

Неорганические материалы, 2021, T. 57, № 8, стр. 886-894
Содержание углерода в сере, летучей и нелетучей фракциях ее дистилляции
М. В. Суханов 1, *, Р. Д. Благин 1, А. П. Вельмужов 1
1 Институт химии высокочистых веществ им. Г.Г. Девятых Российской академии наук
603950 Нижний Новгород, ул. Тропинина, 49, Россия
* E-mail: sukhanov@ihps-nnov.ru
Поступила в редакцию 10.12.2020
После доработки 07.04.2021
Принята к публикации 08.04.2021
Аннотация
Разработана и испытана методика определения общего углерода в малых навесках серы (0.1–1.5 г) и ее летучих и нелетучих примесях. Методика заключается в ИК-спектрометрическом определении углекислого газа, образующегося при окислении оксидом меди(II) пробы серы, нелетучего остатка или газовой фракции примесей. Предел обнаружения углерода составил 0.3–1.5 мкг, погрешность определения – 20%. Определено содержание углерода в образцах серы различного происхождения и в ее летучих и нелетучих примесях.
ВВЕДЕНИЕ
Актуальной задачей при получении халькогенидных оптических материалов является контроль содержания и поведения примеси углерода на всех этапах их производства. Эта примесь в форме CS2, CSe2, COS, CO2, углеводородов и углистых частиц вызывает избыточное селективное поглощение и рассеяние излучения в халькогенидных стеклах [1–3]. Примеси углеводородов в халькогенах являются источниками водорода, дающего полосы селективного поглощения S–H и Se–H в области прозрачности халькогенидных стекол. Основным источником углерод- и водородсодержащих примесей в стеклах на основе сульфидов мышьяка и германия является сера [1, 4]. Содержание углерода в коммерческой сере разных марок достигает величин 10–2–10–3 мас. % [5]. В стеклах, предназначенных для изготовления волоконных световодов с оптическими потерями в среднем ИК-диапазоне на уровне собственных, содержание углерода в сере не должно превышать 10–6 мас. % [1, 6].
В литературе описаны методы определения отдельных форм углерода в сере по их селективному поглощению в ИК-спектрах расплава либо с помощью газохроматографического анализа газовой фракции, полученной при вакуумной экстракции, и некоторые другие [7–9]. Известные методы определения общего углерода в сере основаны на переводе всех углеродсодержащих примесей в удобные для анализа вещества. Эти методы включают окисление углеродсодержащих примесей кислородом, серой или оксидом меди(II) до CO2 и CS2 и их непосредственное газохроматографическое определение или титрование раствора гидроксида бария, поглотившего CO2 [5, 9–13]. Предел обнаружения большинства методик составляет (0.1–1) × 10–4 мас. %. Их недостатками являются относительно сложная система напуска пробы, использование аппаратуры с кранами из политетрафторэтилена и вспомогательных газов, являющихся потенциальными источниками углерода.
Различное происхождение серы, используемой для получения высокочистых халькогенидных стекол, обуславливает различный состав ее углеродсодержащих примесей и их отличающееся поведение в процессах очистки. Для обоснованного выбора эффективного способа очистки серы от примесей, содержащих углерод, или его модификаций, необходимо иметь представление об общем содержании углерода в исходной и очищенной сере, его поведении и распределении в летучих и нелетучих фракциях очистки. Cведений о содержании углерода в разных фракциях очистки серы в литературе не обнаружено. Таким образом, актуальной остается разработка методики определения углерода в сере с широким диапазоном его содержания, а также в разных фракциях ее дистилляционной очистки.
Целью работы было создание методики определения примеси углерода в навесках серы массой 1–2 г и фракциях ее дистилляции в диапазоне концентраций (1–300) × 10–4 мас. %, исследование распределения углерода при однократной дистилляции серы и характеризация образцов серы различных производителей по этим параметрам.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
В работе исследованы образцы газовой серы (С.-Пб., Россия), серы ОСЧ 15-3 (Ленинград, СССР), 6N (All-Chemie Ltd.) и ОСЧ 16-5 (Норильск, Россия). Соответствующие обозначения образцов С1, С2, С3, С4.
Предлагаемая в работе методика основана на высокотемпературном окислении углеродсодержащих примесей в навеске серы, нелетучем остатке и газовой фракции, образующихся при ее отгонке, и последующем ИК-спектрометрическом определении углекислого газа. В качестве окислителя использовали оксид меди(II), обладающий окислительными свойствами и являющийся источником свободного кислорода при нагревании до 800°С [14]. Упрощенно схема окисления углеродсодержащих примесей может быть описана следующими уравнениями реакций:
Для окисления серы, выделения и окисления ее летучих и нелетучих фракций примесей применяли цельнопаяные кварцевые реакторы, предварительно обработанные смесью HCl и HNO3, промытые и прокаленные при 800°С в течение 1 ч для удаления поверхностных углеродсодержащих загрязнений. Оксид меди(II) квалификации “х. ч.” также подвергали прокаливанию при 800°С перед окислением проб.
Система вакуумирования представляла собой турбомолекулярный безмасляный насос Pfeiffer Vacuum HiCube 80 eco и соединенную с ним кварцевую ловушку, помещенную в жидкий азот. Прокаленный реактор с окислителем, кюветой и пробой припаивали к ловушке, и после вакуумирования и отпаивания никакие его внутренние части и содержимое на этапах окисления пробы, концентрирования углекислого газа и ИК-спектроскопических измерений не контактировали с внешней средой.
Регистрацию ИК-спектров образцов проводили в кварцевой микрокювете объемом 0.24 мл на ИК-Фурье-спектрометре Tensor 27 (Bruker, Германия) при спектральном разрешении 4 см–1. Окна микрокюветы изготавливали из тонкого кварцевого стекла с содержанием водорода в форме OH-групп менее 10–4 мас. % для обеспечения ее максимальной прозрачности в спектральных областях поглощения углекислого газа: 3500–3800 и 2200–2450 см–1. При содержании углерода в анализируемой пробе меньше 30 мкг в качестве аналитических линий использовали полосы поглощения CO2 в области 2200–2450 см–1, при большем содержании углерода – полосы поглощения в области 3500–3800 см–1.
Построение градуировочных функций. Для построения градуировочных функций были приготовлены механические смеси фуллерена С60 (99.0 мас. % основного вещества) с порошком прокаленного кварцевого стекла. Точные навески этих смесей, содержащие от 0 до 515 мкг С60 (6 значений), совместно с 0.25 г свежепрокаленного CuO вакуумировали в кварцевых ампулах (рис. 1а), после чего реактор отпаивали и прогревали ампулу 1 в пламени кислородно-пропановой горелки для окисления С60. Выделяющийся CO2 последовательно концентрировали в промежуточной ампуле, затем в кювете путем охлаждения жидким азотом. После этого кювету отпаивали. Полноту сбора продуктов окисления контролировали по отсутствию нового конденсата на внутренних стенках ампулы или отводного капилляра кюветы при их дополнительном погружении в жидкий азот. Воспроизводимые значения аналитического сигнала со стандартным отклонением менее 0.1 при многократном определении углерода в градуировочных смесях (n = 3–5) были получены при времени конденсации продуктов окисления 15 мин.
Рис. 1.
Схема кварцевого реактора для определения общего углерода в сере (а), ее нелетучих (а) и летучих примесях (б): 1 – ампула; 2 – навеска серы или нелетучий остаток, или градуировочная смесь; 3 – CuO; 4 – ампула для промежуточного концентрирования продуктов окисления; 5 – микрокювета; 6 – ампула с летучими примесями; 7 – тонкостенная стеклянная перегородка; 8 – магнитный боек в запаянной кварцевой трубке; 9 – места отпаивания.
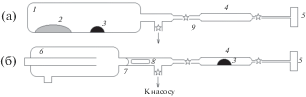
Для регистрации фонового спектра кюветы углекислый газ, сконцентрированный при окислении пробы, намораживали жидким азотом в отводной капилляр. Для этого использовали самодельный кварцевый микрососуд Дьюара. Не меняя положение кюветы в спектрометре, размораживали продукты окисления и записывали их ИК-спектр. Измерения повторяли 3–5 раз.
Определение общего углерода в сере. Для определения общего углерода в сере навеску 0.1–1.5 г вместе с 0.025 г свежепрокаленного CuO помещали в прокаленную кварцевую ампулу 1 (рис. 1а). Ампулу вакуумировали и подвергали термообработке при 800°С в течение 4 ч. Продукты окисления последовательно собирали в промежуточную ампулу 4, охлаждаемую жидким азотом, затем – в кювету. После ее отпаивания проводили ИК-спектрометрическое определение углекислого газа.
Наличие SO2 в продуктах окисления пробы значительно осложняет определение малых количеств углерода. Это обусловлено тем, что полосы поглощения CO2 в области 2200–2450 см–1 недоступны для измерения с достаточной точностью из-за наложения полос поглощения SO2. Для снижения влияния SO2 на предел обнаружения углекислого газа при анализе серы с содержанием углерода менее 2 × × 10–4 мас. % углеродсодержащие примеси в пробе предварительно переводили в CS2 и COS ее нагреванием в вакуумированной ампуле 1 (рис. 1а) при 800°С в течение 4 ч без использования CuO. Согласно [9, 10], степень превращения углеродсодержащих примесей в этих условиях достигает 97%. Образующиеся CS2 и COS собирали и окисляли в отдельной ампуле 4 (рис. 1б), как и при определении углерода в летучих примесях
Количество образующегося SO2 в этом случае было меньше, чем при окислении пробы серы CuO, и не мешало ИК-спектрометрическому определению CO2.
Определение углерода в нелетучей и летучей фракциях примесей серы. Для выделения нелетучих и летучих примесей из серы использовали установку из кварцевого стекла, показанную на рис. 2. Навеску серы 1–1.5 г испаряли при 150°С и непрерывном вакуумировании системы. После полной отгонки серы отпаивали испаритель 1 с нелетучим остатком и приемник 3 с замороженными летучими примесями и проводили определение углерода в них с использованием кварцевых реакторов, схема которых приведена на рис. 1.
Рис. 2.
Схема кварцевого реактора для выделения из серы нелетучих и летучих примесей: 1 – испаритель, 2 – приемник, 3 – ампула для сбора летучих примесей, 4 – сосуд Дьюара с жидким азотом, 5 – навеска серы, 6 – печь сопротивления, 7 – стеклянная перегородка, 8 – места отпаивания.

При анализе легколетучих примесей 0.025 г окислителя помещали в промежуточную ампулу 4 (рис. 1б). После вакуумирования реактора и отпаивания от вакуумного поста разбивали тонкостенную перегородку магнитным бойком, перемораживали легколетучие примеси в промежуточную ампулу 4 и отпаивали. При анализе нелетучих примесей количество окислителя составляло 0.25 г. Окисление нелетучих и летучих примесей проводили нагреванием ампул 1 или 4 соответственно в пламени кислородно-пропановой горелки. Определение CO2 в кювете осуществляли по вышеописанной методике.
Фазовый состав нелетучего остатка после перегонки серы определяли на рентгеновском дифрактометре XRD-6000 фирмы Shimadzu в CuKα-излучении в диапазоне значений 2θ = 10°–60°, с шагом 0.02°. Для идентификации фаз использовали базу данных ICSD PDF2.
РЕЗУЛЬТАТЫ ЭКСПЕРИМЕНТА
Градуировочные функции. На рис. 3 приведены ИК-спектры пропускания продуктов окисления некоторых градуировочных смесей фуллерена с кварцевым стеклом с различным содержанием углерода. Зависимости интегральной интенсивности полос поглощения CO2 в областях 2200–2450 и 3500–3800 см–1 от содержания углерода в смеси описываются уравнениями прямых вида C = kS, где С – содержание углерода в пробе, мкг; k – коэффициент, обратный коэффициенту чувствительности, мкг/см–2; S – интегральная интенсивность соответствующих полос поглощения, см–2 (рис. 4):
Рис. 3.
ИК-спектры пропускания продуктов окисления градуировочных смесей с содержанием фуллерена 0 (холостой опыт) (1), 0.45 (2), 14.9 (3), 117.8 (4), 514 мкг (5).

Рис. 4.
Градуировочные зависимости содержания углерода в навеске от интегральной интенсивности полос поглощения CO2 в области 2200–2450 (1) и 3500–3800 см–1 (2).

Выражение (1) получено для интервала содержания углерода в пробе от 0 до 14.9 мкг, (2) – для интервала от 4.8 до 514 мкг. Доверительный интервал коэффициента пропорциональности приведен для погрешности 95%. Значение холостого опыта – без градуировочной смеси с навеской окислителя – составило 0.1 мкг углерода.
Из-за возможного многократного отражения луча в кювете малых геометрических размеров зависимости (1) и (2) справедливы лишь для конкретной использованной кюветы и конкретного спектрального разрешения.
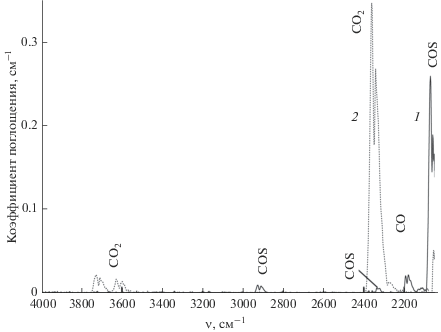
Общее содержание углерода в сере. ИК-спектры продуктов окисления серы и углеродсодержащих примесей приведены на рис. 5. Они содержат полосы поглощения SO2 и CO2, частично перекрывающиеся в области 2200–2450 см–1. Полосы поглощения COS отсутствуют. Расчет содержания углерода в пробе проводили по интегральной интенсивности полос CO2 в области 3500–3800 см–1. Результаты определения углерода в образцах серы приведены в табл. 1.
Рис. 5.
ИК-спектры поглощения продуктов окисления серы C1 (1), C4 (2) и С5 (3) при определении общего углерода.

Таблица 1.
Содержание углерода в нелетучем остатке, легколетучих примесях и общее содержание углерода в сере разных марок
| Сера | Содержание углерода в пересчете на серу ×104, мас. % | ||
|---|---|---|---|
| нелетучий остаток | легколетучие примеси | общее содержание | |
| C1 | 198 ± 44* | 2.3 ± 0.5 | 224 ± 45 |
| C2 | 0.5 ± 0.1 | 0.3 ± 0.1 | 11 ± 2 |
| C3 | 0.4 ± 0.1 | 0.3 ± 0.1 | 13 ± 2 |
| C4 | 0.13 ± 0.03 | 0.4 ± 0.1 | 20 ± 4 |
| С5** | – | – | 0.4 ± 0.2 |
* Среднее четырех определений, для остальных – по одному определению. ** Сера С4, дополнительно очищенная по методике, аналогичной [15], включающей каталитическое окисление на CeO2.
Как было отмечено, присутствие SO2 в продуктах окисления пробы делает невозможным определение малых количеств CO2 в спектральном диапазоне 2200–2450 см–1. Для определения менее 5 мкг углерода (нижний предел построения градуировочной зависимости (2)) по полосам поглощения CO2 в области 3500–3800 см–1, в которой отсутствует поглощение SO2, необходимо использовать пробу массой более 2 г и реакционные ампулы большего объема во избежание превышения избыточного давления при термообработке 2 атм. Для снижения минимального определяемого по градуировочной зависимости (2) содержания углерода и для проверки правильности методики осуществлено определение общего углерода в навесках серы разной массы (0.1–1.6 г). Содержание углерода в разных навесках серы С2–C4, рассчитанное по (2), одинаково в пределах погрешности определения 20% (табл. 2). Это свидетельствует о правильности методики и возможности использования зависимости (2) в расширенном интервале содержания углерода – от 1.5 до 500 мкг.
Таблица 2.
Содержание углерода в разных навесках серы
| Сера | Навеска, г | Найдено углерода | |
|---|---|---|---|
| мкг | ×104, мас. % | ||
| C2 | 0.129 1.590 |
1.6 17.5 |
12 ± 2 11 ± 2 |
| C3 | 0.131 1.601 |
1.4 20.8 |
11 ± 2 13 ± 2 |
| C4 | 0.117 1.428 |
2.7 28.6 |
23 ± 4 20 ± 4 |
Предел обнаружения общего углерода в сере составил 1.5 мкг. Предварительная термообработка серы с целью перевода углеродсодержащих примесей в CS2 и COS и минимизации мешающего влияния SO2 в спектральном диапазоне 2200–2450 см–1 позволила снизить предел обнаружения углерода до 0.4 мкг в сере С5, подвергнутой дополнительной очистке.
Определение углерода в нелетучем остатке. ИК-спектры поглощения и содержание углерода в нелетучем остатке образцов С1–С4 в пересчете на навеску серы приведены на рис. 6 и в табл. 1. Четырехкратное определение углерода в образце С1 позволило рассчитать относительное стандартное отклонение – 14% и доверительный интервал при 95%-ной доверительной вероятности – 22%. Достаточно высокое значение погрешности, по-видимому, связано с неоднородным распределением примесей в исследуемой сере. Предел обнаружения углерода в нелетучем остатке, рассчитанный по утроенному значению холостого эксперимента, составил 0.3 мкг.
Рис. 6.
ИК-спектры поглощения продуктов окисления нелетучего остатка после перегонки 1.8–2.1 г серы: С1 (1), С2 (2), С3 (3), С4 (4).

Рентгенограммы нелетучего остатка серы С1 и С4 свидетельствуют о его одинаковом качественном составе: кварц, ангидрит и пирит (рис. 7). Содержание этих примесей в образце С1 составило 66, 28 и 7 мас. % соответственно, в образце C4 – 21, 46 и 33 мас. %. На рентгенограммах присутствуют малоинтенсивные рефлексы в области 22.0° и 27°–28°, идентифицировать которые затруднительно. Карбонаты, склонные к выделению углекислого газа при прокаливании, в нелетучем остатке отсутствуют. Предположительно, углерод в нелетучем остатке серы содержится в форме рентгеноаморфных фаз – сажи и углеводородов с малым числом атомов водорода.
Рис. 7.
Рентгенограммы нелетучего остатка после дистилляции серы С1 (1) и С4 (2); символами на рентгенограммах обозначены рефлексы отражения SiO2 (3), CaSO4 (4), FeS2 (5); штрих-рентгенограммы SiO2 (6), CaSO4 (7), FeS2 (8) построены по данным ICSD PDF2.

Содержание углерода в летучей фракции серы. На рис. 8 приведен ИК-спектр поглощения легколетучих примесей серы C1 и продуктов их окисления. Эти примеси представлены CO и COS. Результаты определения углерода в летучих примесях серы C1–C4 приведены в табл. 1. Диоксид серы, образующийся при окислении COS, не проявляется в ИК-спектрах и не мешает определению углерода в форме CO2. Предел обнаружения углерода в летучей фракции составил 0.3 мкг.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Разработана методика, позволяющая определять содержание примеси углерода в сере и ее летучей и нелетучей фракциях в широком диапазоне концентраций. Ограничением методики по максимальному содержанию углерода в пробе является возрастающее давление CO2 в кювете. При определении более 500 мкг углерода избыточное давление в ней достигает 3 атм. Для определения больших количеств углерода необходимо уменьшать массу анализируемого образца или использовать кювету большого объема. Предел обнаружения углерода зависит от чувствительности ИК-спектрометрического определения CO2, мешающего влияния SO2, образующегося при окислении серы, мешающего влияния CO2 атмосферы, значения холостого эксперимента. Надежные ИК-спектрометрические измерения содержания CO2 в кювете возможны при многократном превышении его давления над парциальным давлением атмосферного CO2. Для снижения минимальной определяемой массовой доли углерода в сере необходимо увеличивать массу анализируемой навески.
Сравнивая доступность, пределы обнаружения, точность газохроматографического и ИК-спектроскопического методов определения CO2 в продуктах окисления навески серы, можно заключить, что они сопоставимы по этим характеристикам (табл. 3). Преимущество ИК-спектроскопического анализа продуктов окисления с герметичным цельнопаяным реактором заключается в удобстве отбора пробы серы или газовой фазы в процессе ее очистки с соблюдением ее изолированности от внешней среды.
Таблица 3.
Сравнение методик определения углеродсодержащих примесей в сере
| Примесь | Окислитель | Метод | Определяемое вещество | Предел обнаружения С, 10–5 мас. % | Источник |
|---|---|---|---|---|---|
| Σ | Избыток S, O2 | ГХ | CO2, COS | 0.6 | [5] |
| Углеводороды | – | ИКС | –CH2– | 2 | [7] |
| Растворенные газы | – | ГХ | C1–C4, бензол, толуол, CS2, COS, CO2 | 0.001–0.1 | [9] |
| Σ | CuO | ГХ | CO2 | 1 | [10] |
| Σ | O2 | Титрование | BaCO3 | 500 | [11] |
| Σ | O2 | ГХ | CO2 | 150 | [12] |
| Σ | S | ГХ | CS2, COS, CO2 | 1 | [13] |
| Σ | CuO | ИКС | CO2 | 4–15 | Настоящая работа |
| Летучие примеси | 3 | ||||
| Нелетучие примеси | 3 |
Полученные в работе результаты свидетельствуют о существенных различиях в поведении углеродсодержащих примесей в сере разных марок в процессе ее однократной дистилляции. По количеству углерода, извлеченного в нелетучий остаток и отнесенного к общему его содержанию, исследованные образцы серы можно разделить на две группы: со степенью извлечения более 88% (С1) и степенью извлечения менее 3% (С2, С3, С4). Это свидетельствует об отличающемся качественном составе углеродсодержащих примесей. Содержание углерода в образце С1 является типичным (1.0 × 10–2 мас. %) для серы, извлекаемой из природного газа [5]. После одной, двух и трех дистилляций этой серы в нелетучем остатке было найдено 198 × 10–4, 25 × 10–4 и 12 × 10–4 мас. % углерода соответственно, что составляет 84, 10 и 5% от общего содержания углерода. Таким образом, трехкратная дистилляция газовой серы позволяет на 2 порядка снизить содержание в ней примеси углерода. Низкое содержание примеси углерода в летучих и нелетучих фракциях серы по сравнению с его общим количеством в образцах C2, С3 и С4 свидетельствует о малой эффективности однократной перегонки для очистки серы этих марок. Для эффективного удаления углеродсодержащих примесей в этом случае необходимо применять комбинированные способы очистки, включающие химико-термическую обработку либо высокотемпературное каталитическое окисление. Это позволяет снизить содержание углерода в 50 раз (образцы С4 и С5, табл. 1). Полученные закономерности необходимо учитывать при разработке методик глубокой очистки серы от примеси углерода.
ЗАКЛЮЧЕНИЕ
Разработана методика определения общего углерода в навесках серы 0.1–1.5 г и в летучих и нелетучих фракциях ее дистилляции для интервала содержаний 0.3–500 мкг. Предел обнаружения методики составил 0.3–1.5 мкг углерода, погрешность определения – 20%.
Новизной работы является реализация ИК-спектроскопического определения продуктов окисления пробы. Методика является экспрессной, универсальной, позволяет минимизировать воздействие мешающих анализу источников углерода и может быть частично или полностью применена при определении углерода в других халькогенах или при исследовании его распределения в процессе их очистки. Важность разработки такой методики обусловлена необходимостью получения серы с возможно более низким содержанием примеси углерода для сульфидных материалов ИК-оптики. Определено содержание углерода в сере различных марок и ее фракциях. Распределение примеси углерода между летучими и нелетучими фракциями, образующимися при дистилляции серы, существенно зависит от происхождения серы.
Список литературы
Девятых Г.Г., Чурбанов М.Ф. Высокочистые халькогены. Нижний Новгород: Изд-во Нижегородского ун-та, 1997. 243 с.
Shiryaev V.S., Churbanov M.F. Preparation of High-Purity Chalcogenide Glasses // Chalcogenide Glasses Preparation, Properties and Applications / Ed. Adam J.L., Zhong X. Woodhead Publishing Limited, 2014. P. 3–35.
Snopatin G.E., Shiryaev V.S., Plotnichenko V.G., Dianov E.M., Churbanov M.F. High-Purity Chalcogenide Glasses for Fiber Optics // Inorg. Mater. 2009. V. 45. № 13. P. 1439–1460.
Velmuzhov A.P., Sukhanov M.V., Churbanov M.F., Zernova N.S., Ketkova L.A., Sozin A. Yu., Shiryaev V.S., Skripachev I.V., Evdokimov I.I. Sulfur as the Source of Hydrogen Impurity and Heterogeneous Inclusions in the Ge–Ga–S Glasses // Non-Cryst. Solids. 2020. V. 545. P. 120237(1–9).
Малышев А.Ю., Крылов В.А., Буланов А.Д., Адамчик С.А. Газохроматографическое определение углеродсодержащих примесей в высокочистой сере // Журн. аналит. химии. 2000. Т. 55. № 7. С. 770–774.
Девятых Г.Г., Дианов Е.М., Плотниченко В.Г. и др. Волоконные световоды на основе высокочистых халькогенидных стекол // Высокочистые вещества. 1991. № 1. С. 7–36.
Адамчик С.А., Сенников П.Г., Буланов А.Д. Очистка серы от примесей углеводородов термохимическим методом // Неорган. материалы. 2000. Т. 36. № 6. С. 725–729.
Ежелева А.Е., Малыгина Л.С., Чурбанов М.Ф. Газохроматографическое определение растворенных газов и других летучих веществ в халькогенах и халькогенидах // Журн. аналит. химии. 1982. Т. 37. № 8. С. 1502–1504.
Крылов В.А. Определение углерода, кислорода, водорода и азота в халькогенах и мышьяке высокой чистоты // Высокочистые вещества. 1994. № 5. С. 116–120.
Зимаков И.Е., Караванов H.A., Ильина Т.С. Определение примесного углерода в сере, селене, сульфидах и селенидах кадмия и цинка // Изв. вузов. Химия и хим. технология. 1992. Т. 35. Вып. 10. С.45–49.
ГОСТ 30355.1-96. Сера техническая. Определение общего содержания углерода. Титриметрический метод. М.: Изд-во стандартов. 1999. 8 с.
Dowling N.I., Bernard F., Leung J., Lesage K.L. Analytical Procedure for the Determination of Trace Carbon Impurity in Elemental Sulfur // J. Sulfur Chem. 2008. V. 29. № 2. P. 129–137.
Чурбанов М.Ф., Чугунова С.Г., Девятых Г.Г. Газо-хроматографическое определение углерода в сере // Журн. аналит. химии. 1973. Т. 28. № 2. С. 385–387.
Zhu Y., Mimura K., Isshiki M. Oxidation Mechanism of Cu2O to CuO at 600–1050°C // Oxid. Met. 2004. V. 62. № 314. P. 207–222.
Суханов М.В., Сторожева Т.И., Евдокимов И.И., Пименов В.Г., Созин А.Ю., Котерева Т.В. Глубокая очистка моноизотопной серы 32S и 34S // Неорган. материалы. 2017. Т. 53. № 2. С. 126–131.
Дополнительные материалы отсутствуют.
Инструменты
Неорганические материалы