

Неорганические материалы, 2022, T. 58, № 10, стр. 1064-1068
Разработка межатомного потенциала системы Fe–Ni и оценка упругих свойств железоникелевого сплава
М. Ю. Семенов 1, *, И. П. Королев 1, В. А. Панчо-Рамирес 1
1 Московский государственный технический университет
им. Н.Э. Баумана
105005 Москва, ул. 2-я Бауманская, 5/1, Россия
* E-mail: Semenov.m.yu@bmstu.ru
Поступила в редакцию 11.01.2022
После доработки 25.04.2022
Принята к публикации 11.05.2022
- EDN: WBMJWM
- DOI: 10.31857/S0002337X22090111
Аннотация
Разработаны парные межатомные потенциалы γ-Fe, Ni вида Морзе, а также асимметричный парный потенциал взаимодействия атомов Fe и Ni на основе параметров Грюнайзена и энергий сублимации, впервые калиброванных при помощи эмпирических температурных зависимостей модуля сдвига. Впервые для широкого интервала температур от 600 до 1000°С, с возможностью аппроксимации до 1100°С, на основе полученных парных потенциалов в классическом виде Морзе получены расчетные температурные зависимости объемного модуля упругости, которые удовлетворительно сходятся с соответствующими экспериментальными зависимостями для железа, никеля и железоникелевого сплава со структурой ГЦК . Полученные межатомные потенциалы могут быть использованы для анализа энергий активации диффузионных процессов, зарождения вакансий и других эффектов в сплавах системы железо–никель.
ВВЕДЕНИЕ
Как известно, парные межатомные потенциалы открывают возможность с разной степенью точности проводить атомистические расчеты энергетических параметров, характеризующих различные аспекты физических явлений, происходящих в различных веществах, находящихся в конденсированном состоянии. Наиболее распространены простые выражения межатомных потенциалов Леннарт-Джонса в форме степенных функций, а также Морзе в виде экспоненциальных функций [1–3].
Так, в работе [4] при помощи потенциала Ми–Леннарт–Джонса для системы Fe–Pd были рассчитаны температуры фазового превращения (в интервале от 700 до 1100 К), весьма удовлетворительно соответствующие экспериментальным результатам. Этими же авторами при помощи аналогичного парного потенциала для системы Fe–Ni выполнены расчеты фазового равновесия между неупорядоченными фазами FeNi3 и FeNi в температурном интервале от 400 до 800 К, условно удовлетворительно соответствующие эмпирическим данным [5].
С другой стороны, по мнению автора [6], именно потенциал Морзе в наибольшей мере отражает физическую природу межатомного взаимодействия в переходных металлах.
В предшествующих работах нами разрабатывались парные потенциалы типа Морзе для железа, хрома и марганца [7] с использованием значений атомной сжимаемости, представленных в [8], с помощью которых были выполнены адекватные экспериментальным данным оценки значений объемного модуля упругости и энергетического порога полиморфного превращения. Аналогичные потенциалы были разработаны для никеля и кобальта [9] на основе выбора значений параметра Грюнайзена из экспериментально определенных различными методами.
В последнее время опубликованы многочисленные работы, в которых разрабатывали межатомные потенциалы в системах Fe–Ni, Fe–Ni–Cr и др., как правило, методом погруженного атома, основанным на теории функционала плотности. Так, в [10] определены значения коэффициентов самодиффузии Fe, Ni и Cr в тройной системе при температуре 1373 К. В [11] разработан межатомный потенциал для системы Fe–Ni при 0 К. В [12] определены константы упругости для различных сплавов системы Fe–Ni–Cr при комнатной температуре, удовлетворительно соотносящиеся с экспериментальными данными. Поскольку в указанных работах применяли межатомные потенциалы для конкретных температур (как правило, низких), то и температурные распределения констант упругости не рассчитывались.
Таким образом, как потенциалы, учитывающие многочастичное взаимодействие (например, метод погруженного атома), так и более простые выражения (например, Морзе) как правило приводят к результатам, точность которых достаточна для большинства прикладных задач. Несмотря на большую физическую строгость потенциалов, учитывающих многочастичное взаимодействие, на наш взгляд, использование более простых парных потенциалов также обосновано и целесообразно, особенно при моделировании процессов, происходящих в широких температурных интервалах, когда возникает необходимость учета влияния температуры на константы упругости, для чего приходится прибегать к различным искусственным методам по причине недостатка экспериментальных данных [7].
Следует отметить, что полные экспериментальные данные о взаимном влиянии Fe, Ni и Cr на кинетику диффузии на всем интервале температур, имеющем практическое значение (т.е. примерно до 1100–1250°С), отсутствуют. Имеются лишь разрозненные эмпирические данные [13].
Целью настоящей работы явилось построение адекватного эмпирическим данным межатомного потенциала в системе Fe–Ni (а в перспективе и Fe–Ni–Cr) для моделирования диффузионных процессов, зарождения вакансий и иных схожих явлений.
МЕТОДИКА РАЗРАБОТКИ ПАРНЫХ ПОТЕНЦИАЛОВ
Разрабатывали парный потенциал Морзе в форме
(1)
$\begin{gathered} U = {{U}_{b}} + {{U}_{r}} = \\ = \,\,{{U}_{0}}\left\{ {\exp \left[ { - 2\varphi \left( {\frac{d}{{{{d}_{0}}}} - 1} \right)} \right] - 2\exp \left[ { - \varphi \left( {\frac{d}{{{{d}_{0}}}} - 1} \right)} \right]} \right\}, \\ \end{gathered} $Равновесное межатомное расстояние d0 в никеле и железе приняли равным 0.248 и 0.252 нм, соответственно.
Значение параметра Грюнайзена никеля приняли равным 1.72 [9]. Значение параметра Грюнайзена железа существенно зависит от кристаллической решетки [14]. Так, при комнатной температуре и нормальном давлении параметр Грюнайзена ОЦК-железа равен 1.74, ГЦК-железа – 2.20. С увеличением температуры до 900 К значение параметра Грюнайзена ОЦК-железа незначительно возрастает – до 1.79. При этом значение параметра Грюнайзена ГЦК-железа практически не изменяется [14].
Поскольку целью настоящей работы является исследование диффузионных процессов и превращений в системе Fe–Ni, в которой при температурах выше 600°C и содержании более 20 ат. % Ni образуется твердый раствор с ГЦК-решеткой [15], то при разработке межатомного потенциала для железа значение параметра Грюнайзена принималось равным 2.20.
Минимум межатомного потенциала U = U0 при равновесном межатомном расстоянии d = d0 равен энергии сублимации металла:
Температурная зависимость энергии сублимации представляет собой сумму
где Hs – энтальпия сублимации, Ss – энтропия сублимации при 450–1250 К.Энтальпию сублимации никеля при комнатной температуре приняли равной 421 кДж/моль [16], железа – 415 кДж/моль [17].
Величину Ss определяли аналогично энтропии активации диффузии по формуле [18]
где G – модуль сдвига, G0 – модуль сдвига при T = = 0 К.Калибровку энергии сублимации проводили при помощи формулы (4) по температурной зависимости модуля сдвига
где ξ – коэффициент пропорциональности, который находили путем регрессионного анализа экспериментальных данных.В результате регрессионного анализа экспериментальных данных из [19] выражение (5) для никеля приобрело вид
Используя данные [19] для железа, выражение (5) преобразовали в
Полученные при помощи выражения (1) значения энергий взаимного притяжения и отталкивания атомов никеля и ГЦК-железа использовали для решения асимметричной задачи, т.е. расчета энергий взаимного притяжения и отталкивания пары атомов Fe и Ni ($U_{b}^{{{\text{Fe}} - {\text{Ni}}}}$(d) и $U_{r}^{{{\text{Fe}} - {\text{Ni}}}}$(d) соответственно) при произвольном межатомном расстоянии d согласно выражению [8]
(8)
$U_{b}^{{{\text{Fe}} - {\text{Ni}}}}(d) = - {{W}^{{{\text{Fe}} - {\text{Ni}}}}}(d)N_{d}^{{{\text{Fe}} - {\text{Ni}}}}(10 - N_{d}^{{{\text{Fe}} - {\text{Ni}}}}){\text{/}}20,$(9)
${{W}^{{{\text{Fe}} - {\text{Ni}}}}}(d) = {{\left\{ {\left[ {{{W}^{{{\text{Fe}}}}}{{{(d)}}^{2}} + {{W}^{{{\text{Ni}}}}}{{{(d)}}^{2}}} \right]{\text{/}}2} \right\}}^{{1/2}}},$(11)
$U_{r}^{{{\text{Fe}} - {\text{Ni}}}}(d) = {{W}^{{{\text{Fe}} - {\text{Ni}}}}}(d)N_{d}^{{{\text{Fe}} - {\text{Ni}}}}(10 - N_{d}^{{{\text{Fe}} - {\text{Ni}}}}){\text{/}}40,$Тогда межатомный потенциал в системе железо–никель при межатомном расстоянии d может быть определен по уравнению [9]
РЕЗУЛЬТАТЫ РАСЧЕТОВ И ОБСУЖДЕНИЕ
По выражениям (1)—(12) выполнили расчеты энергий взаимного притяжения и отталкивания атомов никеля и ГЦК-железа в рамках симметричных атомных потенциалов Fe, γ-Ni, а также асимметричных для системы Ni–γ-Fe.
Адекватность разработанных выражений межатомных парных потенциалов оценили путем сравнения расчетного значения объемного модуля упругости при равновесном межатомном расстоянии с имеющимися в литературе данными.
Расчетное значение объемного модуля упругости при равновесном межатомном расстоянии определяли путем численного дифференцирования полученных выражений парного потенциала Морзе U в точке, соответствующей равновесному межатомному расстоянию d = d0, по известной формуле [8, 20 и др.]
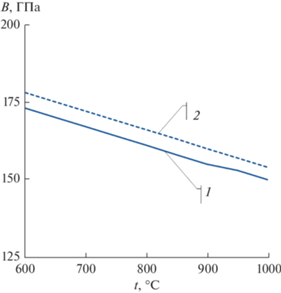
где Ω – атомный объем.Полученную температурную зависимость объемного модуля упругости никеля сопоставили с зависимостью, полученной из экспериментальных температурных зависимостей нормального модуля упругости и модуля сдвига [21, 22]. Значение коэффициента Пуассона приняли в соответствии с данными [19] равным 0.33. Расхождение между расчетными и экспериментальными значениями объемного модуля упругости Ni при 600–1000°C минимально (рис. 1).
Рис. 1.
Температурные зависимости объемного модуля упругости никеля, полученные расчетным путем из разработанного межатомного потенциала (1) и из экспериментальных данных [19] (2).
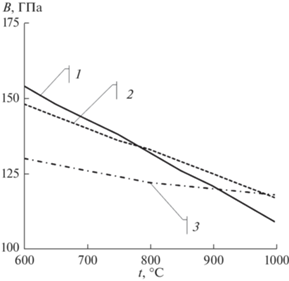
Аналогичным образом полученную температурную зависимость объемного модуля упругости ГЦК-железа сопоставили с зависимостью, полученной по экспериментальным значениям модуля сдвига [23, 24]. Известны данные о коэффициенте Пуассона железа только до 800 К включительно: от 0.25 до 0.45. По нашему мнению, такой большой разброс значений коэффициента Пуассона в указанном температурном интервале обусловлен магнитным переходом в железе из ферромагнитного в парамагнитное состояние. В этой связи, учитывая сходство электронного строения атомов железа и никеля, мы для определения объемного модуля ГЦК-железа использовали значение коэффициента Пуассона ГЦК-никеля, т.е., как указано выше, 0.33. Расхождение между расчетными и экспериментальными значениями объемного модуля упругости Fe при 600–1000°C мало (рис. 2). Несколько больше расхождение между полученным нами значением объемного модуля упругости и данными, рассчитанными на основе решения уравнения состояния в работе [14].
Рис. 2.
Температурные зависимости объемного модуля упругости ГЦК-железа, полученные расчетным путем из разработанного межатомного потенциала (1), из экспериментальных данных (2), расчетным путем на основе решения уравнения состояния [14] (3).
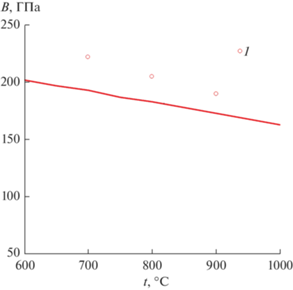
Из полученного потенциала взаимодействия атомов Ni и Fe получили расчетную зависимость объемного модуля упругости при 600–1000°C (рис. 3). Эта зависимость удовлетворительно совпадает с величинами объемного модуля упругости, полученными из экспериментальных значений нормального модуля упругости и коэффициента Пуассона ГЦК-сплава, состоящего из 51% Fe и 49% Ni [19, 25].
Таким образом, на основе парного межатомного потенциала Морзе (1) с использованием данных о параметре Грюнайзена для никеля и ГЦК-железа разработаны адекватные межатомные потенциалы никеля и железа, а также асимметричный межатомный потенциал для системы Fe–Ni. Полученные межатомные потенциалы могут быть использованы для расчетов энергии активации зарождения вакансий, диффузионных скачков, а также иных эффектов для сплавов системы железо–никель.
ЗАКЛЮЧЕНИЕ
Разработаны межатомные потенциалы в виде Морзе для ГЦК-железа и никеля на основе калибровки экспериментальных данных об энергии сублимации по температуре с использованием значений параметра Грюнайзена, имеющихся в литературе. Удовлетворительная адекватность полученных межатомных потенциалов проверена по экспериментальным и другим данным о температурных зависимостях констант упругости и коэффициента Пуассона в температурном интервале от 600 до 1000°C.
На основе полученных межатомных потенциалов для никеля и ГЦК-железа разработан асимметричный парный потенциал для атомов железа и никеля, удовлетворительная адекватность которого в температурном интервале от 600 до 1000°C проверена по температурным зависимостям нормального модуля упругости и коэффициента Пуассона железоникелевого сплава, содержащего 49% Ni.
Список литературы
Jones J.E. On the Determination of Molecular Fields. II. From the Equation of State of a Gas // Proc. R. Soc. London, A: Math., Phys. Eng. Sci. 1924. V. 106. № 738. P. 463–477.
Morse P.M. Diatomic Molecules According to the Wave Mechanics. II. Vibrational Levels // Phys. Rev. 1929. V. 34. № 1. P. 57–64.
Рехвиашвили С.Ш., Бухурова М.М., Сокуров А.А. Определение парного взаимодействия атомов по взаимодействию адатома с графеном // Журн. неорган. химии. 2020. Т. 65. № 9. С. 1229–1233.
Horiuchi T., Uzawa H., Igarashi M., Abe F., Mohri T. Determination of Lennard-Jones Type Potential for Fe–Pd Phase // Calphad. 2002. V. 26. № 1. P. 3–14.
Horiuchi T., Igarashi M., Abe F., Mohri T. Phenomenological Calculation of Phase Equilibria in the Fe–Ni System // Calphad. 2002. V. 26. № 4. P. 591–597.
Pettifor D.G. Bonding and Structure of Molecules and Solids. Oxford: Claredon, 1996. 296 p.
Семенов М.Ю., Крапошин В.С., Талис А.Л., Симич-Лафицкий Н.Д. Переброска диагоналей в ромбе – элементарный акт полиморфного превращения. Расчет энергетического порога в металлах // Металловедение и термическая обработка металлов. 2020. № 2(776). С. 8–17.
Pettifor D.G. Electron Theory of Metals / Ed. Cahn R.W. Haasen P. // Physical Metallurgy. Amsterdam: North-Holland, 1996. V. 1. P. 47–133.
Семенов М.Ю., Королев И.П., Арестов В. Построение парных потенциалов Морзе для никеля и кобальта на основе уточнения значений параметра Грюнайзена и атомной сжимаемости // Изв. РАН. Сер. физ. 2021. Т. 85. № 7. С. 948–952.
Bonny G., Castin N., Terentyev D. Interatomic Potential for Studying Ageing under Irradiation in Stainless Steels: the FeNiCr Model Alloy // Modelling Simulat Mater. Sci. Eng. 2013. V. 21. № 8. AN 085004.
Wu C., Lee B. J., Su X. Modified Embedded-Atom Interatomic Potential for Fe–Ni, Cr–Ni and Fe–Cr–Ni Systems // Calphad. 2017. V. 57. P. 98–106.
Tong K., Ye F., Gao M., Lei M.K., Zhang C. Interatomic Potential for Fe–Cr–Ni–N System Based on the Second Nearest-Neighbor Modified Embedded-Atom Method // Mol. Simulat. 2016. V. 42(15). P. 1256–1262.
Duyananda M.A. Diffusion in Ternary Alloys //Diffusion in Solid Metals and Alloys / Ed. Mehrer H. // Landolt-Bornstein New Series. V. III (26). Springer-Verlag, 1990. P. 372–436.
Dorogokupets P.I., Dymshits A.M., Litasov K.D., Sokolova T.S. Thermodynamics and Equations of State of Iron to 350 GPa and 6000 K // Sci. Rep. 2017. V. 7. № 1. P. 1–11.
Cacciamani G., De Keyzer J., Ferro R., Klotz U.E., Lacaze J., Wollants P. Critical Evaluation of the Fe–Ni, Fe–Ti and Fe–Ni–Ti Alloy Systems // Intermetallics. 2006. V. 14. № 10–11. P. 1312–1325.
Kant A. Dissociation Energies of Diatomic Molecules of the Transition Elements. I. Nickel // J. Chem. Phys. 1964. V. 41. № 6. P. 1872–1876.
Desai P.D. Thermodynamic Properties of Iron and Silicon // J. Phys. Chem. Ref. Data. 1986. V. 15. № 3. P. 967–983.
Zener C. Theory of D0 for Atomic Diffusion in Metals // J. Appl. Phys. 1951. V. 22. № 4. P. 372–375.
Ledbetter H.M., Reed R.P. Elastic Properties of Metals and Alloys, I. Iron, Nickel, and Iron-Nickel Alloys // J. Phys. Chem. Ref. Data. 1973. V. 2. № 3. P. 531–618.
Harrison W.A. Elementary Electronic Structure. Singapore: World Scientific,1999. XXI + 819 p.
Koch K.R., Dannecker C. Die Elastizität einiger Metalle und Legierungen bis zu Temperaturen, die ihrem Schmelzpunkt naheliegen // Ann. Phys. 1915. V. 352. № 10. S. 197–226.
Köster W. Die Tenperaturabhängïgkeit des Elastizitätsmoduls reiner Metalle // Z. Metallkd. 1948. V. 39. S. 1–12.
Neuhaus J., Leitner M., Nicolaus K., Petry W., Hennion B., Hiess A. Role of Vibrational Entropy in the Stabilization of the High-Temperature Phases of Iron // Phys. Rev. B. 2014. V. 89. № 18. AN. 184302.
Lindgren L.E., Back J.G. Elastic Properties of Ferrite and Austenite in Low Alloy Steels versus Temperature and Alloying // Materialia. 2019. V. 5. AN. 100193.
Chevenard P., Crussard C. Influence des traitements thermiques et mécaniques sur le coefficient de Poisson des métaux et alliages // C. R. Acad. Sci. 1943. V. 216. P. 685.
Дополнительные материалы отсутствуют.
Инструменты
Неорганические материалы