–48121.
Sen R., Baltimore D. // Cell. 1986. V. 47. P. 921–928.
Beg A.A., Sha W.C., Bronson R.T., Ghosh S., Baltimore D. // Nature. 1995. V. 376. P. 167‑170.
Mattson M.P., Camandola S. // J. Clin. Invest. 2001. V. 107. P. 247–254.
Hayden M.S., Ghosh S. // Genes Dev. 2012. V. 26. P. 203–234.
Kaltschmidt B., Widera D., Kaltschmidt C. // Biochim. Biophys. Acta – Mol. Cell Res. 2005. V. 1745. P. 287–299.
Ledoux A.C., Perkins N.D. // Biochem. Soc. Trans. 2014. V. 42. P. 76–81.
Mattson M.P. // Neurochem. Res. 2005. V. 30. P. 883–893.
Gorbacheva L., Strukova S., Pinelis V., Ishiwata S., Stricker R., Reiser G. // Neurochem. Int. 2013. V. 63. P. 101–111.
Gorbacheva L., Pinelis V., Ishiwata S., Strukova S., Reiser G. // Neuroscience. 2010. V. 165. P. 1138–1146.
Fridmacher V., Kaltschmidt B., Goudeau B., Ndiaye D., Rossi F.M., Pfeiffer J., Kaltschmidt C., Israël A., Mémet S. // J. Neurosci. 2003. V. 23. P. 9403–9408.
Herrmann O., Baumann B., De Lorenzi R., Muhammad S., Zhang W., Kleesiek J., Malfertheiner M., Köhrmann M., Potrovita I., Maegele I., Beyer C., Burke J.R., Hasan M.T., Bujard H., Wirth T., Pasparakis M., Schwaninger M. // Nat. Med. 2005. V. 11. P. 1322–1329.
Kaltschmidt B., Kaltschmidt C. // Front. Mol. Neurosci. 2015. V. 8. P. 1–11.
Duckworth E.A.M., Butler T., Collier L., Collier S., Pennypacker K.R. // Brain Res. 2006. V. 1088. P. 167–175.
Widera D., Klenke C., Nair D., Heidbreder M., Malkusch S., Sibarita J.-B., Choquet D., Kaltschmidt B., Heilemann M., Kaltschmidt C. // Neurophotonics. 2016. V. 3. P. 041804.
Okun E., Griffioen K.J., Lathia J.D., Tang S.C., Mattson M.P., Arumugam T. V. // Brain Research Reviews. 2009. V. 59. P. 278–292.
Mettang M., Reichel S.N., Lattke M., Palmer A., Abaei A., Rasche V., Huber–Lang M., Baumann B., Wirth T. // FASEB J. 2018. V. 32. P. 1916–1932.
Lian H., Shim D.J., Gaddam S.S., Rodriguez-Rivera J., Bitner B.R., Pautler R.G., Robertson C.S., Zheng H. // Mol. Neurodegener. 2012. V. 7. P. 1–11.
Dresselhaus E.C., Meffert M.K. // Frontiers in Immunology. 2019. V. 10 P. 1043.
aldwin A.S. // Annu. Rev. Immunol. 1996. V. 14. P. 649–681
Ghosh S., May M.J., Kopp E.B. // Annu. Rev. Immunol. 1998. V. 16. P. 225–260.
Heissmeyer V., Krappmann D., Wulczyn F.G., Scheidereit C. // EMBO J. 1999. V. 18. P. 4766–4778.
Hayakawa M., Miyashita H., Sakamoto I., Kitagawa M., Tanaka H., Yasuda H., Karin M., Kikugawa K. // EMBO J. 2003. V. 22. P. 3356–3366.
Ray P., Zhang D.H., Elias J.A., Ray A. // J. Biol. Chem. 1995. V. 270. P. 10680–10685.
Chen L.W., Egan L., Li Z.W., Greten F.R., Kagnoff M.F., Karin M. // Nat. Med. 2003. V. 9. P. 575–581.
D’Acquisto F., May M.J., Ghosh S. // Mol. Interv. 2002. V. 2. P. 22–35.
Guttridge D.C., Albanese C., Reuther J.Y., Pestell R.G., Baldwin A.S. // Mol. Cell. Biol. 1999. V. 19. P. 5785–5799.
Solan N.J., Miyoshi H., Carmona E.M., Bren G.D., Paya C.V. // J. Biol. Chem. 2002. V. 277. P. 1405–1418.
Baeuerle P.A., Henkel T. // Annu. Rev. Immunol. 1994. V. 12. P. 141–179.
Chen Z.J. // Nature Cell Biology. 2005. V. 7. P. 758–765.
O’Dea E., Hoffmann A. // Wiley Interdiscip. Rev. Syst. Biol. Med. 2009. V. 1. P. 107–115.
Gyrd-Hansen M., Meier P. // Nat. Rev. Cancer 2010. V. 10. P. 561–574.
Oeckinghaus A., Hayden M.S., Ghosh S. // Nat. Immunol. 2011. V. 12. P. 695–708.
Liu X., He F., Pang R., Zhao D., Qiu W., Shan K., Zhang J., Lu Y., Li Y., Wang Y. // J. Biol. Chem. 2014. V. 289. P. 28971–28986.
Wajant H., Scheurich P. // FEBS J. 2011. V. 278. P. 862–876.
Jha P., Das H. // Int. J. Mol. Sci. 2017. V. 18. P. 2383.
Nayak L., Goduni L., Takami Y., Sharma N., Kapil P., Jain M.K., Mahabeleshwar G.H. // Am. J. Pathol. 2013. V. 182. P. 1696–1704.
Perkins N.D., Schmid R.M., Duckett C.S., Leung K., Rice N.R., Nabel G.J. // Proc. Natl. Acad. Sci. USA. 1992. V. 89. P. 1529–1533.
Perkins N.D. // International J. Biochemistry and Cell Biology. 1997. V. 29. P. 1433–1448.
Beg A.A., Baldwin A.S. // Oncogene. 1994. V. 9. P. 1487–1492.
Yeh J.X., Park E., Schultz K.L.W., Griffin D.E. // J. Virol. 2019. V. 93. P. 1–82.
Колпакова А.Ф., Шарипов Р.Н., Колпаков Ф.А. // Сибирское медицинское обозрение 2009. Т. 57.
Skaug B., Jiang X., Chen Z.J. // Annu. Rev. Biochem. 2009. V. 78. P. 769–796.
Huang W.-C., Hung M.-C. // J. Biomed. Sci. 2013. V. 20. P. 1–13.
Diamant G., Dikstein R. // Biochimica et Biophysica Acta – Gene Regulatory Mechanisms. 2013. V. 1829. P. 937–945.
Sizemore N., Lerner N., Dombrowski N., Sakurai H., Stark G.R. // J. Biol. Chem. 2002. V. 277. P. 3863–3869.
Haegeman G. // Eur. Respir. Journal, Suppl. 2003. V. 22. P. 16s–19s.
Guha M., Mackman N. // J. Biol. Chem. 2002. V. 277. P. 32124–32132.
Poletto A.C., Furuya D.T., David-Silva A., Ebersbach-Silva P., Corrêa-Giannella M.L., Passarelli M., Machado U.F. // Mol. Cell. Endocrinol. 2015. V. 401. P. 65–72.
Carta S., Semino C., Sitia R., Rubartelli A. // Front. Immunol. 2017. V. 8. P. 345.
Cai D., Yuan M., Frantz D.F., Melendez P.A., Hansen L., Lee J., Shoelson S.E. // Nat. Med. 2005. V. 11. P. 183–190.
Patel H., McIntire J., Ryan S., Dunah A., Loring R. // J. Neuroinflammation. 2017. V. 14. P. 1–15.
Kim J.E., You D.J., Lee C., Ahn C., Seong J.Y., Hwang J.I. // Cell. Signal. 2010. V. 22. P. 1645–1654.
Yu M., Li H., Liu Q., Liu F., Tang L., Li C., Yuan Y., Zhan Y., Xu W., Li W., Chen H., Ge C., Wang J., Yang X. // Cell. Signal. 2011. V. 23. P. 883–892.
Liu G.H., Qu J., Shen X. // Biochim. Biophys. Acta – Mol. Cell Res. 2008. V. 1783. P. 713–727.
Sivandzade F., Prasad S., Bhalerao A., Cucullo L. // Redox Biology. 2019. V. 21 P. 101059.
Jha N.K., Jha S.K., Kar R., Nand P., Swati K., Goswami V.K. // J. Neurochem. 2019. V. 150. P. 113–137.
Nicolas C.S., Amici M., Bortolotto Z.A., Doherty A., Csaba Z., Fafouri A., Dournaud P., Gressens P., Collingridge G.L., Peineau S. // Jak-Stat. 2013. V. 2. P. e22925.
Guzzo C., Mat N.F.C., Gee K. // J. Biol. Chem. 2010. V. 285. P. 24404–24411.
Deng Z., Jin J., Wang Z., Wang Y., Gao Q., Zhao J. // Int. J. Nanomedicine. 2017. V. 12. P. 3617–3636.
Boutant M., Cantó C. // Mol. Metab. 2014. V. 3. P. 5–18.
Yeung F., Hoberg J.E., Ramsey C.S., Keller M.D., Jones D.R., Frye R.A., Mayo M.W. // EMBO J. 2004. V. 23. P. 2369–2380.
Chen L.F., Greene W.C. // J. Mol. Med. 2003. V. 81. P. 549–557.
Park S.Y., Kim H.Y., Park H.J., Shin H.K., Hong K.W., Kim C.D. // PLoS One. 2016. V. 11. P. e0168286–e0168286.
Meyer K.C. // Inflammation, Advancing Age and Nutrition: Research and Clinical Interventions Elsevier Inc., 2013. P. 29–38.
Harry G.J., Kraft A.D. // Expert Opinion on Drug Metabolism and Toxicology. 2008. V. 4. P. 1265–1277.
Giatti S., Boraso M., Melcangi R.C., Viviani B. // J. Molecular Endocrinology. 2012. V. 49. P. 125–134.
Bradl M., Hohlfeld R. // J. Neurology, Neurosurgery and Psychiatry. 2003. V. 74. P. 1364–1370.
Lyman M., Lloyd D.G., Ji X., Vizcaychipi M.P., Ma D. // Neuroscience Research. 2014. V. 79. P. 1–12.
Shastri A., Bonifati D.M., Kishore U. // Mediators of Inflammation. 2013. V. 2013.
Su F., Bai F., Zhou H., Zhang Z. // Brain, Behavior, and Immunity. 2016. V. 52. P. 187–198.
Han L.P., Li C.J., Sun B., Xie Y., Guan Y., Ma Z.J., Chen L.M. // J. Diabetes Res. 2016. V. 2016.
Thawkar B.S., Kaur G. // J. Neuroimmunol. 2019. V. 326. P. 62–74.
Agalave N.M., Larsson M., Abdelmoaty S., Su J., Baharpoor A., Lundbäck P., Palmblad K., Andersson U., Harris H., Svensson C.I. // Pain 2014. V. 155. P. 1802–1813.
Chen X., Wu S., Chen C., Xie B., Fang Z., Hu W., Chen J., Fu H., He H. // J. Neuroinflammation. 2017. V. 14. P. 1–12.
Lv R., Du L., Liu X., Zhou F., Zhang Z., Zhang L. // Life Sci. 2019. V. 223. P. 158–165.
Fang P., Schachner M., Shen Y.Q. // Mol. Neurobiol. 2012. V. 45. P. 499–506.
Park J.S., Svetkauskaite D., He Q., Kim J.Y., Strassheim D., Ishizaka A., Abraham E. // J. Biol. Chem. 2004. V. 279. P. 7370–7377.
Wang L., Zhang X., Liu L., Yang R., Cui L., Li M. // Neurosci. Lett. 2010. V. 471. P. 152–156.
Zheng C., Liu C., Wang W., Tang G., Dong L., Zhou J., Zhong Z. // Am. J. Transl. Res. 2016. V. 8. P. 5016–5024.
Sun L., Li M., Ma X., Feng H., Song J., Lv C., He Y. // J. Neuroinflammation. 2017. V. 14. P. 231.
Nitsch R., Bechmann I., Deisz R.A., Haas D., Lehmann T.N., Wendling U., Zipp F. // Lancet 2000. V. 356. P. 827–828.
Blamire A.M., Anthony D.C., Rajagopalan B., Sibson N.R., Perry V.H., Styles P. // J. Neurosci. 2000. V. 20. P. 8153–8159.
Swaroop S., Sengupta N., Suryawanshi A.R., Adlakha Y.K., Basu A. // J. Neuroinflammation. 2016. V. 13. P. 1–19.
Sriram K., Matheson J.M., Benkovic S.A., Miller D.B., Luster M.I., O’Callaghan J.P. // FASEB J. 2006. V. 20. P. 670–682.
Ghosh N., Mitra S., Sinha P., Chakrabarti N., Bhattacharyya A. // Neurosci. Res. 2018. V. 137. P. 36–42.
Beyer E.M., MacBeath G. // Mol. Cell. Proteomics. 2012. V. 11. P. 1–14.
Hayden M.S., Ghosh S. // Semin. Immunol. 2014. V. 26. P. 253–266.
Cao J.P., Wang H.J., Yu J.K., Liu H.M., Gao D.S. // Brain Res. Bull. 2008. V. 76. P. 505–511.
Marchetti L., Klein M., Schlett K., Pfizenmaier K., Eisel U.L.M. // J. Biol. Chem. 2004. V. 279. P. 32869–32881.
Kaushal V., Schlichter L.C. // J. Neurosci. 2008. V. 28. P. 2221–2230.
Walsh M.C., Choi Y. // Front. Immunol. 2014. V. 5. P. 511.
Xiu Y., Xu H., Zhao C., Li J., Morita Y., Yao Z., Xing L., Boyce B.F. // J. Clin. Invest. 2014. V. 124. P. 297–310.
Naik E., Dixit V.M. // Journal of Experimental Medicine. 2011. V. 208. P. 417–420.
Bahar E., Kim J.Y., Yoon H. // Int. J. Mol. Sci. 2017. V. 18. P. 1989.
Terazawa R., Akimoto N., Kato T., Itoh T., Fujita Y., Hamada N., Deguchi T., Iinuma M., Noda M., Nozawa Y., Ito M. // Pharmacol. Res. 2013. V. 71. P. 34–43.
Ouyang Y., Kantor D., Harris K.M., Schuman E.M., Kennedy M.B. // J. Neurosci. 1997. V. 17. P. 5416–5427.
Meffert M.K., Chang J.M., Wiltgen B.J., Fanselow M.S., Baltimore D. // Nat. Neurosci. 2003. V. 6. P. 1072–1078.
Speese S.D., Trotta N., Rodesch C.K., Aravamudan B., Broadie K. // Curr. Biol. 2003. V. 13. P. 899–910.
Ninan I., Arancio O. // Neuron. 2004. V. 42. P. 129–141.
Snow W.M., Albensi B.C. // Frontiers in Molecular Neuroscience. 2016. V. 9. P. 118.
Wang R., Chen S., Liu Y., Diao S., Xue Y., You X., Park E.A., Liao F.F. // J. Biol. Chem. 2015. V. 290. P. 22532–22542.
Wang W.Y., Tan M.S., Yu J.T., Tan L. // Annals of Translational Medicine. 2015. V. 3. P. 136.
Camandola S., Mattson M.P. // Expert Opinion on Therapeutic Targets. 2007. V. 11. P. 123–132.
Khasnavis S., Jana A., Roy A., Mazumder M., Bhushan B., Wood T., Ghosh S., Watson R., Pahan K. // J. Biol. Chem. 2012. V. 287. P. 29529–29542.
Listwak S.J., Rathore P., Herkenham M. // Neuroscience 2013. V. 250. P. 282–299.
Lanzillotta A., Porrini V., Bellucci A., Benarese M., Branca C., Parrella E., Spano P.F., Pizzi M. // Front. Neurol. 2015. V. 6.
Meffert M.K., Baltimore D. // Trends in Neurosciences. 2005. V. 28. P. 37–43.
Kaltschmidt B., Ndiaye D., Korte M., Pothion S., Arbibe L., Prüllage M., Pfeiffer J., Lindecke A., Staiger V., Israël A., Kaltschmidt C., Mémet S. // Mol. Cell. Biol. 2006. V. 26. P. 2936–2946.
Mémet S. // Biochem. Pharmacol. 2006. V. 72. P. 1180–1195.
Perkins N.D. // Nat. Rev. Mol. Cell Biol. 2007. V. 8. P. 49–62.
Engelmann C., Weih F., Haenold R. // Neural Regen. Res. V. 2014. 9. P. 707–711.
Helmy A., De Simoni M.G., Guilfoyle M.R., Carpenter K.L.H., Hutchinson P.J. // Prog. Neurobiol. 2011. V. 95. P. 352–372.
Pennypacker K.R., Kassed C.A., Eidizadeh S., O’callaghan J.P. // Brain injury: prolonged induction of transcription factors. // Acta Neurobiol. Exp. 2000, V. 60.
Lattke M., Magnutzki A., Walther P., Wirth T., Baumann B. // J. Neurosci. 2012. V. 32. P. 11511–11523.
Lattke M., Reichel S.N., Magnutzki A., Abaei A., Rasche V., Walther P., Calado D.P., Ferger B., Wirth T., Baumann B. // Mol. Neurodegener. 2017. V. 12. P. 1–20.
Schmeisser M.J., Baumann B., Johannsen S., Vindedal G.F., Jensen V., Hvalby O.C., Sprengel R., Seither J., Maqbool A., Magnutzki A., Lattke M., Oswald F., Boeckers T.M., Wirth T. // J. Neurosci. 2012. V. 32. P. 5688–5703.
Pennypacker K.R., Kassed C.A., Eidizadeh S., Saporta S., Sanberg P.R., Willing A.E. // Exp. Neurol. 2001. V. 172. P. 307–319.
Nonaka M., Chen X.H., Pierce J.E.S., Leoni M.J., McIntosh T.K., Wolf J.A., Smith D.H. // J. Neurotrauma. 1999. V. 16. P. 1023–1034.
Acarin L., González B., Castellano B. // J. Neuropathol. Exp. Neurol. 2000. 59. P. 151–163.
Wang D.B., Kinoshita C., Kinoshita Y., Morrison R.S. // Biochim. Biophys. Acta – Mol. Basis Dis. 2014. 1842. P. 1186–1197.
Mattson M.P., Goodman Y., Luo H., Fu W., Furukawa K. // J. Neurosci. Res. 1997. V. 49. P. 681–697.
Qin Z.H., Tao L.Y., Chen X. // Acta Pharmacol. Sin. 2007. V. 28. P. 1859–1872.
Yang L., Tao L.Y., Chen X.P. // Neurosci. Bull. 2007. V. 23. P. 307–313.
Brambilla R., Bracchi-Ricard V., Hu W.H., Frydel B., Bramwell A., Karmally S., Green E.J., Bethea J.R. // J. Exp. Med. 2005. V. 202. P. 145–156.
Jayakumar A.R., Tong X.Y., Ruiz-Cordero R., Bregy A., Bethea J.R., Bramlett H.M., Norenberg M.D. // J. Neurotrauma 2014. V. 31. P. 1249–1257.
Lagraoui M., Sukumar G., Latoche J.R., Maynard S.K., Dalgard C.L., Schaefer B.C. // Brain. Behav. Immun. 2017. V. 61. P. 96–109.
Tamatani M., Mitsuda N., Matsuzaki H., Okado H., Miyake S.I., Vitek M.P., Yamaguchi A., Tohyama M. // J. Neurochem. 2000. V. 75. P. 683–693.
Zhang H., Zhang D., Li H., Yan H., Zhang Z., Zhou C., Chen Q., Ye Z., Hang C. // Int. J. Mol. Med. 2018. V. 41. P. 3203–3210.
Nijboer C.H., Heijnen C.J., Groenendaal F., May M.J., Van Bel F., Kavelaars A. // Stroke 2008. V. 39. P. 2578–2586.
Pizzi M., Goffi F., Boroni F., Benarese M., Perkins S.E., Liouand H.C., Spano P. // J. Biol. Chem. 2002. V. 277. P. 20717–20723.
Delekta P.C., Apel I.J., Gu S., Siu K., Hattori Y., McAllister-Lucas L.M., Lucas P.C. // J. Biol. Chem. 2010. V. 285. P. 41432–41442.
Дополнительные материалы отсутствуют.