

Нейрохимия, 2022, T. 39, № 1, стр. 80-88
Экспрессия генов регуляции синаптической пластичности в гиппокампе и пространственное обучение у крыс разного возраста в условиях моделирования стрептозотоцинового диабета
Г. В. Карантыш 1, А. М. Менджерицкий 2, В. Н. Прокофьев 1, О. В. Лянгасова 1, М. П. Фоменко 1
1 ФГАОУ ВО “Южный федеральный университет”
Ростов-на-Дону, Россия
2 ФГБОУ ВО “Донской государственный технический университет”
Ростов-на-Дону, Россия
Поступила в редакцию 01.01.2021
После доработки 21.07.2021
Принята к публикации 21.08.2021
- EDN: XNFXDK
- DOI: 10.31857/S1027813322010071
Аннотация
В данном исследовании представлены результаты сравнительного анализа экспрессии генов регуляции синаптической пластичности, способности к пространственному обучению и сохранению выработанного навыка у животных 3-х и 18-месячного возраста в модели стрептозотоцинового диабета. Оценку пространственного обучения проводили в лабиринте Морриса. В гиппокампе исследовали относительный уровень траскриптов генов c-fos, c-jun, Egr-1 и Cyt-c с использованием метода полимеразной цепной реакции в реальном времени. На 3-й день тестирования в водном лабиринте Морриса время выхода на платформу у 18-месячных крыс превышало значения 3-месячных животных, как и после моделирования стрептозотоцинового диабета. Уровни экспрессии генов c-fos, c-jun и Egr-1 у животных 3-месячного возраста повышались как в ответ на выработку пространственного обучения, так и моделирование диабета, у 18-месячных крыс наблюдали снижение уровня экспрессии данного гена в модели стрептозотоцинового диабета. В обеих группах отмечали повышение уровня экспрессии гена Cyt-c в модели диабета относительно контроля, но у 18-месячных животных выявлено более выраженное увеличение данного показателя.
ВВЕДЕНИЕ
Сахарный диабет (СД) представляет собой серьезную проблему современного общества. Распространенность сахарного диабета во всем мире резко возросла за последние годы и составляет 422 млн человек [1]. Риск заболеваемости СД увеличивается с возрастом [2]. Развивающийся при этом метаболический синдром приводит к полиогранным нарушениям, формирующимися на фоне ожирения, гипертонии, гипергликемии, дислипидемии и хронического окислительного стресса [3–6 и др.]. СД повышает риск сердечно-сосудистых заболеваний, деменции, способствует снижению продолжительности жизни и т.д. [7–9 и др.].
Механизмы, лежащие в основе развития деменции у больных сахарным диабетом, исследованы недостаточно, хотя о связи, например, болезни Альцгеймера (БА) с сахарным диабетом сообщают уже на протяжении нескольких десятилетий. Известно, что сахарный диабет провоцирует развитие микроангиопатий на фоне окислительного стресса и нейровоспалительных процессов [10–12 и др.]. Также при нейродегенеративных процессах нарушается регуляция клеточного цикла, что вместе с окислительным стрессом инициирует патологический каскад реакций в гиппокампе при БА [13–15 и др.].
В условиях СД изменяется экспрессия генов в клетках мозга, что приводит к дисфункции нейронов и глиальных клеток на уровне нарушений передачи сигнала и клеточного цикла, связанных с развитием деменции и болезни Альцгеймера [16]. В процессе старения снижается способность к обучению, хотя молекулярные механизмы, лежащие в основе дефицита памяти, до сих пор не до конца исследованы. Также не до конца определены механизмы, лежащие в основе снижения способности к сохранению ранее выработанных навыков в условиях развития сахарного диабета.
Целью данного исследования явилось выявление возрастных изменений способности к пространственному обучению и сохранности выработанного навыка, а также сравнение экспрессии генов регуляции синаптической пластичности (c-fos, c-jun, Egr-1 и Cyt-c) в гиппокампе у 3-х и 18-месячных крыс в модели стрептозотоцинового диабета.
МЕТОДЫ ИССЛЕДОВАНИЯ
Все экспериментальные процедуры проводились в соответствии с директивами Европейского Союза 86/609/ЕЕС по использованию экспериментальных животных и местным законодательством по этике экспериментов на животных. Протоколы для животных были оценены и одобрены Комитетом по уходу и использованию животных Южного федерального университета (разрешение № 02/2014, 12 февраля 2014 г.).
Эксперимент проведен на беспородных белых крысах-самцах 3-х и 18-месячного возраста. Крыс делили на группы: 1 – контрольная (оценивали только уровень экспрессии генов), 2 – моделирование пространственного обучения в лабиринте Морриса (ПО), 3 – моделирование стрептозотоцинового диабета, 4 – моделирование стрептозотоцинового диабета и пространственного обучения (диабет + ПО). Количество животных в каждой группе представлено в табл. 1, дизайн эксперимента – в табл. 2.
Таблица 1.
Количество животных в экспериментальных группах
| Группы животных | 3-месячные крысы | 18-месячные крысы | |
|---|---|---|---|
| Контроль (К) | n = 8 | n = 8 | |
| Пространственное обучение (ПО) | 3 сутки эксперимента (ПО_3) | n = 8 | n = 6 |
| 24 сутки эксперимента (ПО_24) | n = 8 | n = 6 | |
| Стрептозотоциновый диабет (диабет) | n = 8 | n = 8 | |
| Пространственное обучение + стрептозотоциновый диабет + (ПО+диабет) | 24 сутки эксперимента (ПО+диабет_24) |
n = 8 | n = 8 |
Таблица 2.
Дизайн эксперимента
| Группа УР (выработка условного рефлекса) |
Основная группа |
|---|---|
| Тестирование в лабиринте Морриса (1–3 дни эксперимента) (n = 24) | |
| + | + |
| Модель стрептозотоцинового диабета (4–24 дни эксперимента) (n = 40) | |
| – | + |
| Оценка сохранности выработанного навыка в лабиринте Морриса (24 день эксперимента) (n = 32) | |
| + | + |
Экспериментальный диабет вызывали путем однократного введения крысам раствора стрептозотоцина (Sigma) в 0.4 мл цитратного буфера в дозе 50 мг/кг массы тела после 18-часового голодания [17]. На 21 сут после введения стрептозотоцина проводили декапитацию; в крови определяли уровень гликозилированного гемоглобина (HbA1, %) с использованием анализатора DS5 Glycomat (Великобритания).
Изучение пространственного обучения проводили с использованием лабиринта Морриса [18], представляющего круглый бассейн диаметром 100 см, заполненного непрозрачной белой водой (t = 24–25°С) с разведенным сухим молоком. В центре лабиринта располагали платформу из оргстекла площадью 25 см2, погруженную на 5 см ниже поверхности воды. Платформа находилась в одном и том же положении в бассейне на всем протяжении эксперимента. Вокруг бассейна помещали два черно-белых рисунка на плотной бумаге (визуальные ориентиры), которые располагали в одинаковом положении вокруг бассейна в течении всего эксперимента. При каждом тестировании животное помещали в лабиринт Морриса из разных положений относительно визуальных ориентиров. Тестирование проводили 3 дня подряд: максимальное нахождение в лабиринте Морриса составляло 120 с. После нахождения платформы животное извлекали из бассейна, вручную обсушивали полотенцем и помещали в отдельную клетку с подогревом. На 24 день после первого тестирования у крыс оценивали сохранность пространственной памяти. Учитывали средние показатели времени трех повторов тестирования в водном лабиринте Морриса на 1-й, 2-й, 3-й и 24-й дни эксперимента (между каждым тестом в день эксперимента было не менее 1.0 ч). Критерием обучения служило снижение времени выхода на платформу относительно показателя в 1-й день тестирования в лабиринте Морриса.
Статистическую обработку результатов исследования пространственного обучения проводили с использованием программы Stаtistiсa 10.0 (Statsoft, США). Проверку на нормальность распределения экспериментальных данных проводили с использованием критерия Колмагорова-Смирнова. Для оценки межгрупповых различий использовали t-критерий Стьюдента. Данные представлены в виде средних значений и стандартных ошибок среднего (M ± SEM). Достоверными считали различия при уровне значимости р < 0.05.
Относительный уровень траскриптов изучаемых генов исследовали с использованием метода полимеразной цепной реакции в реальном времени (ПЦР-РВ) после обратной траскрипции.
Образцы тканей (гиппокампа) крыс забирали через 1 ч после последнего тестирования в лабиринте Морриса, мгновенно замораживали в жидком азоте и хранили при –80°C. Количество анализируемых образцов в каждой группе соответствовало количеству животных (табл. 1).
Чтобы избежать деградации, образцы размораживали только один раз. Тотальную РНК из тканей коры и гиппокампа выделяли при помощи набора Aurum Total RNA fatty and fibrous tissue kit (BioRad). Количество и чистоту выделенной РНК оценивали спектрофотометрически на приборе NanoPhotometer (Implen). OD260/280 образцов РНК колебалось от 1.80 до 2.00. Тотальную РНК (0.5 мкг) подвергали обратной транскрипции в кДНК, в объеме смеси 25 мкл. Реакцию обратной транскрипции проводили c использованием реагентов фирмы Евроген (Россия), в соответствии с инструкцией производителя при 40°С в течение 40 мин, после чего инактивировали при 92°С в течение 10 мин. Дизайн прямых и обратных праймеров для проведения ПЦР-РВ проводили в программе Primer3 с последующей проверкой в IDT. Праймеры были синтезированы фирмой Синтол (Россия). Последовательности праймеров приведены в табл. 3. ПЦР-РВ была проведена на амплификаторе CFX-96 фирмы Bio-Rad c использованием готовой реакционной смеси iQ BioRad в объеме 25 мкл, включая по 50 пмоль прямого и обратного праймеров и 1 мкл кДНК. Условия для проведения ПЦР: первичную денатурацию проводили при 95°С 3 мин. Проводили 35 циклов амплификации: 20 с при 95°С, 10 с при 60°С, 20 с при 72°С. Для проверки специфичности после окончания циклов амплификации строили кривую плавления от 65 до 95°С с шагом 0.5°С. В качестве красителя использовали SYBR Green I. Реакцию амплификации для каждого исследуемого гена проводили отдельно в дупликатах. В качестве референсного гена был использован ген HPRT1, кодирующий фермент гипоксантин фосфорибозилтрансферазу 1.
Таблица 3.
Последовательности праймеров
| Ген | Прямой праймер | Обратный праймер | t плавления, °C |
|---|---|---|---|
| EGR-1 | Сac-ctg-acc-aca-gag-tcc-tttt | Aag-tgt-tgc-cac-tgt-tgg-gt | 60 |
| c-fos | Ggc-aga-agg-ggc-aaa-gta-ga | Agt-tga-tct-gtc-tcc-gct-tgg | 60 |
| c-jun | Ctc-cgg-gct-gtt-cat-ctg-tt | Ccg-gga-ctt-gtg-agc-ttc-tt | 60 |
| Cyt c | Ctt-ggg-cta-gag-agc-ggg-a | Tta-aat-tcg-gtc-cgg-gct-gg | 60 |
| HPRT1 | Tcc-tcc-tca-gac-cgc-ttt-tc | Atc-act-aat-cac-gac-gct-ggg | 60 |
Расчет относительной экспрессии проводили, оценивая величины Ct (Threshold cycle), получаемые в ходе проведения ПЦР-РВ. Все данные представлены в виде относительных единиц (отн. ед.) уровня мРНК изучаемых генов в сравнении с референсным геном (HPRT1), расчет которых проводили по отношению к контрольной группе с использованием метода 2–ΔΔCt. Статистическую обработку результатов исследования экспрессии генов проводили с помощью программы Statistica 10.0 с использованием критериев Колмогорова–Смирнова и Ливиня, а также с помощью однофакторного дисперсионного анализа ANOVA. Статистически значимыми считали различия при p < 0.05.
РЕЗУЛЬТАТЫ ИССЛЕДОВАНИЯ
Животных, у которых уровень HbA1 после моделирования стрептозотоцинового диабета оставался в пределах контрольных значений, исключали из эксперимента: пять крыс 3-месячного возраста и три крысы 18-месячного возраста. Анализ результатов исследования уровня HbA1 показал, что данный показатель после моделирования диабета был значительно выше исходных значений как у 3-месячных, так и, особенно, 18-месячных крыс (p < 0.001) (табл. 4).
Таблица 4.
Уровень HbA1 (%) в крови у 3-х и 18-месячных крыс в модели стрептозотоцинового диабета, M ± SEM
| группы | 3-месячные крысы | 18-месячные крысы |
|---|---|---|
| Контрольная | 6.68 ± 0.29 (n = 8) | 8.65 ± 0.38*# (n = 8) |
| Основная | 18.54 ± 1.37* (n = 15) | 24.48 ± 1.79*# (n = 17) |
В динамике трех дней тестирования в лабиринте Морриса было установлено снижение времени платформы как у 3-месячных, так и 18-месячных животных. Достоверных различий показателей времени выхода на платформу в каждый день тестирования на этапе обучения в группе 3-месячных животных не установлено, как и в группе 18-месячных крыс (табл. 5).
Таблица 5.
Время выхода на платформу (с) у 3-х и 18-месячных крыс в модели стрептозотоцинового диабета, M ± SEM
| Дни эксперимента/возраст крыс | 3-месячные | 18-месячные | |
|---|---|---|---|
| Пространственное обучение | 1 | 18.48 ± 1.02 | 25.87 ± 1.43# |
| 2 | 11.63 ± 0.64* | 17.52 ± 0.94*# | |
| 3 | 6.35 ± 0.37* | 8.43 ± 0.48*# | |
| 24 | 8.76 ± 0.45* | 13.69 ± 0.84*# | |
| Пространственное обучение + диабет | 1 | 17.59 ± 0.93 | 23.85 ± 1.16# |
| 2 | 12.06 ± 0.71* | 15.39 ± 0.82*# | |
| 3 | 6.71 ± 0.39* | 8.10 ± 0.41* | |
| 24 | 12.85 ± 0.69*§ | 26.98 ± 0.96# | |
* достоверные отличия времени выхода на платформу относительно 1 дня эксперимента; § достоверные отличия показателей в группе крыс в модели пространственного обучения и стрептозотоцинового диабета относительно значений в группе животных, которым моделировали только пространственное обучение (при p < 0.05), # достоверные отличия показателей в группах 18-месячных животных относительно значений в группах 3-месячных крыс (при p < 0.05)
В контрольной группе на 3-й день тестировании в лабиринте Морриса среднее значение времени выхода на платформу у 18-месячных крыс было на 32% (p < 0.05) выше относительно 3-месячных животных, а в модели ПО + диабет_3 – на 21% (0.05 < p < 0.1). Сходная динамика показателей времени выхода на платформу в ходе пространственного обучения в обеих группах (контроль и ПО + диабет_3) объясняется тем, что моделирование диабета во втором случае осуществляли после 3-х дней тестирования в лабиринте Морриса.
В результате проведения оценки сохранности пространственного обучения в лабиринте Морриса установлено, что на 24-й день эксперимента у животных 3-месячного возраста контрольной группы и в модели ПО + диабет_24 время выхода на платформу увеличивалось на 38% (p < 0.05) и 92% (p < 0.05), соответственно. У 18-месячных крыс наблюдали еще более значительное возрастание времени выхода на платформу относительно значений на 3 день эксперимента: в контрольной группе увеличение данного показателя составило 74% (p < 0.01), а в модели ПО + диабет_24 – на 233% (p < 0.0001) (табл. 5). Это подтверждает существующее представление о том, что с возрастом снижается способность к обучению, в частности к пространственному обучению [19].
Анализ экспрессии генов регуляции синаптической пластичности показал, что на 3-й день эксперимента у 3-месячных крыс происходило возрастание уровня экспрессии гена c-fos (рис. 1). Ген c-fos связан с различными сигнальными каскадами, участвующими в процессах пластичности нейронов, роста клеток и митозе. При этом уровень экспрессии данного гена был выше контрольных значений у 3-месячных крыс и на 24-й день эксперимента, а также в модели ПО + диабет_24. У 18-месячных крыс наблюдали сниженные значения относительной экспрессии гена c-fos по сравнению с 3-месячными животными; после моделирования стрепотозотоцинового диабета и, в меньшей степени, в модели ПО + диабет_24 у 18‑месячных животных установлено достоверное уменьшение уровня экспрессии данного гена относительно контрольных показателей у животных этого возраста. Повышение экспрессии гена c-fos в гиппокампе после тестирования в водном лабиринте Морриса, а также в модели ПО + диабет_24 у 3-месячных животных в отличии от 18-месячных крыс может отражать возрастные различия уровня экспрессии данного гена в ответ на предъявление стрессовых воздействий разной интенсивности. Следует отметить, что у 3-месячных в модели ПО + диабет_24 время выхода на платформу было ниже значения в первый день обучения, что наблюдали на фоне роста уровня экспрессии гена c-fos. В тоже время у 18-месячных животных время выхода на платформу в модели ПО + диабет_24 не отличалось от показателя на 1-й день тестирования в лабиринте Морриса, и у этих животных уровень экспрессии гена c-fos был ниже контрольных показателей (табл. 5, рис. 1).
Рис. 1.
Относительная экспрессия гена c-fos в гиппокампе у 3-х (n = 40) и 18-месячных крыс (n = 36) в модели стептозотоцинового диабета. Примечание: К – контрольная группа; ПО – группа животных в модели пространственного обучения (ПО_3 – 3-й день эксперимента, ПО_24 – 24-й день эксперимента); диабет – модель стрептозотоцинового диабета (24 день эксперимента); ПО + диабет_24 – группа крыс в модели стрептозотоцинового диабета после пространственного обучения (24 день эксперимента). * достоверные отличия показателей относительно контрольных значений; § достоверные отличия показателей относительно значений в группе УР_3; # достоверные отличия у 18-месячных животных показателей относительно значений у 3-месячных крыс (при р < 0.05).
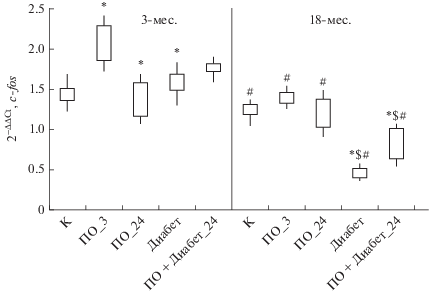
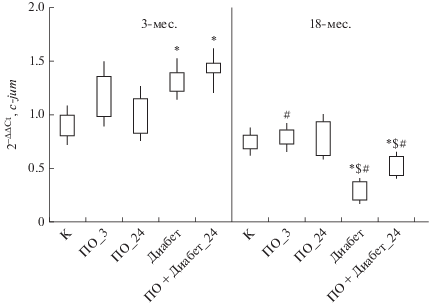
Известно, что ген c-fos кодирует ядерный фосфопротеин Fos, который образует димерный комплекс с белком Jun, имеющим участки связывания последовательности ДНК для белка-активатора фактора транскрипции-1 (AP-1) [20]. При изучении уровня экспрессии гена c-jun установлено, что у 3-месячных животных данный показатель значительно повышается в моделях стрептозотоцинового диабета и ПО + диабет_24, тогда как у 18-месячных крыс, напротив, в данных моделях, особенно, в модели стрептозотоцинового диабета, уровень его экспрессии снижается относительно контрольных значений. Также показано, что у этих крыс уровень экспрессии гена c-jun снижен на 3-й день эксперимента по сравнению с 3-месячными животными (рис. 2).
Рис. 2.
Относительная экспрессия гена c-jun в гиппокампе у 3-х (n = 40) и 18-месячных крыс (n = 36) в модели стептозотоцинового диабета. Примечание: см. рис. 1.
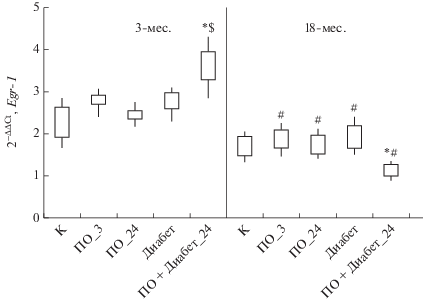
Анализ результатов исследования экспрессии гена Egr-1, связанного с синаптической активностью зрелых нейронов, индуктором транскрипции которого является активация НМДА-рецепторов [21], показал следующее. У 3-х и 18-месячных крыс изменение уровня экспрессии данного гена выявлено только в модели ПО + диабет_24: у 3-месячных животных наблюдали повышение экспрессии Egr-1, а у животных 18-месячного возраста, напротив, ее снижение по сравнению с контрольными значениями. Также установлено, что после тестирования в лабиринте Морриса и в модели ПО + диабет_24 у 18-месячных крыс уровень экспрессии был ниже относительно 3-месячных животных в этих же экспериментальных моделях (рис. 3).
Рис. 3.
Относительная экспрессия гена Egr-1 в гиппокампе у 3-х (n = 40) и 18-месячных крыс (n = 36) в модели стептозотоцинового диабета. Примечание: см. рис. 1.
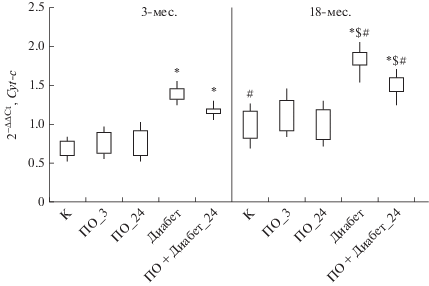
При изучении уровня экспрессии гена Cyt-c, кодирующего белок цитохром с, наблюдали повышение его экспрессии в гиппокампе у 3-х и, особенно, 18-месячных животных в модели ПО + диабет_24 и, особенно, в модели стрептозотоцинового диабета. Также установлено, что у 18-месячных крыс уровень экспрессии данного гена превышал значения у 3-месячных животных как в контрольной группе, так и в модели ПО + диабет_24 (рис. 4). Повышение экспрессии гена Cyt-c, как известно, усиливает активность каспаз-3 и -9, что сопровождается активацией апоптоза клеток мозга, а также выходом цитохрома-с из мембран и развитием окислительного стресса [22].
Рис. 4.
Относительная экспрессия гена Cyt-c в гиппокампе у 3-х (n = 40) и 18-месячных крыс (n = 36) в модели стептозотоцинового диабета. Примечание: см. рис. 1.
Таким образом, у 18-месячных крыс снижение обучаемости в лабиринте Морриса, а также нарушение сохранности ранее выработанного пространственного обучения, особенно, при моделировании стрептозотоцинового диабета, сопровождается изменением экспрессии генов регуляции синаптической пластичности относительно 3-месячных животных.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ ИССЛЕДОВАНИЯ
Механизмы, происходящие в мозге при развитии сахарного диабета, остаются недостаточно исследованными. В настоящее время известны следующие факторы, влияющие на функции инсулина в мозге. Инсулин способен проникать через гематоэнцефалический барьер (ГЭБ) [23, 24] с помощью переносчика. Данный процесс является регулируемым, зависит от уровня периферического инсулина: если его уровень повышается, это приводит к росту уровня инсулина в спиномозговой жидкости, но в условиях периферической гиперинсулинемии наблюдается подавление связывания рецепторов инсулина в клетках ГЭБ с инсулином, что препятствует транспорту инсулина в мозг [25, 26]. Периферический инсулин может проникать в ЦНС и непосредственно через area postrema и околожелудочковую область, что обеспечивает свободную диффузию растворимых в плазме веществ непосредственно в эту область [27].
В мозге выявлено множество сигнальных путей инсулина, которые принимают участие в регуляции метаболизма мозга, дифференцировки и роста нейронов, а также нейромодуляции [28]. При недостаточности инсулина в мозге может приводить к нейрональной гибели путем апоптоза [29]. Таким образом, эти данные, а также тот факт, что недостаточность инсулина может быть связана с развитием болезни Альцгеймера, доказывают участие инсулина в пластических процессах, связанных с поведением.
В настоящее время насчитывают семь различных путей инициации болезни Альцгеймера в результате развития сахарного диабета. Один из них – это повышение активности инсулин-деградирующего фермента в областях мозга, чувствительных к инсулину, в том числе, в гиппокампе [30]. Данный фермент также способен снижать экспрессию белков, участвующих в синаптической пластичности [31]. В то же время повышение уровня инсулиноподобного фактора роста может приводить к развитию окислительного стресса [32] в тканях мозга и как результат, стать одним из факторов накопления бета-амилоида [24].
В последние десятилетия открытие инсулиновых рецепторов в областях мозга, связанных с обучением и памятью, привело к формированию понимания роли инсулина в процессе старения мозга и развитии нейродегенеративных заболеваний, и, таким образом, о его влиянии на пластические процессы [33, 34].
Модификация активности синапсов и изменение их количества является функциональным и структурным субстратом для пластических процессов, лежащих в основе обучения и памяти, а также закрепления поведенческих изменений [35]. Стимуляция инсулином нейронов гиппокампа вызывает как пресинаптические, так и постсинаптические эффекты. Инсулин повышает высвобождение нейротрансмиттеров из пресинаптических терминалей [36], поэтому в условиях недостатка инсулина в мозге, как например, при сахарном диабете, возможно снижение способности к обучению.
В моделях диабета, чаще всего индуцированных стрептозоцином, у животных, выявляется незначительное снижение объема памяти [37, 38]. Это подтверждается полученными в данном исследовании результатами. Наиболее выражено снижение способности сохранять ранее выработанный навык установлен у 18-месячных крыс. Это сопровождается изменением уровня экспрессии генов регуляторов синаптической пластичности.
Снижение уровней экспрессии ранних генов c-fos и c-jun, связанных с сигнальными каскадами, участвующими в процессах пластичности нейронов, роста клеток и митозе, установлено у 18-месячных крыс, особенно, в модели ПО + диабет_24. Семейство белков Jun и родственные им Fos-белки образуют многочисленное количество комбинаций, контролирующих поздние гены и белки, связанные с процессами памяти, моторики и обучения [39]. Гены c-fos и c-jun являются важными компонентами взаимодействий между генами и стимулами из окружающей среды, поскольку они обеспечивают молекулярную основу для быстрого и динамического ответа нейронов и открывают возможность для длительной и устойчивой адаптации посредством регуляции экспрессии широкого спектра поздних генов. Таким образом, ранние гены тесно связаны с изменением активности нейронов, а также с различными процессами, регулируемыми центральной нервной системой, такими как обучение, память и т.д. [40]. Тем не менее, регуляция экспрессии этих двух генов может различаться при действии разных факторов [41]. В данном исследовании у крыс, которых тестировали в лабиринте Морриса, изменения уровня экспрессии генов c-fos и c-jun у 3‑месячных крыс не были сонаправлены: возрастание уровня экспрессии гена c-fos происходило на фоне отсутствия изменений экспрессии гена c-jun. В тоже время у 18-месячных животных в модели ПО + диабет_24 снижалась экспрессия обоих генов.
Разнонаправленное изменение экспрессии гена Egr-1 у 3-х и 18-месячных животных может отражать возрастные изменения его регуляции в условиях одинаковых внешних воздействий на организм. Согласно существующим представлениям экспрессия гена Egr-1 может активироваться при большом разнообразии стимулов, что отражается в его повышающей регуляции после увеличения уровня внутриклеточного кальция в нейронах гиппокампа. Это дало основание предполагать, что возрастание уровня экспрессии этого гена в структурах мозга в выше перечисленных экспериментальных моделях может лежать в основе повышения нейрональной активности, нейроадаптационных реакций, лежащих в основе обучения и памяти, а также реакции на стресс [42, 43]. Кроме того, известно, что электрическая стимуляция нейронов, приводящая к развитию длительной потенциации, способствует возрастанию уровня мРНК Egr-1, индуктором экспрессии которой является активация НМДА-рецепторов [21]. Поэтому предполагают, что у экспериментальных животных, у которых в структурах мозга наблюдается возрастание экспрессии данного гена происходит активация НМДА-рецепторов [44].
Повышение уровня экспрессии гена Cyt-c в гиппокампе у 18-месячных крыс, особенно, в модели ПО + диабет_24 предположительно может сопровождается и ростом уровня цитозольного цитохрома с, который играет ключевую роль в активации апоптоза нейронов [45] через запуск каскада каспаз. При этом показано, что инсулин способен подавлять высвобождение цитохрома с из митохондрий посредством PI3-K-зависимого пути и оказывать нейропротекторные эффекты в условиях окислительного стресса [46].
Таким образом, в основе возрастных изменений обучения и памяти лежат молекулярные механизмы, регуляция которых, вероятно, изменяется на фоне развивающегося окислительного стресса в структурах мозга и сопутствующих патологий, в частности, развития сахарного диабета.
ЗАКЛЮЧЕНИЕ
Способность к пространственному обучению и сохранности выработанного навыка в лабиринте Морриса выше у 3-месячных крыс относительно 18-месячных животных. В модели стрептозотоцинового диабета после обучения в лабиринте Морриса снижение сохранности выработанного навыка у 18-месячных крыс наблюдается на фоне значительного повышения экспрессии гена Cyt-c и понижении экспрессии генов c-fos, c-jun и Egr-1.
Список литературы
Global report on diabetes. World Health Organization 2016. (Globally, an estimated 422 million adults were living with diabetes in 2014, compared to 108 million in 1980. The global prevalence (age-standardized) of diabetes has nearly doubled since 1980, rising from 4.7% to 8.5% in the adult population.)
Dal Canto E., Ceriello A., Rydén L., Ferrini M., Hansen T.B., Schnell O., Standl E., Beulens J.W. // Eur. J. Prev. Cardiol. 2019. V. 26. Suppl 2. P. 25–32.
Feldman E.L., Callaghan B.C., Pop-Busui R., Zochodne D.W., Wright D.E., Bennett D.L., Bril V., Russell J.W., Viswanathan V. // Nat Rev Dis Primers. 2019. V. 5. № 1. e41.
Hedayati N., Bemani N.M., Mohammadinejad A., Mohajeri S.A. // Phytother. Res. 2019. V. 33. № 12. P. 3040–3053.
Huang X., Liu G., Guo J., Su Z. // Int. J. Biol. Sci. 2018. V. 14. № 11. P. 1483–1496.
Verhulst M.J.L., Loos B.G., Gerdes V.E.A., Teeuw W.J. // Front. Endocrinol. (Lausanne). 2019. V. 10. e56.
Bharadwaj P., Wijesekara N., Liyanapathirana M., Newsholme P., Ittner L., Fraser P., Verdile G. // J. Alzheimers Dis. 2017. V. 59. P. 421–432.
Sutherland G.T., Lim J., Srikanth V., Bruce D. // J. Alzheimers Dis. 2017. V. 59. P. 393–403.
Batkulwar K., Godbole R., Banarjee R., Kassasr O., Williams R., Kulkarni M. // ACS Chem. Neurosci. 2018. V. 9. P. 988–1000.
Pugazhenthi S., Qin L., Reddy P.H. // Biochim Biophys Acta Mol. Basis Dis. 2017. V. 1863. № 5. P. 1037–1045.
Umeno A., Biju V., Yoshida Y. // Free Radic. Res. 2017. V. 51(4). P. 413–427.
Sun Y., Ma C., Sun H., Wang H., Peng W., Zhou Z., Wang H., Pi C., Shi Y., He X. // J. Diabetes Res. 2020. V. 2020. e4981814.
Webber K.M., Raina A.K., Marlatt M.W., Zhu X., Prat M.I., Morelli L., Casadesus G., Perry G., Smith M.A. // Mech. Ageing Dev. 2005. V. 126. № 10. P. 1019–1025.
Koseoglu M.M., Norambuena A., Sharlow E.R., Lazo J.S., Bloom G.S. // J. Alzheimers Dis. 2019. V. 67. 1. P. 1–11.
Ippati S., Deng Y., van der Hoven J., Heu C., van Hummel A., Chua S.W., Paric E., Chan G., Feiten A., Fath T., Ke Y.D., Haass N.K., Ittner L.M. // Proc. Natl. Acad. Sci. USA. 2021. V. 118. № 12. e2011876118.
Bury J.J., Chambers A., Heath P.R., Ince P.G., Shaw P.J., Matthews F.E., Brayne C., Simpson J.E., Wharton S.B. // Cognitive Function and Ageing Study. Acta Neuropathol. Commun. 2021. V. 9. № 1. e5.
Szkudelski T. // Ibed. 2001. V. 50. № 6. P. 536–546.
Morris R.G. // J. Neurosci. Meth. 1984. V. 11. P. 47–60.
Coppola V.J., Bingman V.P. // Neurobiol. Aging. 2020. V. 87. P. 98–107.
Barros V.N., Mundim M., Galindo L.T., Bittencourt S., Porcionatto M., Mello L.E. // Front. Cell. Neurosci. 2015. V. 9. e72.
Qin X., Jiang Y., Tse Y.C., Wang Y., Wong T.P., Paudel H.K. // J. Biol. Chem. 2015. V. 290. № 49. P. e29603–e29616.
Jiang W., Chen Y., Li B., Gao S. // Mol. Biosyst. 2017. V. 13(9). P. 1863–1873.
Laron Z. // Archives of Physiology and Biochemistry. 2009. V. 115. № 2. P. 112–116.
Mittal Kh., Mani R.J., Katare D.P. // Sci. Rep. 2016. V. 6. № 6. e25589.
Moreira P. I., Duarte A. I., Santos M. S., Rego A. C., Oliveira C. R. // J. Alzheimer’s Disease. 2009. V. 16. № 4. P. 741–761.
Duarte A.I., Moreira P.I., Oliveira C.R. // J. Aging Research. 2012. V. 2021. e384017.
Sankar R., Thamotharan S., Shin D., Moley K.H., Devaskar S.U. // Molecular Brain Research. 2002. V. 107. № 2. P. 157–165.
Gasparini L., Xu H. // Trends in Neurosciences. 2003. V. 26(8). P. 404–406.
Huang T.J., Verkhratsky A., Fernyhough P. // Molecular and Cellular Neuroscience. 2005. V. 28. № 1. P. 42–54.
Ren H. // Mol. Metab. 2014. V. 3. P. 452–459.
Ding L., Becker A.B., Suzuki A., Roth R.A. // J. Biol. Chem. 1992. V. 267. P. 2414–2420.
Lin T., Wang D., Nagpal M. L., Chang W., Calkins J.H. // Endocrinology. 1992. V. 130. P. 1217–1224.
Mainardi M., Fusco S., Grassi C. // Neural Plast. 2015. V. 2015. e657928.
Kullmann S., Heni M., Hallschmid M., Fritsche A., Preissl H., Haring H.U. // Physiol. Rev. 2016. V. 96. P. 1169–1209.
Nakahata Y., Yasuda R. // Front. Synaptic Neurosci. 2018. V. 10. e29.
Lee C.C., Huang C.C., Hsu K.S. // Neuropharmacology. 2011. V. 61. P. 867–879.
Biessels G.J., Kamal A., Urban I.J., Spruijt B.M., Erkelens D.W., Gispen W.H. // Brain Res. 1998. V. 800. P. 125–135.
Popovic M., Biessels G.J., Isaacson R.L., Gispen W.H. // Behav. Brain Res. 2001. V. 122. P. 201–207.
Varela P., Escosteguy-Neto J.C., Coelho C.T., Mello L.E., da Silveira D.X., Santos-Junior J.G. // Int. J. Neuropsychopharmacol. 2014. V. 17. № 11. P. 1815–1830.
Bahrami S., Drabløs F. // Adv. Biol. Regul. 2016. V. 62. P. 37–49.
Knox D., Stanfield B.R., Staib J.M., David N.P., DePietro T., Chamness M., Schneider E.K., Keller S.M., Lawless C. // Behav. Brain Res. 2018. V. 341. P. 189–197.
Hendrickx A., Pierrot N., Tasiaux B., Schakman O., Kienlen-Campard P., De Smet C., Octave J.-N. // PLoS One. 2014. V. 9. e99467.
Rusconi F., Grillo B., Ponzoni L., Bassani S., Toffolo E., Paganini L., Mallei A., Braida D., Passafaro M., Popoli M., Sala M., Battaglioli E. // Proc. Natl. Acad. Sci. USA. 2016. V. 113. P. 3651–3656.
Duclot F., Kabbaj M. // Genome Biology. 2015. V. 16. e256.
Cao G., Xing J., Xiao X., Liou A.K., Gao Y., Yin X.M., Clark R.S., Graham S.H., Chen J. // J. Neurosci. 2007. V. 27. P. 9278–9293.
Guerra-Castellano A., Díaz-Quintana A., Pérez-Mejías G., Elena-Real C.A., González-Arzola K., García-Mauriño S.M., De la Rosa M.A., Díaz-Moreno I. // PNAS. 2018. V. 115(31). P. 7955–7960.
Дополнительные материалы отсутствуют.