

Неорганические материалы, 2020, T. 56, № 12, стр. 1328-1335
Синтез и свойства карбоната иттрия-аммония
В. А. Матвеев 1, *, К. А. Яковлев 1, В. Я. Кузнецов 1, О. А. Залкинд 1
1 Институт химии и технологии редких элементов и минерального сырья им. И.В. Тананаева
184209 Мурманская область, Апатиты, Академгородок, 26а, Россия
* E-mail: v.matveev@ksc.ru
Поступила в редакцию 10.03.2020
После доработки 29.06.2020
Принята к публикации 30.06.2020
Аннотация
Карбонат иттрия-аммония состава NH4Y(CO3)2 · H2O синтезирован взаимодействием гидратированного оксида иттрия и карбоната аммония при 80°С. Установлено, что процесс протекает через кристаллизацию двух промежуточных фаз, представляющих собой гидроксокарбонаты иттрия и аммония различного состава. Полученные соединения охарактеризованы методами РФА, ИКС, ДСК–ТГ и химического анализа. Впервые приведены рентгенографические характеристики и кристаллографические данные элементарной ячейки NH4Y(CO3)2 ∙ H2O. Установлено, что при термическом разложении карбоната иттрия-аммония образуется кубическая модификация оксида иттрия в виде тонкодисперсного порошка.
ВВЕДЕНИЕ
Оксид иттрия (Y2O3) является наиболее распространенным соединением иттрия с очень широким диапазоном применения. Это один из самых перспективных прекурсоров для получения лазерной керамики и люминофоров [1–4]. Оксид иттрия используется также для покрытия тиглей и форм, предотвращая агрессивное химическое воздействие расплавленных металлов, солей, шлаков и стекла при высоких температурах [5], в качестве антиоксиданта [6], как высокоэффективная добавка в функциональные композиционные материалы на основе оксидов алюминия и циркония [7–9], в производстве катализаторов [10].
Для получения оксида иттрия предложено использовать различные способы, в том числе такие высокотехнологичные, как самораспространяющийся высокотемпературный синтез (СВС) и плазменно-распылительный пиролиз (FSP) [11, 12]. Однако обычно порошки оксида иттрия получают термическим разложением малорастворимых соединений иттрия, в качестве которых используют гидроксид, гидроксонитрат, оксалат, салицилат, карбонаты иттрия [13–21].
На свойства получаемых оксидных материалов существенное влияние оказывает структура исходных порошков оксида иттрия, поэтому основной задачей является получение возможно более тонкого оксида иттрия в виде низкоагломерированного порошка. Лучшие результаты с этой точки зрения достигаются при термическом разложении основного и среднего карбонатов иттрия [22–24], что свидетельствует о перспективности использования новых карбонатных соединений иттрия в качестве предшественников оксида иттрия, в частности карбоната иттрия-аммония. Сведения об этом соединении обнаружены в двух источниках. В работе [25] сообщается о получении смеси состава NH4Y(CO3)2 ∙ H2O осаждением из раствора хлорида иттрия в течение 6 ч при 25°С под действием гидрокарбоната аммония NH4HCO3 при его молярном отношении к Y3+, равном 15. Данных, подтверждающих получение кристаллического соединения состава NH4Y(CO3)2 ∙ H2O, не приводится. В работе [26] сообщается о синтезе двойного карбоната иттрия и аммония, который в общем виде протекает в соответствии с уравнением
Согласно материалам публикации, на первом этапе получали карбонат иттрия осаждением из раствора нитрата иттрия гидрокарбонатом аммония. Далее карбонат иттрия подвергался автоклавной обработке при 160°С в течение 24 ч. По мнению авторов, механизм реакции основан на проникновения ионов ${\text{NH}}_{{\text{4}}}^{ + }$ и ${\text{CO}}_{{\text{3}}}^{{{\text{2}} - }}$ в наноканалы, имеющиеся в структуре карбоната иттрия.
Настоящее исследование посвящено изучению возможности синтеза карбоната иттрия-аммония из гидратированного оксида иттрия взаимодействием с карбонатом аммония, а также изучению ряда свойств полученного соединения.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
В качестве исходных реагентов для получения гидратированного оксида иттрия были использованы Y(NO3)3 ⋅ 6H2O и аммиак водный квалификации “ч. д. а.”. В эксикатор емкостью 5 л заливали 500 мл аммиака водного, содержащего 28.7% NH3, а на решетчатом поддоне размещали фарфоровую чашку с 25 г нитрата иттрия, распределенного тонким слоем. Выдержку в атмосфере большого избытка аммиака вели в течение суток при периодическом перемешивании. При этом протекала химическая реакция в соответствии с уравнением
Полученную реакционную массу выщелачивали водой при 50°С в течение 20 мин. Осадок отфильтровывали, промывали и высушивали до постоянной массы при 105°C. Далее полученный гидратированный оксид иттрия обрабатывали раствором карбоната аммония с концентрацией соли ~160 г/л при молярном отношении (NH4)2CO3 к оксиду иттрия, равном 5–6, и температуре 80°C. Для исследования происходящих фазовых превращений продолжительность взаимодействия изменяли от 5 до 360 мин. Синтез в течение 5–60 мин вели при атмосферном давлении, а при продолжительности 120–360 мин – в автоклаве (во избежание улетучивания карбоната аммония). Полученные осадки отфильтровывали, промывали, сушили при 105°C до постоянной массы и анализировали. Часть синтезированного продукта подвергали термической обработке при 800°С в течение 1 ч с получением порошка оксида иттрия.
Содержание в образцах иттрия определяли атомно-эмиссионным методом на спектрометре OPTIMA 8300, иона аммония – методом обратного титрования, углерода – инфракрасной абсорбцией на анализаторе ELTRA CS-2000. Кристаллооптический анализ выполняли с помощью петрографического микроскопа со специализированной цифровой камерой Leica DM 2500P. Рентгенофазовый анализ (РФА) синтезированных соединений проводили на приборе SHIMADZU XRD-6000, а также на дифрактометре Дрон-2 с CuKα-излучением и графитовым монохроматором (в качестве внутреннего стандарта использовали Si). ИК-спектроскопические исследования выполняли на приборе Nicolet 6700 (образцы таблетировали с KBr). Анализ ДСК–ТГ проводили на синхронном термоанализаторе STA 409 фирмы Netzsch. Морфологию образцов фиксировали методом сканирующей электронной микроскопии с использованием цифровых сканирующих электронных микроскопов SEM LEO-420 и Quanta 650 FEG. Дисперсность порошка оксида иттрия определяли методом лазерной дифракции на приборе SHIMADZU SALD-201V. Удельную поверхность определяли методом БЭТ с использованием низкотемпературной адсорбции азота на анализаторе удельной поверхности и пористости TriStar 3020.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
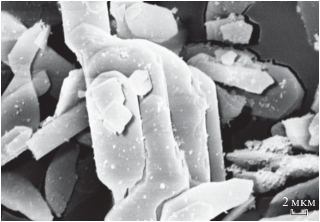
Полученный гидратированный оксид иттрия (ГОИ) представляет собой рентгеноаморфное вещество с содержанием 61.1% Y2O3, состоящее из агломератов более мелких частиц пластинчатой формы (рис. 1).
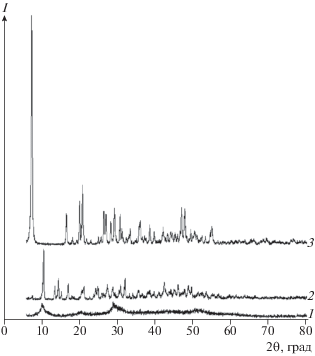
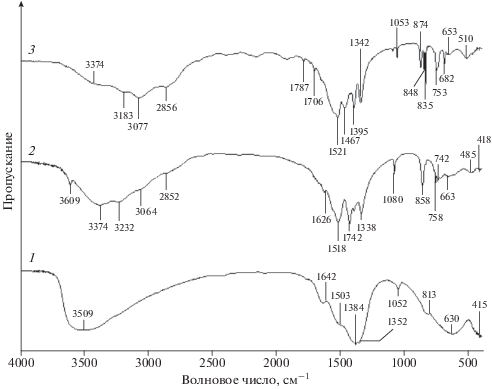
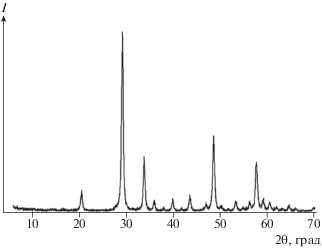
Анализ рентгенограмм продуктов, полученных при различной продолжительности синтеза, показывает (рис. 2), что уже через 5 мин обработки ГОИ в небольшом количестве образуется неизвестная кристаллическая фаза, что говорит о высокой реакционной способности ГОИ по отношению к карбонату аммония. При увеличении продолжительности синтеза до 15–60 мин кристаллизуется вторая неизвестная фаза, а после 120 и более мин – третья, которая в дальнейшем остается без изменений. Анализ ИК-спектров этих же продуктов (рис. 3) подтвердил последовательное образование трех фаз. При этом уже на спектре 1 в области волновых чисел 3200 см–1 отмечается перегиб, что говорит о наличии в структуре катиона аммония ${\text{NH}}_{{\text{4}}}^{ + }.$ Это означает, что синтез с самого начала идет через непосредственное присоединение ионов ${\text{NH}}_{{\text{4}}}^{ + }$ и ${\text{СО}}_{{\text{3}}}^{{{\text{2}} - }}$ к гидратированному оксиду иттрия.
Рис. 2.
Дифрактограммы продуктов при различной продолжительности синтеза, мин: 1 – 5, 2 – 15–60, 3 – 120–360.
Химический состав синтезированных соединений (табл. 1) показывает, что обе промежуточные фазы представляют собой гидроксокарбонаты иттрия и аммония различного состава.
Таблица 1.
Химический состав продуктов реакции
| Фаза | Содержание, мас. % | Формула | ||
|---|---|---|---|---|
| ${\text{NH}}_{4}^{ + }$ | Y+ | ${\text{CO}}_{3}^{{2 - }}$ | ||
| 1 | 0.65 | 40.2 | 37.5 | (NH4)0.08Y(CO3)1.38(OH)0.32 · 2.36H2O |
| 2 | 5.94 | 33.0 | 42.3 | (NH4)0.89Y(CO3)1.9(OH)0.09 · 2.73H2O |
| 3 | 7.33 | 36.3 | 49.1 | NH4Y(CO3)2 · H2O |
Двойной карбонат иттрия-аммония состоит из анизотропных кристаллов в форме пластин (рис. 4), т.е. наследует морфологию исходного гидратированного оксида иттрия, и имеет показатель преломления Ng = 1.580 + 0.003 и Np = 1.548 + 0.003. В связи с отсутствием дифракционных данных для NH4Y(CO3)2 ∙ H2O в базе ICDD для индицирования полученной рентгенограммы были использованы данные для соединения NH4Gd(CO3)2 ∙ H2O [27]. Выбор его обусловлен тем, что соединения иттрия и РЗЭ группы самарий–тулий изоструктурны вследствие близости их катионных радиусов [28]. Значения углов 2θ, межплоскостных расстояний d, интенсивности I/Io и индексы отражений hkl для соединения NH4Y(CO3)2 ∙ H2O приведены в табл. 2, кристаллографические данные элементарной ячейки – в табл. 3.
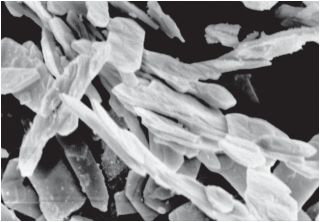
Таблица 2.
Кристаллографические данные элементарной ячейки NH4Y(CO3)2 ∙ H2O
| Сингония | Орторомбическая |
| Параметры ячейки | a = 6.057 Å, b = 9.139 (4) Å, c = 24.685 (9) Å, V = 1366 (2) Å3 |
| Пр. гр. | D152h = Pbca (61) |
| Число молекул в ячейке | Z = 8 |
| Расчетная плотность, г/см3 | 2.395 |
Таблица 3.
Рентгенографические характеристики NH4Y(CO3)2 · H2O
| № п/п | 2θ, град | d, Å | I/Iо, % | hkl |
|---|---|---|---|---|
| 1 | 7.19 | 12.2840 | 100 | 002 |
| 2 | 16.26 | 5.4470 | 21 | 102 |
| 3 | 17.92 | 4.9460 | 3 | 110 |
| 4 | 19.66 | 4.5120 | 22 | 021 |
| 5 | 20.49 | 4.3310 | 50 | 104 |
| 6 | 21.53 | 4.1240 | 5 | 006 |
| 7 | 22.64 | 3.9241 | 4 | 114 |
| 8 | 24.66 | 3.6070 | 6 | 121 |
| 9 | 25.43 | 3.4995 | 7 | 122 |
| 10 | 26.19 | 3.3997 | 29 | 106 |
| 11 | 26.69 | 3.3371 | 28 | 123 |
| 12 | 27.95 | 3.1895 | 15 | 116 |
| 13 | 28.91 | 3.0857 | 45 | 008 |
| 14 | 30.46 | 2.9321 | 25 | 125 |
| 15 | 30.83 | 2.8978 | 22 | 117 |
| 16 | 32.03 | 2.7919 | 3 | 027 |
| 17 | 32.69 | 2.7370 | 12 | 126 |
| 18 | 33.11 | 2.7033 | 12 | 131 |
| 19 | 35.66 | 2.5156 | 25 | 221 |
| 20 | 36.33 | 2.4707 | 6 | 222 |
| 21 | 36.86 | 2.4364 | 6 | 204 |
| 22 | 37.36 | 2.4049 | 5 | 119 |
| 23 | 38.28 | 2.3492 | 18 | 029 |
| 24 | 39.46 | 2.8120 | 14 | 1.0.10 |
| 25 | 40.63 | 2.2186 | 3 | 1.1.10 |
| 26 | 41.79 | 2.1597 | 17 | 208 |
| 27 | 43.03 | 2.1002 | 6 | 218 |
| 28 | 43.97 | 2.0575 | 11 | 0.0.12 |
| 29 | 44.14 | 2.0500 | 11 | 1.1.11 |
| 30 | 45.03 | 2.0115 | 12 | 0.2.11 |
| 31 | 45.64 | 1.9860 | 3 | 219 |
| 32 | 46.68 | 1.9442 | 44 | 312 |
| 33 | 47.56 | 1.9102 | 40 | 1.2.11 |
| 34 | 49.14 | 1.8524 | 14 | 229 |
| 35 | 50.14 | 1.8178 | 13 | 241 |
| 36 | 50.69 | 1.7994 | 7 | 323 |
| 37 | 52.14 | 1.7527 | 7 | 0.2.13 |
| 38 | 53.03 | 1.7254 | 5 | 325 |
| 39 | 54.53 | 1.6814 | 33 | 154.331 |
Полосы поглощения NH4Y(CO3)2 ∙ H2O (рис. 3, спектр 3) характеризуются малой полушириной, что свидетельствует об образовании его структуры в виде мелких кристаллов без признаков аморфизации. Хорошо просматриваются полосы поглощения в области 3300–2700 см–1, относящиеся к ионам аммония. Анализ спектра относительно карбонат-иона проводился при рассмотрении двух областей поглощения: ν1 – полносимметричное колебание (1100–1000 см–1) и ν3 – симметричное валентное колебание (1800–1300 см–1). В области ν1 наблюдаются три полосы поглощения разной интенсивности. Как известно [29], это колебание синглетно. Это позволяет сделать вывод о наличии трех типов карбонат-иона, при этом по интенсивности можно судить о степени симметрии. Анализ области ν3 указывает на тип симметрии: наличие пяти полос поглощения карбонат-иона говорит в пользу существования высокосимметричного карбонат-иона и двух типов низкосимметричного, т.к. 4 полосы поглощения в области ν3 свидетельствуют о снятии вырождения колебания ν3 с двух типов карбонат-ионов. Полоса поглощения при 752 см–1, по-видимому, свидетельствует о наличии сильной водородной связи М–О…ОН. Причем эта связь обусловлена разными (по структуре) кислородами, т.к. на спектре основной полосы видно плечо в области 745 см–1.
Результаты термического анализа показывают (рис. 5), что разложение NH4Y(CO3)2 ∙ H2O происходит в несколько стадий. При нагревании до 164°С удаляется незначительное количество физически сорбированной воды. При дальнейшем повышении температуры до ~320°С происходит быстрая потеря ~17% массы, обусловленная удалением кристаллизационной воды, значительной части аммиака и небольшого количества диоксида углерода. С возрастанием температуры до 580°С происходит полное удаление аммиака и примерно половины содержащегося в исходном соединении диоксида углерода с образованием основного карбоната иттрия. При дальнейшем увеличении температуры происходит разложение этого соединения (эндоэффект с максимумом при 623°С) с импульсным выделением диоксида углерода [1]. Потеря массы завершается в районе 800°С. Остаточная масса составляет 46.12%, что практически соответствует содержанию оксида иттрия в соединении NH4Y(CO3)2 ∙ H2O.
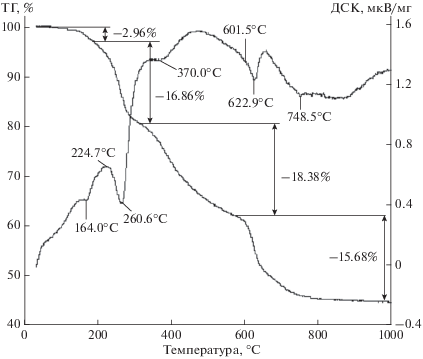
Продукт прокаливания двойного карбоната иттрия и аммония при 800°С в течение 1 ч представляет собой кубическую модификацию оксида иттрия (рис. 6), средний размер кристаллитов которого, рассчитанный с использованием формулы Дебая–Шеррера по двум наиболее интенсивным пикам (2θ = 29.16° и 2θ = 48.54°), составляет 38.47 ± 2.20 нм.
В свою очередь, размер кристаллитов, найденный по величине удельной поверхности порошка оксида иттрия 15.4 м2/г, определенной для исследуемого образца (плотность 4.84 г/см3), составил 80.5 нм. Судя по микрофотографии высокого разрешения (рис. 7), порошок оксида иттрия состоит из агломерированных сферических кристаллитов, имеющих размер, по крайней мере, менее 100 нм.

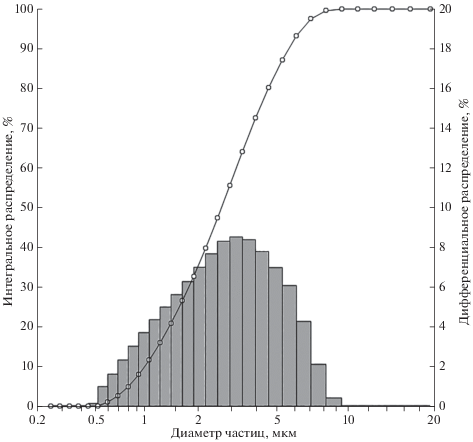
Из приведенной на рис. 8 гистограммы порошка оксида иттрия следует, что размер подавляющей массы агрегатов не превышает 8 мкм.
ЗАКЛЮЧЕНИЕ
Впервые при взаимодействии гидратированного оксида иттрия с карбонатом аммония получен двойной карбонат иттрия-аммония. Установлено, что синтез протекает через кристаллизацию двух промежуточных фаз, представляющих собой гидроксокарбонаты иттрия и аммония различного состава. Впервые приведены рентгенографические характеристики и кристаллографические данные элементарной ячейки NH4Y(CO3)2 ∙ H2O. Показана перспективность использования этого соединения для получения тонкодисперсных порошков оксида иттрия.
Список литературы
Федоров П.П., Ильин Н.В. Термолиз карбоната иттрия // Журн. неорган. химии. 2012. Т. 57. № 2. С. 282.
Федоров П.П., Ткаченко Е.А., Кузнецов С.В. и др. Получение нанопорошков оксида иттрия из карбонатных прекурсоров // Журн. неорган. химии. 2010. Т. 55. № 6. С. 883.
Watanabe T., Iso Y., Isobe T. Synthesis of Y2O3:Bi3+,Eu3+ Nanosheets from Layered Yttrium Hydroxide Precursor and Their Photoluminescence Properties // RSC Adv. 2017. V. 7. P. 14107.
Shivaramu N.J., Lakshminarasappa B.N., Nagabhushana K.R., Singh F. Synthesis Characterization and Luminescence Studies of Gamma Irradiated Nanocrystalline Yttrium Oxide // Spectrochim. Acta. Part A. 2016. V. 154. P. 220.
Larimi Z.M., Amirabadizadeh A., Zelati A. Synthesis of Y2O3 Nanoparticles by Modified Transient Morphology Method // Int. Conf. on Chemistry and Chemical Process IPCBEE. 2011. V. 10. P. 86.
Mellado-Vázquez R., García-Hernández M., López-Marure A. et al. Sol-Gel Synthesis and Antioxidant Properties of Yttrium Oxide Nanocrystallites // Materials. 2014. V. 7. P. 6768. https://doi.org/10.3390/ma7096768
Kannan S.K., Sundrarajan M. Biosynthesis of Yttrium Oxide Nanoparticles Using Acalypha Indica Leaf Extract // Bull. Mater. Sci. V. 38. № 4. P. 945–950.
Плетнев П.М., Непочатов Ю.К., Маликова Е.В. и др. Технология получения корундовой бронекерамики, модифицированной сложными добавками // Изв. Томского политехнического ун-та. 2015. Т. 326. № 3. С. 40.
Тельнова Г.Б., Коломиец Т.Ю., Коновалов А.А. и др. Фазовые превращения при синтезе Y3Al5O12:Nd // Журн. неорган. химии. 2015. Т. 60. № 2. С. 163.
Basavegowda N., Mishra K., Thomba R.S. et al. Sonochemical Green Synthesis of Yttrium Oxide (Y2O3) Nanoparticles as a Novel Heterogeneous Catalyst for the Construction of Biologically Interesting 1,3-Thiazolidin-4-ones // Catal. Lett. 2017. V. 147. № 10. P. 2630.
Балабанов С.С., Гаврищук Е.М., Дроботенко В.В. и др. Получение наноразмерных порошков оксида иттрия методом самораспространяющегося высокотемпературного синтеза // Вестн. Нижегородского ун-та им. Н.И. Лобачевского. 2011. № 2(1). С. 91.
Jae S.L., Jinhyung L., Han H. et al. Single-Step Synthesis of Cubic Y2O3:Eu3+ Nanophosphor by Flame Spray Pyrolysis // J. Therm. Spray Technol. 2016. V. 25. № 8. P. 1570.
Япрынцев А.Д., Скогарева Л.С., Гольдт А.Е. и др. Синтез пероксопроизводного слоистого гидроксида иттрия // Журн. неорган. химии. 2015. Т. 60. № 9. С. 1131.
Ermakov R.P., Voronov V.V., Fedorov P.P. et al. X-Ray Diffraction Study of the Phase and Morphology Changes in Yttrium Compound Nanoparticles // Nanosyst.: Phys., Chem., Math. 2013. № 4(2). P. 196.
Abdulghani A.J., Waleed M. Al-Ogedy Preparation and Characterization of Yttrium Oxide Nanoparticles at Different Calcination Temperatures from Yttrium Hydroxide Prepared by Hydrothermal and Hydrothermal Microwave Methods // Iraqi J. Sci. 2015. V. 56. № 2. P. 1572.
Титов А.А., Клименко М.А., Горячева Е.Г. и др. Получение нанокристаллических порошков оксидов церия и иттрия при термическом разложении оксалатов, карбонатов и гидроксидов // Неорган. материалы. 2008. Т. 44. № 10. С. 1229.
Ranjbar M., Mannan S., Yousefi M. Yttria Nanoparticles Prepared from Salicylic Acid-y(iii) Nanocomposite as a New Precursor // Am. Chem. Sci. J. 2013. V. 3. № 1. P. 1–10.
Gong H., Tang D.-Y., Huang H., Zhang T.-S. Effect of Grain Size on the Sinterability of Yttria Nanopowders Synthesized Bycarbonate-Precipitation Process // Mater. Chem. Phys. 2008. V. 112. P. 423.
Afzali E. Preparation Methods of Nanostructures Synthesis with Different Size and Shape // Bull. Env. Pharmacol. Life Sci. 2014. V. 4. № 1. P. 129.
Chen J., Huang B., Huang C. et al. Preparation of Nanoscaled Yttrium Oxide by Citrate Precipitation Method // J. Rare Earths. 2017. V. 35. № 1. P. 79.
Юдина Е.П., Фролова А.В., Кривцов И.В. и др. Анализ продуктов гидротермальной обработки нитрата и хлорида иттрия // Бюлл. Северного уральского государственного ун-та. Серия: Химия. 2015. Т. 7. № 1. С. 51.
Иванов В.К., Баранчиков А.Е., Ванецев А.С. и др. Влияние гидротермальной и гидротермально-ультразвуковой обработки на фазовый состав и микроморфологию гидроксокарбоната иттрия // Журн. неорган. химии. 2007. Т. 52. № 9. С. 1413.
Горячева Е.Г., Вдовина Л.В., Карманников В.П. Способ получения мелкодисперсного порошка оксида иттрия: Пат. РФ № 2194014, C01F. Опубл. 10.12.2002.
Тельнова Г.Б., Поликанова А.С., Солнцев К.А. Способ получения нанопорошков оксида иттрия: Пат. РФ № 2354610 C01F/00. Опубл. 10.05.2009.
Li Y.-X., Li M., He X.-B., Gu Z.-Y., Hu P.-G., Zhou X.-Z. Composition and Crystalline Phase Type of Products Obtained by Precipitating Yttrium Ion with Ammonium Bicarbonate // Chin. J. Inorg. Chem. 2002. V. 18.№ 11. P. 1138–1142.
Liu Y., Qin X., Yang Y., Zeng Z., Xin H., Dai Z., Song C., Zhu X., Li D., Zhang J. & Kawazoe Y. Chemical Reaction Directed Oriented Attachment: from Precursor Particles to New Substances. (2015) [Электронный ресурс] // портал ResearchGate.net URL: https:// www.researchgate.net/publication/285648654_Chemi-cal_reaction_directed_oriented_attachment_from_precursor_particles_to_new_substances (дата обращения 25.11.2019)
Rogow D.L., Swanson C.H., Oliver A.G. et al. Two Related Gadolinium Aquo Carbonate 2-d and 3-d Structures and Their Thermal, Spectroscopic, and Paramagnetic Properties // Inorg. Chem. 2009. V. 48. № 4. P. 1533.
McDonald T.R.R., Spink J.M. The Crystal Structure of a Double Oxalate of Yttrium and Ammonium, NH4Y(C2O4)2 · H2O // Acta Crystallogr. 1967. V. 23. P. 944.
Накамото К. ИК спектры и спектры КР неорганических и координационных соединений: Пер. с англ. М.: Мир, 1991. 505 с.
Дополнительные материалы отсутствуют.
Инструменты
Неорганические материалы