

Неорганические материалы, 2019, T. 55, № 10, стр. 1101-1107
Исследование кристаллизационной устойчивости оптически активных стекол GeSx〈Bi〉
А. М. Кутьин 1, А. Д. Плехович 1, *, М. В. Суханов 1, К. В. Балуева 1
1 Институт химии высокочистых веществ им. Г.Г. Девятых Российской академии наук
603951 Нижний Новгород, Тропинина ул., 49, Россия
* E-mail: plehovich@gmail.com
Поступила в редакцию 28.12.2018
После доработки 12.02.2019
Принята к публикации 28.02.2019
Аннотация
Методом дифференциальной сканирующей калориметрии (ДСК) в области 320−1000 К изучена кинетика кристаллизации особочистых стекол системы Ge–S, легированных висмутом GeSx〈nBi〉 (1.25 ≤ х ≤ 1.6, n = 1 ат. %). Развитая методика обработки кристаллизационных пиков дает возможность их описания, не уступающего полуэмпирической модели Ерофеева при аналитической форме степени кристаллизации α (t, Т), развивающей упрощенный вариант теории Колмогорова–Мейла–Джонсона. Найденные по методике кинетические характеристики кристаллизации стекла в зависимости α (t, Т) и ее графическом представлении в виде ТТТ (Time–Temperature–Transition)-диаграммы позволяют определить оптимальные температурно-временные условия изготовления волоконных световодов.
ВВЕДЕНИЕ
Халькогенидные германийсодержащие стекла, легированные висмутом, рассматриваются как перспективные материалы для нелинейной оптики, оптоэлектроники и высококонтрастной фотолитографии благодаря наличию ультраширокой полосы люминесценции [1, 2]. Они прозрачны в спектральном диапазоне от 0.6 до 8.0 мкм, имеют высокие температуры стеклования, низкие энергии фононов, характеризуются высокой растворимостью редкоземельных элементов [3].
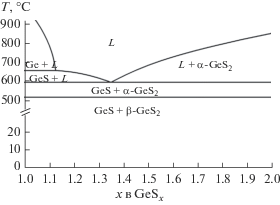
На фазовой диаграмме базовой стеклообразующей системы Ge–S [4] (ее часть представлена на рис. 1) отмечены эвтектика при х = 1.35 как фактор, сопутствующий стеклообразованию, и две стеклообразующие области [5–7] при х = 2−9 и х = 1.31−1.5. Определены области стеклообразования указанных стекол с добавками висмута. По данным работы [8], такая область расположена вдоль линии Ge20S80 – x−Bix до 15 ат. % Bi, а в работе [9] – вдоль линии GeS2−Bi2S3 до 25 ат. % Bi.
Изучение кристаллизации в температурной области переохлажденного расплава Ge–S [10, 11] показало, что кинетика кристаллизации хорошо описывается моделью Шестака–Берггрена, в основе которой полуэмпирическая модель Ерофеева. Однако отсутствие аналитически выраженной температурно-временной зависимости степени кристаллизации затрудняет ее практическое использование, в том числе учет неизотермичности измерений методом дифференциальной сканирующей калориметрии (ДСК).
Введение редкоземельных элементов или Bi в качестве оптически активных добавок в бинарную основу GeSx ставит перед производителями оптических элементов дополнительные задачи, связанные с исследованием кристаллизационной устойчивости новых по составу стекол для получения волоконных и планарных устройств с необходимой фазовой чистотой.
В статье [12] в связи с природой люминесценции висмута изучено его межфазное распределение. Было показано, что легирующая примесь Bi не включается в решетку кристаллов сульфида германия, а в стекле находится в растворенном виде. Содержание висмута более 1 ат. % повышает вероятность образования индивидуальной металлической фазы. Кинетика кристаллизации легированных висмутом стекол GeSx в настоящее время не изучена.
Целью настоящей работы явилось исследование кристаллизационных процессов в синтезированных высокочистых образцах стекол, легированных висмутом GeSx〈nBi〉 (1.25 ≤ х ≤ 1.6, n = 1 aт. %), методом ДСК, а также разработка методики обработки ДСК-пиков посредством унифицированной модели кристаллизации и ее использование для минимизации кристаллизации при получении оптических стекол и световодов с достаточной фазовой чистотой.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Исследовали кристаллизацию стекол составов: GeS1.25 + 0.01Bi, GeS1.35 + 0.01Bi, GeS1.4 + 0.01Bi, GeS1.6 + 0.01Bi. Методика их получения и сведения о чистоте образцов приведены в работах [12, 13].
Для измерений готовилась серия образцов массой 30–40 мг. Контактирующие с дном тигля поверхности образцов подвергались шлифовке для обеспечения воспроизводимого контакта с последующими ультразвуковой очисткой в дистиллированной воде и сушкой. Для измерений использовались предварительно отожженные алюминиевые тигли как наиболее химически инертные по отношению к стеклу Ge–S.
ДСК-кривые для четырех изученных образцов (рис. 2) в ходе линейного повышения температуры последовательно представляют участки стеклообразного состояния, заканчивающиеся соответствующими “ступенями” расстекловывания в интервале температур 351–381 К, и следующие за ними интервалы вязкотекучего или переохлажденного жидкого состояния, в которых “проявляются” соответствующие пики кристаллизации. Результаты стандартной обработки ДСК-кривых (температура стеклования определялась по точке перегиба в интервале стеклования, а температуры кристаллизации и плавления – по началу пика, определяемого пересечением касательной переднего фронта пика с линией основания пика) представлены в табл. 1. Табличные данные демонстрируют также установленную зависимость температур кристаллизации от состава переохлажденного расплава GeSx + 0.01Bi. При х = 1.25, 1.35, 1.4 последовательно кристаллизуются GeS и GeS2, а при х = 1.6 образуется только кристалл GeS2. Такой характер кристаллизации соответствует фазовой диаграмме Ge–S. Возможное фазовое превращение β-GeS2 в α-GeS2 не наблюдается [12].
Рис. 2.
ДСК-кривые сульфид-германатных стекол: 1 – GeS1.25 + 0.01Bi, 2 – GeS1.35 + 0.01Bi, 3 – GeS1.4 + + 0.01Bi, 4 – GeS1.6 + 0.01Bi при скорости нагревания 10 К/мин.
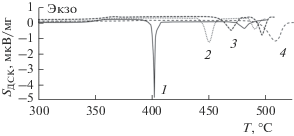
Таблица 1.
Температуры стеклования (Tg) в стеклообразующем расплаве GeSx + 0.01Bi в зависимости от содержания серы (х), а также температуры кристаллизации (Тcr) GeS и GeS2 из переохлажденных расплавов (скорость нагревания 10 К/мин)
| x | Tg, °С | Тcr, °С | ΔT = Tcr – Tg | ||
|---|---|---|---|---|---|
| GeS | GeS2 | GeS | GeS2 | ||
| 1.25 | 350.9 | 401.1 | 472.9 | 50.2 | 122 |
| 1.35 | 355.9 | 443.2 | 488.8 | 87.3 | 132.9 |
| 1.40 | 356.3 | 460.6 | 491.2 | 104.3 | 134.9 |
| 1.5* | 135** | 135** | |||
| 1.60 | 381.3 | – | 489.1 | – | 107.8 |
Определяемые методом ДСК температуры стеклования и кристаллизации часто используются для качественной оценки стабильности стекла к кристаллизации посредством эмпирического критерия ΔT = Tcr – Tg (табл. 1), величина которого пропорциональна кристаллизационной устойчивости. Однако детальную и полную информацию о скоростях кристаллизационных процессов можно получить после анализа и обработки участков ДСК-кривых, соответствующих пикам кристаллизации.
МЕТОДИКА ОБРАБОТКИ КРИСТАЛЛИЗАЦИОННЫХ ДСК-ПИКОВ
Кристаллизацию стекла А можно представить квазихимической реакцией А → *А, которая определяет текущее изменение энтальпии процесса ∆Н [14] мольной координатой ξ и энтальпией реакции кристаллизации ∆rH
(1)
$\Delta Н = {\text{ }}\xi {{\Delta }_{r}}H = {\text{ }}\alpha {{n}_{0}}{{\Delta }_{r}}H = {\text{ }}\alpha {{m}_{0}}({{{{\Delta }_{r}}H} \mathord{\left/ {\vphantom {{{{\Delta }_{r}}H} M}} \right. \kern-0em} M}).$Второе и третье выражения в цепочке (1) записаны с использованием степени кристаллизации α = ξ/n0 как доли текущего числа молей ξ от начального количества n0 = m0/M исходной (маточной) фазы; m0 и М – соответственно начальная и мольная масса кристаллизующегося вещества.
Деление уравнения (1) на m0 определяет взаимосвязь указанных энтальпий и теплот в удельных, т.е. отнесенных к единице массы, величинах:
Второе из уравнений (1') связывает текущую удельную теплоту Qm = –∆Н/m0 с удельной теплотой реакции А → *А QrM = –∆rH/M.
В определенных пределах независимым от массы образца в приборах “ДСК теплового потока” принято [15] считать сигнал, пропорциональный удельному тепловому потоку
(2)
${{S}_{{{\text{ДСК}}}}} = C{{\dot {Q}}_{m}} = С\dot {\alpha }{{Q}_{{rM}}} = {{C}_{N}}\dot {\alpha },$Подобные рассуждения справедливы и для пиков плавления, по которым производится калибровка прибора. Поэтому для калиброванного прибора (С = 1) значение и размерность приборной или нормировочной константы ${{С}_{N}}$ = $С{{Q}_{{rM}}}$ соответствуют величине и размерности удельной теплоты процесса ${{Q}_{{rM}}}$ (Дж/г), обеспечивая размерность ДСК-сигнала (Дж/(г с)) как удельного потока тепла или удельной мощности.
При размерности сигнала прибора [SДСК] = = мкВ/мг размерность нормировочной константы равна $\left[ {{{С}_{N}}} \right]$ = $\left[ {\frac{{{{S}_{{{\text{ДСК}}}}}}}{{\dot {\alpha }}}} \right]$ = $\frac{{{\text{мкВ с}}}}{{{\text{мг}}}}.$
Температурная и временная зависимость степени кристаллизации в наиболее распространенной модели Джонсона–Мейла–Аврами (ДМА), которая является значительным упрощением теории Колмогорова–Джонсона–Мейла, имеет вид
Здесь τ = kt, k = A exp(–Ea/(RT)) – константа скорости кристаллизации в представлении Аррениуса, A – предэкспоненциальный множитель, Ea – энергия активации, n – степенной параметр ДМА, качественно соответствующий т. н. размерному параметру теории Колмогорова–Джонсона–Мейла. Интегральной форме модели ДМА (3) соответствует ее дифференциальная форма(4)
$\begin{gathered} \dot {\alpha } = \frac{{d\alpha }}{{dt}} = k\alpha _{{\tau }}^{'}(\alpha ); \\ \alpha _{{\tau }}^{'}(\alpha ) = n(1 - \alpha ){{[ - \ln (1 - \alpha )]}^{{1 - {1 \mathord{\left/ {\vphantom {1 n}} \right. \kern-0em} n}}}}, \\ \end{gathered} $Шестак и Берггрен (Sestak–Berggren) [16], а вслед за ними Малек [17] показали, что полуэмпирическая модель кристаллизации Ерофеева [18] является более точной для обработки ДСК-пиков кристаллизации по сравнению с моделью ДМА. Дифференциальная форма этой модели имеет вид
Однако отсутствие решения α (t, Т) дифференциального уравнения (5) в аналитической форме осложняет численную обработку результатов эксперимента, учет неизотермичности ДСК-измерений и, как следствие, заметно ограничивает предсказательные возможности модели. Другим существенным недостатком полуэмпирической модели является скрытое в эмпирических степенных параметрах a и b смысловое содержание, а также эффективный характер константы скорости kэф. Последние факторы затрудняют выявление деталей механизма кристаллизации с позиции представлений Колмогорова–Джонсона–Мейла.
Отмеченные недостатки устраняются в предлагаемой унифицированной модели кристаллизации (УМК), интегральная и дифференциальная форма которой имеют вид
(6)
$\alpha (t,T) = \frac{1}{{{{{(1 + {1 \mathord{\left/ {\vphantom {1 {{{\tau }^{n}}}}} \right. \kern-0em} {{{\tau }^{n}}}})}}^{p}}}},$(7)
$\dot {\alpha } = \alpha _{{\tau }}^{'}\tau _{t}^{'} = k\alpha _{{\tau }}^{'} = knp{{\alpha }^{{1 - {1 \mathord{\left/ {\vphantom {1 {(np)}}} \right. \kern-0em} {(np)}}}}}{{(1 - {{\alpha }^{{{1 \mathord{\left/ {\vphantom {1 p}} \right. \kern-0em} p}}}})}^{{1 + {1 \mathord{\left/ {\vphantom {1 n}} \right. \kern-0em} n}}}}.$Сопоставление формул (3), (4) и (6), (7) показывает, что при едином способе определения температурно-временной зависимости α (t, Т) только через произведение τ = kt роль важного для модели ДМА параметра n, эффективным образом определяющего механизм кристаллизации, в УМК переходит к произведению np. Наконец, сравнение формул (5) и (7) показывает, что УМК раскрывает содержание как степенных параметров a и b полуэмпирической модели, так и эффективной константы скорости kэф.
При нагревании с постоянной скоростью β температура измерительной ячейки, отсчитываемая от начала пика кристаллизации Т0, является линейной функцией времени:
Результат дифференцирования в формулах (4) и (7) с учетом (8) при аррениусовой форме k
(9)
$\dot {\alpha } = \frac{{{\text{d}}\alpha }}{{{\text{d}}t}} = \alpha _{{\tau }}^{'}\tau _{t}^{'} = \alpha _{{\tau }}^{'}k\left( {1 + \frac{{(T - {{T}_{0}})\theta }}{{{{T}^{2}}}}} \right)$отличается от выражений (4) и (7) только сомножителем в скобках и при естественном отнесении его к “неизотермической” константе скорости
повторяет их с заменой $k$ на $\bar {k}.$
Наличие в УМК явного выражения для степени кристаллизации существенно упрощает решение задачи параметризации при обработке кристаллизационных ДСК-пиков, сводя ее к нелинейной регрессии со стандартными процедурами минимизации среднеквадратичного отклонения экспериментальных и рассчитанных по уравнениям (2)–(4), (6), (7), (10) значений ДСК-сигнала.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Методика обработки данных ДСК должна учитывать изменение температуры во время измерения. Игнорирование неизотермичности условий определения кинетических констант, как показано в работе [19], может привести к погрешностям определения температурно-временной зависимости степени кристаллизации α (T, t) 10% и более.
Обработка методом наименьших квадратов (нелинейная регрессия) кристаллизационных пиков (рис. 3) по отражающим неизотермичность измерений уравнениям (6)–(10) дает параметры (табл. 2), в которых вычисленная энергия активации Ea дополнительно контролировалась уравнением Киссинджера. Кроме “технических” констант (Т0, CN), кинетические параметры кристаллизации не зависят от скорости сканирования, что в свою очередь повышает достоверность определения α (T, t).
Рис. 3.
Измеренные (значки) и рассчитанные (линии) по уравнениям (2), (6)–(10) значения ДСК-сигнала при скоростях нагревания 10 К/мин: (×) и (- - -) и 15 К/мин: (⚪) и (· - · -); пикам кристаллизации соответствуют синхронизированные со временем температурные зависимости степени кристаллизации (α) для стеклообразующих расплавов GeSx + 0.01Bi с х = = 1.25 (a), 1.35 (б), 1.4 (в), 1.6 (г).
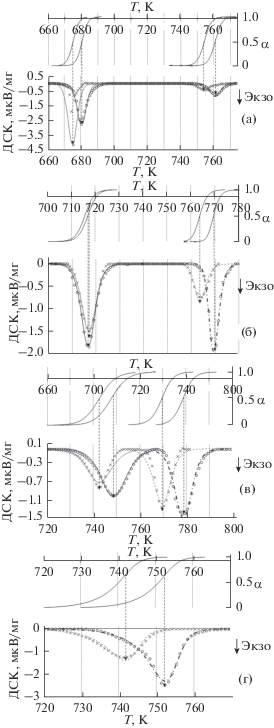
Таблица 2.
Кинетические параметры УМК стекол GeSx + 0.01Bi (Ea – энергия активации, s – натуральный логарифм предэкспоненциального множителя, n, p – степенные параметры)
| х | Ea, кДж/моль | s = ln(A/с–1) | n | p | Т0 | CN, мкВ/мг | ||
|---|---|---|---|---|---|---|---|---|
| 10* | 15* | 10* | 15* | |||||
| Образование фазы GeS | ||||||||
| 1.25 | 324.8 | 52.82 | 4.64 | 0.86 | 650.1 | 656.6 | 180.42 | 81.72 |
| 1.35 | 483.1 | 74.90 | 4.15 | 0.91 | 640.94 | 663.97 | 71.61 | 56.93 |
| 1.40 | 322.2 | 45.88 | 3.50 | 1.01 | 651.1 | 658.2 | 60.57 | 58.48 |
| Образование фазы GeS2 | ||||||||
| 1.25 | 494.6 | 72.38 | 4.28 | 0.93 | 646.9 | 678.6 | 24.60 | 29.65 |
| 1.35 | 644.9 | 97.05 | 3.13 | 1.28 | 747.0 | 757.7 | 31.26 | 42.82 |
| 1.40 | 417.4 | 58.63 | 4.64 | 0.86 | 652.9 | 697.1 | 67.21 | 56.08 |
| 1.60 | 312.3 | 44.41 | 6.57 | 0.32 | 670.6 | 699.8 | 98.24 | 104.65 |
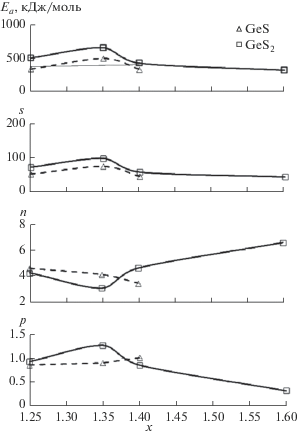
Примечание. Произведение np по форме вхождения в уравнение (7) соответствует параметру n в модели ДМА (см. уравнение (4)); составы стекол, близкие к эвтектическому, характеризуются более высокой энергией активации. * Скорость нагревания, К/мин.
Графическая зависимость кристаллизационных параметров Ea, s, n, p от состава стекол представлена на рис. 4. Рассчитанные по этим параметрам температурно-временные диаграммы α (t, Т) (рис. 5) – основа прогнозирования температурно-временных режимов обработки стекол, минимизирующих кристаллизацию, а значит ее вклад в оптические потери.
Рис. 4.
Зависимости кинетических параметров кристаллизации (табл. 2) Ea, s, n, p фаз GeS и GeS2 от содержания серы в стеклообразующем расплаве GeSx + + 0.01Bi (x = 1.25–1.6).
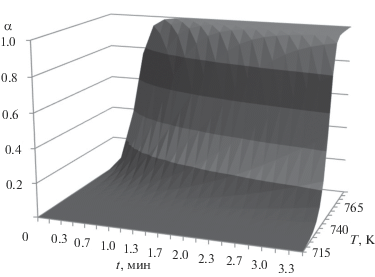
Кристаллизация из переохлажденного расплава GeSx + 0.01Bi (x = 1.25–1.6) с небольшими, в первом приближении не влияющими на процесс, добавками Bi происходит при температурах ниже линии солидуса диаграммы Ge–S. Конечным состоянием в этом случае является смесь кристаллов GeS + GeS2. В отличие от обычных переходов, соответствующих диаграмме плавкости, кристаллизация из переохлажденного расплава для ряда исследованных стекол: слева от эвтектического (х = 1.25), эвтектического (х = 1.35) и следующего за ним (х = 1.40) происходит следующим образом. При повышении температуры после расстекловывания следующие друг за другом пики кристаллизации соответствуют образованию рентгенографически подтвержденных кристаллов GeS, а затем GeS2. Температурный интервал между пиками при их общем сдвиге в область высоких температур уменьшается, по оценке стремясь к совпадению при x = 1.5 (рис. 2, 3а–3в). При х > 1.5, как это наблюдается для текла с x = 1.60, кристаллизуется индивидуальная фаза GeS2 (рис. 2, 3г). Возвращаясь к табл. 1 и примечанию к ней, отметим, что при максимальном содержании серы x = 1.5, при котором совмещаются кристаллизационные пики образования GeS и GeS2, судя по соответствующему значению эмпирического критерия ΔT = Tcr – Tg, достигается максимальная устойчивость к кристаллизации. Для стекла с x = 1.60, в котором образуется единственная фаза GeS2 и наблюдается один пик кристаллизации, критерий ΔT(x) уменьшается от ΔT(1.5) = = 135 К до ΔT(1.6) = 107.8 К (см. табл. 1).
ЗАКЛЮЧЕНИЕ
По эмпирическому критерию с использованием полученных температур стеклования и кристаллизации в стеклообразующих расплавах GeSx + 0.01Bi (х = 1.25, 1.35, 1.4, 1.6) выявлен максимально устойчивый к кристаллизации состав стекла с содержанием серы х = 1.5. Менее стойкими к кристаллизации оказываются стекла как с меньшим содержанием серы, так и с бóльшим. С повышением температуры при х ≤ 1.5 поочередно выделяются кристаллы GeS и GeS2. При x ≥ 1.5 наблюдается единственный пик кристаллизации с образованием фазы GeS2.
Обработка кристаллизационных пиков по методике, использующей УМК, позволила определить инвариантные к скорости термосканирования параметры кристаллизации фаз GeS и GeS2 для всех образцов GeSx + 0.01Bi (x = 1.25, 1.35, 1.4, 1.6). При этом набор четырех основных кинетических параметров Ea, s, n, p определяет температурно-временную зависимость доли (степени) α (t, Т) образования каждой из кристаллизующихся фаз – GeS и GeS2. Полученные зависимости дают возможность прогнозирования параметров кристаллизации, а значит, и α (t, Т) для стекол неизученных составов.
Графическое представление степени кристаллизации α (t, Т) – Time–Temperature–Transition (ТТТ)-диаграмма, которая определяет режим вытяжки волокна в виде линии пересечения поверхности ТТТ-диаграммы с горизонтальной плоскостью на уровне α, соответствующем требуемой фазовой чистоте.
Список литературы
Plotnichenko V.G., Philippovskiy D.V., Sokolov V.O., Sukhanov M.V., Velmuzhov A.P., Churbanov M.F., Dianov E.M. Infrared Luminescence in Bi-Doped Ge–S and As–Ge–S Chalcogenide Glasses and Fibers // Opt. Mater. Express. 2014. V. 4. № 2. P. 366–374. https://doi.org/10.1364/OME.4.000366
Dianov E.M. Amplification in Extended Transmission Bands Using Bismuth-Doped Optical Fibers // J. Lightwave Technol. 2013. V. 31. № 4. P. 681–688. https://doi.org/10.1109/JLT.2012.2211569
Simons D.R., Faber A.J., Waal H. GeSx Glass for Pr3+-Doped Fiber Amplifiers at 1.3 m // J. Non-Cryst. Solids. 1995 V. 185 P. 283–288. https://doi.org/10.1016/0022-3093(94)00700-4
Kawamoto Y., Tsuchinashi S. Thermal Analysis of Ge–S Glasses // J. Am. Ceram. Soc. 1971. V. 54. P. 526–527. https://doi.org/10.1111/j.1151-2916.1971.tb12194.x
Borisova Z.U. Glassy semiconductors. New York: Plenum, 1981. P. 505. https://doi.org/10.1007/978-1-4757-0851-6
Виноградова Г.З. Стеклообразование и фазовые равновесия в халькогенидных системах. Двойные и тройные системы. М.: Наука, 1984. 176 с.
Liu C.H., Pashinkin A.S., Novoselova A.V. An Investigation of the Germanium-Sulphur System // Dokl. Akad. Nauk SSSR. 1963. V. 151. P. 1335–1338.
Frumar M., Ticha H., Koudelka L., Faimon J. Semiconducting Glasses of the Ge–Bi–S System // Mater. Res. Bull. 1976. V. 11. P. 1389–1396. https://doi.org/10.1016/0025-5408(76)90050-7
Tichy L., Ticha H., Benes L., Malek J. The Glass-Forming Region and Electrical Conductivity of Ge–Bi–S Glasses // J. Non-Cryst. Solids. 1990. V. 116. P. 206–218. https://doi.org/10.1016/0022-3093(90)90694-H
Voigt B. Über Glasbildung und Eigenschaften von Chalkogenidsystemen. XVII. Zur Glaschemie des Germaniumdisulfides // Z. Anorg. Allg. Chem. 1978. V. 447. P. 153–160. https://doi.org/10.1002/zaac.19784470117
Malek J., Klikorka J. Crystallization Kinetics of Glassy GeS2 // J. Therm. Anal. 1987. V. 32. P. 1883–1893. https://doi.org/10.1007/BF01913981
Iskhakova L.D., Ermakov R.P., Sukhanov M.V., Voronov V.V., Velmuzhov A.P., Philippovskiy D.V., Plotnichenko V.G., Churbanov M.F. The Study of Phase Formation Processes in GeSx:Bi (1 < x < 2) Chalcogenide Glasses // J. Non-Cryst. Solids. 2015. V. 428. P. 132–137. https://doi.org/10.1016/j.jnoncrysol.2015.08.021
Plotnichenko V.G., Philippovskiy D.V., Sokolov V.O., Sukhanov M.V., Velmuzhov A.P., Churbanov M.F., Dianov E.M. Infrared Luminescence in Bi-Doped Ge–S and As–Ge–S Chalcogenide Glasses and Fibers // Opt. Mater. Express. 2014. V. 4. P. 366–373. https://doi.org/10.1364/OME.4.000366
Пригожин И., Кондепуди Д. Современная термодинамика. От тепловых двигателей до диссипативных структур: Пер. с англ. Данилова Ю.А. и Белого В.В. М.: Мир, 2002. С. 461.
Borchard H.J., Daniels F. The Application of Differential Thermal Analysis to the Study of Reaction Kinetics // J. Am. Chem. Soc. 1957. V. 79. P. 41–46. https://doi.org/10.1021/ja01558a009
Sestaik J., Berggren G. Study of the Kinetics of Mechanism of Solid-State Reactions at Increasing Temperatures // Thermochim. Acta. 1971. V. 3. P. 1–12. https://doi.org/10.1016/0040-6031(71)85051-7
Malek J., Zmrhalova Z., Bartak J., Honcova P. A Novel Method to Study Crystallization of Glasses // Thermochim. Acta. 2010. V. 511. P. 67–73. https://doi.org/10.1016/j.tca.2010.07.026
Янг Д. Кинетика разложения твердых веществ: Пер. с англ. Печерской К.А.; Под ред. Ерофеева Б.В. М.: Мир, 1969. С. 263.
Malek J. The Kinetic Analysis of Non-Isothermal Data // Thermochim. Acta. 1992. V. 200. P. 257–269. https://doi.org/10.1016/0040-6031(92)85118-F
Дополнительные материалы отсутствуют.
Инструменты
Неорганические материалы