

Неорганические материалы, 2022, T. 58, № 4, стр. 442-450
Расчетная оценка адсорбционно-десорбционного поведения продуктов пиролиза при получении GaAsxP1 – x в условиях МОС-гидридной эпитаксии
А. Д. Максимов 1, *, М. А. Давыдкин 1, Т. А. Багаев 2, 3, А. Ю. Андреев 2, И. В. Яроцкая 2, М. А. Ладугин 2, А. А. Мармалюк 2
1 МИРЭА – Российский технологический университет
119454 Москва, пр. Вернадского, 78, Россия
2 АО “НИИ “Полюс” им. М.Ф. Стельмаха”
117342 Москва, ул. Введенского, 3, к. 1, Россия
3 Российский университет дружбы народов
117198 Москва, ул. Миклухо-Маклая, 6, Россия
* E-mail: maksimov_a@mirea.ru
Поступила в редакцию 27.10.2021
После доработки 27.01.2022
Принята к публикации 04.02.2022
- EDN: QOYHTC
- DOI: 10.31857/S0002337X2204011X
Аннотация
Методом расчета функционала электронной плотности определены потенциалы адсорбции и десорбции продуктов пиролиза с подложкой в процессе роста GaAsxP1– x/GaAs в условиях МОС-гидридной эпитаксии. На основании полученных данных произведена симуляция поведения продуктов пиролиза на поверхности роста с учетом адсорбции, десорбции и диффузии кинетическим методом Монте-Карло. Результаты расчетной оценки хорошо согласуются с экспериментальными данными. Представленный алгоритм расчета может быть использован для исследования адсорбционно-десорбционного поведения материалов других систем.
ВВЕДЕНИЕ
Твердые растворы GaAsхP1– х широко применяются в качестве материала активной области квантоворазмерных полупроводниковых лазеров, излучающих в ближнем инфракрасном диапазоне спектра [1–6]. Одним из основных методов промышленного производства гетероструктур для таких лазеров является МОС-гидридная эпитаксия. В качестве источников элементов V группы Периодической системы при выращивании твердых растворов GaAsхP1– х данным методом применяются арсин и фосфин. Из-за значительного различия температур полного разложения этих соединений (~500°С для арсина и ~900°С для фосфина [7]), а также из-за того, что рост эпитаксиальных слоев в методе МОС-гидридной эпитаксии проводится при температурах 600–800°С, возникают трудности управления составом материала активной области GaAsхP1– х, необходимого для точного контроля длины волны излучения [8–10].
В работе [11] предложена модель расчета состава твердого раствора GaAsхP1– х, учитывающая кинетику разложения гидридов и адсорбционно-десорбционное взаимодействие продуктов пиролиза с поверхностью роста. Эта методика показала хорошее соответствие расчета результатам проведенных экспериментов в широком диапазоне параметров роста (температура в реакторе, парциальные давления компонентов и общее давление в реакторе). Однако в данной модели для учета адсорбционно-десорбционного поведения продуктов пиролиза с поверхностью роста применялась экспериментальная температурная зависимость отношения константы адсорбции к константе десорбции, полученная в работе [12]. Применение таких экспериментальных зависимостей значительно ограничивает гибкость модели в силу того, что они получены при конкретных условиях роста. Кроме того, подобные зависимости получены для ограниченного набора материалов.
В настоящей работе поставлена задача определения потенциалов адсорбции и десорбции продуктов пиролиза в условиях роста. Для расчетов выбрана гетеросистема GaAsxP1 – x на подложке GaAs, так как поведение продуктов пиролиза фосфина и арсина при совместном осаждении досконально не изучено. На основании полученных результатов проведена симуляция поведения продуктов пиролиза на поверхности роста с учетом адсорбции, десорбции и диффузии по кинетическому методу Монте-Карло (КМК).
ТЕОРЕТИЧЕСКИЙ АНАЛИЗ
В основу расчета была положена работа [13], в которой использовался неэмпирический подход, основанный на нахождении свободной энергии компонентов в газовой фазе. Полагается, что частица, падающая на подложку, будет адсорбироваться, если ее химический потенциал в газовой фазе (μgas) больше энергии ее адсорбции на поверхность. И наоборот, если ее потенциал в газовой фазе будет меньше, чем энергия адсорбции, то частица будет десорбироваться с поверхности. Схематически данная модель показана на рис. 1.
Рис. 1.
Схематичное изображение модели исследования адсорбционно-десорбционного поведения компонентов на поверхности роста.
Для нахождения энергии адсорбции компонентов газовой фазы на поверхность роста необходимо построить и рассчитать основное состояние (с минимумом свободной энергии) двух систем: поверхность с атомом в газовой фазе и поверхность с адсорбированным на ней атомом. По разнице энергий между двумя системами можно оценить энергию адсорбции компонента на поверхности:
где Ead – энергия адсорбции, ΣEreact – сумма энергий свободной частицы и поверхности по отдельности, ΣEprod – энергия системы поверхности с адсорбированным атомом.Химический потенциал компонента в газовой фазе определялся по уравнению
(2)
${{\mu }_{{gas}}} = - kT\ln \left( {\frac{{gkT}}{p} \times {{f}_{{trans}}}{{f}_{{tot}}}{{f}_{{vibr}}}} \right),$где
(3)
${{f}_{{trans}}} = {{\left( {\frac{{2\pi MkT}}{{{{h}^{2}}}}} \right)}^{{{3 \mathord{\left/ {\vphantom {3 2}} \right. \kern-0em} 2}}}},$(4)
${{f}_{{rot}}} = \left( {\frac{1}{{\pi \sigma }}} \right){{\left[ {\frac{{8{{\pi }^{3}}{{{({{I}_{A}}{{I}_{B}} \ldots )}}^{{{1 \mathord{\left/ {\vphantom {1 n}} \right. \kern-0em} n}}}}kT}}{{{{h}^{2}}}}} \right]}^{{{n \mathord{\left/ {\vphantom {n 2}} \right. \kern-0em} 2}}}},$(5)
${{f}_{{vibr}}} = {{\prod\limits_i^{3N - 3 - n} {\left[ {1 - \exp \left( {\frac{{ - h{{\mu }_{i}}}}{{kT}}} \right)} \right]} }^{{ - 1}}}.$Здесь ftrans, frot, fvibr – суммы состояний поступательного, вращательного и колебательного движений молекул соответственно, k – постоянная Больцмана, T – температура, g – фактор вырождения энергетических электронных уровней [11], p – парциальное давление, M – масса частицы, h – постоянная Планка, σ – фактор симметрии, Ij – момент инерции по одной степени свободы, n – количество вращательных степеней свободы, N – количество атомов в частице, i – количество колебательных степеней свободы, νi – частота колебаний молекулы. Момент инерции равен
где m – масса атома, r – радиус вращения молекулы вокруг оси.Как видно из уравнений (1)–(6), неизвестными являются νi и r. Для их нахождения в данной работе используются квантово-химические вычисления по теории “Функционала электронной плотности” (Density Functional Theory – DFT) [14]. В основе метода лежит определение функции электронов в системе не через решение уравнения Шредингера с определением волновой функции, а через нахождение функции электронной плотности этой системы. Методы теории функционала плотности имеют высокую вычислительную способность при незначительном снижении точности по сравнению с неэмпирическими и полуэмпирическими методами вычислений. При этом DFT идеально подходит для больших непериодических и периодических (кристаллических) систем [15]. Согласно DFT, радиус вращения вокруг оси может быть получен как результат оптимизации молекулы, заключающейся в определении геометрической формы молекулы, соответствующей основному энергетическому состоянию. Частота колебаний рассчитывается по результатам вибрационного анализа молекулы.
На основе рассчитанных таким образом потенциалов молекул в газовой фазе и энергий адсорбции можно произвести симуляцию поведения продуктов пиролиза на поверхности роста с учетом адсорбции, десорбции и диффузии с помощью КМК [16]. Согласно данной методике, изменение вероятности Pi(τ) системы находиться в состоянии i (адсорбированное, десорбированное состояние, диффузия) в момент времени τ зависит только от вероятностей перехода из текущего состояния i в любое другое состояние j, kij и от вероятностей перехода в состояние i из любого другого состояния j, kji. С точки зрения химической кинетики, эти вероятности прыжков выражаются как константы скорости элементарных процессов с размерностью [с–1]. Таким образом, общее изменение Pi(τ) регулируется уравнением баланса (основное уравнение Маркова), которое содержит только следующие константы скорости:
(7)
$\frac{{d{{P}_{i}}(t)}}{{dt}} = - \sum\limits_{j \ne i} {{{k}_{{ij}}}} {{P}_{i}}(t) + \sum\limits_{j \ne i} {{{k}_{{ij}}}{{P}_{j}}} (t).$С математической точки зрения уравнение представляет собой систему связанных дифференциальных уравнений скорости. В случае поверхностных явлений константы скорости kij и kji можно рассчитать с помощью теории переходных состояний (ТПС), применяя уравнение Эйринга–Поляни [17]
(8)
$k_{{ij}}^{{{\text{ТПС}}}} = {{k}_{0}}\frac{{{{k}_{B}}T}}{h}\exp \left( { - \frac{{ - \Delta {{E}_{{ij}}}}}{{{{k}_{B}}T}}} \right),$(9)
$\Delta E \cong {{c}_{1}}({{E}_{{{\text{к}}{\text{.с}}}}} - {{E}_{{{\text{н}}{\text{.с}}}}}) - {{c}_{2}},$(10)
$D = \frac{{\left\langle {{\text{CКC}}(\tau ) - {\text{CК}}{{{\text{C}}}_{0}}} \right\rangle }}{{2d\tau }},$Расчеты по методу DFT проведены в программе Cp2k [20], написанной на языке Fortran 2008. Симуляция КМК проведена при помощи кода, написанного на языке программирования Python. Детали симуляции изложены при обсуждении результатов.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Экспериментальные образцы изготовлены на установке МОС-гидридной эпитаксии горизонтального типа в атмосфере высокочистого водорода при пониженном давлении. Для роста использовались подложки n-GaAs〈Si〉 с кристаллографической ориентацией (100), разориентированные на 10° к направлению 〈111〉, с концентрацией основных носителей заряда на уровне (1–3) × 1018 см–3. Активная область GaAsxP1– x, расположенная между барьерными слоями AlGaAs, во всех случаях имела толщину 100 Å. Измерение толщин активной области проводилось на просвечивающем электронном микроскопе Jeol JEM-2100F с предельным разрешением не хуже 2 Å. Состав квантовой ямы GaAsxP1 – x рассчитывался из измерения спектра фотолюминесценции при комнатной температуре. Однако следует указать, что GaAsxP1 – x являлся материалом квантовой ямы. Очевидно, что в этом случае яма будет напряженной и упругие напряжения могут влиять на поверхностные процессы [21], что в предлагаемой модели не учитывается.
РЕЗУЛЬТАТЫ РАСЧЕТА
Ранее определено [11], что основными компонентами в реакторе МОС-гидридной эпитаксии при получении GaAsxP1– x являются тетрамеры и неразложившиеся гидриды мышьяка и фосфора. Концентрация димеров при типичных температурах роста гораздо ниже, а мономера и сложных соединений вида AsxPy – пренебрежимо мала [22]. Выбор базисных наборов функций для расчетов был проведен на основе оптимизации геометрии ячейки арсенида галлия из 8 атомов. Выбраны базисы DZVP-MOLOPT-SR-GTH-q(N), а функции псевдопотенциалов – GTH-BLYP-q(N). Для проведения расчетов по методу DFT построена модель поверхности GaAs размером 3 × 3 элементарных ячейки и толщиной в одну ячейку.
Молекулы реагентов и продуктов пиролиза, для которых было определено основное состояние методом геометрической оптимизации, были размещены на поверхности арсенида галлия ориентации (100). Выбранная ориентация поверхности типична при проведении роста по методу МОС-гидридной эпитаксии. При этом моделировалась нереконструированная поверхность, так как при типичных температурах роста (t ≥ 500°С) и предварительном отжиге в атмосфере газа-носителя локально упорядоченная структура поверхности разрушается [23]. Результаты расчетов энергий систем “молекула в газовой фазе”, “поверхность” и “адсорбированная молекула + поверхность”, а также рассчитанные по ним энергии адсорбции (в атомных единицах – единицах Хартри, и в электронвольтах) представлены в табл. 1.
Таблица 1.
Энергия поверхности, рассчитанная методом DFT и энергии адсорбции компонентов
| Молекула | Энергия молекулы в свободном состоянии*, а.е. | Энергия системы “поверхность + адмолекула”, а.е. | Ead**, а.е. | Ead**, эВ |
|---|---|---|---|---|
| As2 | –25.11 | –4182.29 | –0.043 | –1.17 |
| As4 | –12.49 | –4169.69 | –0.075 | –2.04 |
| AsH3 | –7.97 | –4165.11 | –0.011 | –0.30 |
| P2 | –26.22 | –4183.38 | –0.033 | –0.90 |
| P4 | –13.05 | –4170.24 | –0.064 | –1.73 |
| PH3 | –8.27 | –4165.42 | –0.022 | –0.59 |
Для нахождения частотных характеристик молекул (νi) проведен вибрационный анализ. Результаты вычислений и литературные данные представлены в табл. 2. Видно, что используемый в настоящей работе подход применим для определения частот колебаний молекул.
Таблица 2.
Результаты вибрационного анализа
| Молекула | ν, см–1 | |
|---|---|---|
| настоящая работа | данные [13] | |
| As2 | 417 | 446 |
| P2 | 777 | 792 |
| As4 | 343 | – |
| P4 | 602 | – |
| AsH3 | 2313 | – |
| PH3 | 2456 | – |
По данным табл. 2 с использованием выражения (2) построены зависимости потенциала тетрамеров мышьяка и фосфора в газовой фазе от температуры при разных значениях парциального давления (10, 102, 103 Па) (рис. 2).
Рис. 2.
Температурные зависимости потенциала тетрамеров мышьяка и фосфора в газовой фазе при разных значениях парциального давления.
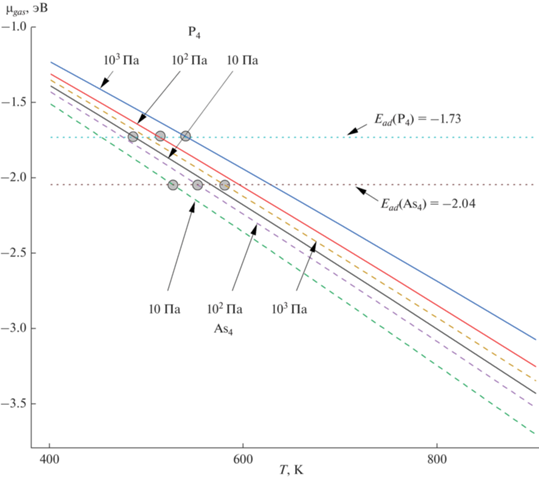
На рис. 2 горизонтальная линия показывает рассчитанный потенциал адсорбции (табл. 2). Из хода зависимостей видно, что тетрамер фосфора при равенстве парциальных давлений проявляет бóльшую склонность к десорбции. Это хорошо согласуется с экспериментом [23]. В точках пересечения с линией потенциала в газовой фазе (выделены окружностями) наблюдается равновесие адсорбции–десорбции. Таким образом построена фазовая диаграмма состояния равновесия адсорбции–десорбции для тетрамеров мышьяка и фосфора (рис. 3).
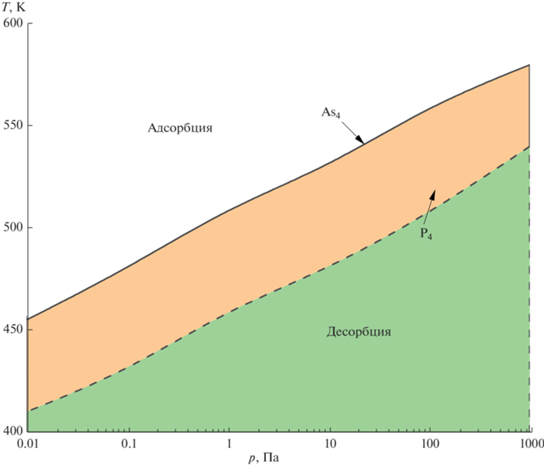
Для симуляции КМК активационные барьеры определены по уравнению (9) следующим образом:
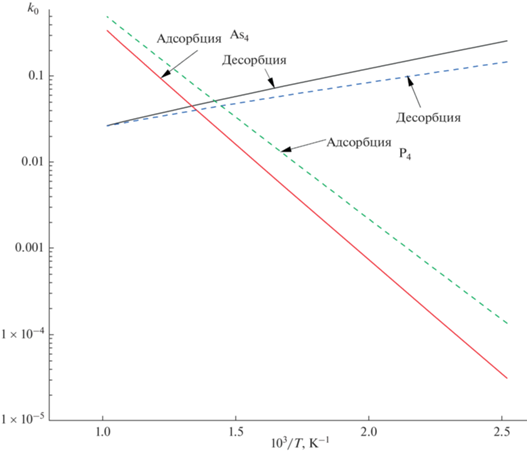
где значения Ead получены по методу DFT (табл. 2), значение μgas рассчитано по уравнению (2), а Ed = = –Ead. Значение активационного барьера диффузии принято равным 0.4 эВ [13]. Пример полученной зависимости констант скорости адсорбции и десорбции для тетрамеров фосфора и мышьяка при парциальном давлении 10 Па без учета коэффициента ${{k}_{0}}\frac{{{{k}_{B}}T}}{h}$ представлен на рис. 4. Видно, что интенсивность адсорбции убывает с ростом температуры значительно быстрее роста интенсивности десорбции, которая является активационным процессом.Рис. 4.
Температурные зависимости констант скорости адсорбции и десорбции для тетрамеров мышьяка и фосфора при парциальном давлении 10 Па: пунктиром обозначены данные для P4, сплошной линией – для As4.
Адсорбция симулировалась по КМК для 10 000 частиц, изначально расположенных в газовой фазе. При этом в цепи Маркова (выражение (8)) учитывались также темпы десорбции и диффузии. Аналогично, симуляция десорбции проведена для 10 000 частиц, изначально находящихся в адсорбированном состоянии. Результаты симуляции для As4 и P4 представлены на рис. 5.
Рис. 5.
Результаты симуляции адсорбционно-десорбционного поведения As4 и P4 на поверхности GaAs(100).
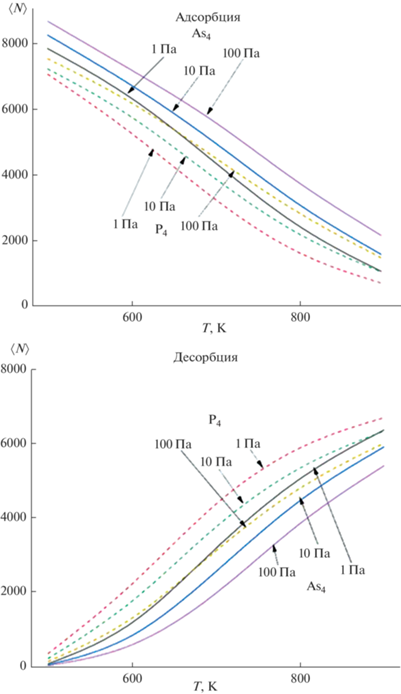
Из результатов симуляции видно, что при равной температуре для достижения одинакового количества адсорбированных атомов тетрамеров мышьяка и фосфора необходимо не менее чем десятикратное увеличение парциального давления последнего. При этом адсорбированные молекулы фосфора значительно легче десорбируются, что также требует порядка десятикратного повышения парциального давления для получения требуемых концентраций тетрамеров на поверхности. Это хорошо согласуется с практическим опытом [24, 25]. Исходя из того, что количество молекул на поверхности есть произведение результатов адсорбции и десорбции (в долях от общего количества), результаты симуляции можно представить в виде кривой Аррениуса, которая позволяет оценить энергию активации процесса осаждения. Это уравнение Аррениуса, полученное из первых принципов, применено вместо эмпирической зависимости в расчетной методике работы [11] по определению состава твердого раствора GaAsxP1 – x, получаемого в условиях МОС-гидридной эпитаксии. Результаты представлены на рис. 6.
Рис. 6.
Зависимости изопериодного с GaAs состава твердого раствора GaAsxP1 –x от исходного отношения фосфина и арсина при температурах роста от 650 до 800°С: линиями обозначены результаты расчета, экспериментальные данные представлены маркерами.
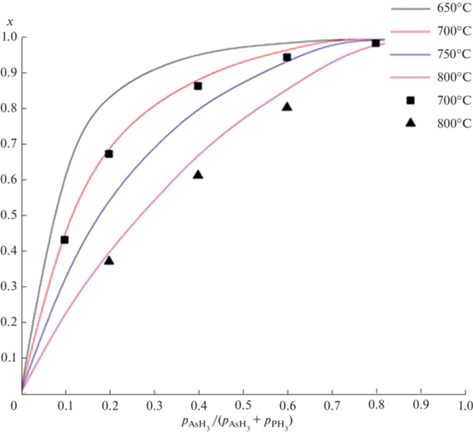
Из данных рис. 6 видно, что применение результатов симуляции адсорбции и десорбции для расчета состава твердых растворов GaAsxP1– x показывает высокую точность. Относительная погрешность не превышает 10%.
ЗАКЛЮЧЕНИЕ
Методом DFT определены потенциалы адсорбции и десорбции продуктов пиролиза с подложкой в процессе роста GaAsxP1 – x/GaAs в условиях МОС-гидридной эпитаксии. Проведена симуляция поведения продуктов пиролиза на поверхности роста с учетом адсорбции, десорбции и диффузии кинетическим методом Монте-Карло. Рассчитан состав твердых растворов GaAsxP1– x для заданных потоков источников мышьяка и фосфора, значений температуры и давления парогазовой смеси с учетом результатов симуляции. Относительная погрешность при определении состава не превышает 10%. Предложенный алгоритм применим для расчета составов и в других многокомпонентных системах.
Список литературы
Crump P., Dong W., Grimshaw M., Wang J., Patterson S., Wise D., DeFranza M., Elim S., Zhang S., Bougher M., Patterson J., Das S., Bell J., Farmer J., DeVito M., Martinsen R. 100-W+ Diode Laser Bars Show > 71% Power Conversion from 790-nm to 1000-nm and Have Clear Route to >85% // Proc. SPIE. 2007. V. 6456. P. 64560M-1-11. https://doi.org/10.1117/12.704496
Wang Y., Yang Y., Qin L., Wang C., Yao D., Liu Y., Wang L. 808nm High-Power High-Efficiency GaAsP/GaInP Laser Bars // Proc. SPIE–Int. Soc. Opt. Eng. 2008. V. 7135. P. 71350N-1-8. https://doi.org/10.1117/12.803301
Li P., Jiang K., Zhang X., Tang Q., Xia W., Li S., Ren Z., Xu X. 20.8W TM Polarized GaAsP Laser Diodes of 808nm Wavelength // Proc. SPIE. 2013. V. 8605. P. 860510-1-5. https://doi.org/10.1117/12.2002983
Безотосный В.В., Васильева В.В., Винокуров Д.А., Капитонов В.А., Крохин О.Н., Лешко А.Ю., Лютецкий А.В., Мурашова А.В., Налет Т.А., Николаев Д.Н., Пихтин Н.А., Попов Ю.М., Слипченко С.О., Станкевич А.Л., Фетисова Н.В., Шамахов В.В., Тарасов И.С. Мощные лазерные диоды с длиной волны излучения 808 нм на основе различных типов асимметричных гетероструктур со сверхшироким волноводом // Физика и техника полупроводников. 2008. Т. 42. № 3. С. 357–360.
Дегтярева Н.С., Кондаков С.А., Микаелян Г.Т., Горлачук П.В., Ладугин М.А., Мармалюк А.А., Рябоштан Ю.Л., Яроцкая И.В. Непрерывные мощные лазерные линейки спектрального диапазона 750–790 нм // Квантовая электроника. 2013. Т. 43. № 6. С. 509–511.
Levy M., Berk Y., Karni Y. Effect of Compressive and Tensile Strain on the Performance of 808 nm QW High Power Laser Diodes // Proc. SPIE. 2006. V. 6104. P. 93–104. https://doi.org/10.1117/12.645815
Stringfellow G.B. Organometallic Vapor-Phase Epitaxy. Theory and Practice. Cambridge: Academic Press, 1999. P. 572.
Fukui T., Kobayashi N. Vapor-Solid Distribution Relation in MOCVD GaAsxP1 – x and InAsxP1 – x // J. Cryst. Growth. 1985. V. 71. № 1. P. 9–11.
Zhong L., Ma X., Wang S., Liu S. 808 nm GaAsP/GaInP Laser Diode Arrays Grown by MOCVD Using AsH3 and TBP // IEEE 2008 International Nano-Optoelectronics Workshop (i-Now). Tokyo, 2008. V. 12. P. 237–238.
Chen D., Cheng G., Hicks R.F., Noori A.M., Hayashi S.L., Goorsky M.S., Kanjolia R., Odedra R. Metalorganic Vapor-Phase Epitaxy of III/V Phosphides with Tertiarybutylphosphine and Tertiarybutylarsine // J. Cryst. Growth. 2004. V. 270. P. 322–328.
Максимов А.Д., Эйстрих-Геллер В.Ю., Мармалюк А.А., Ладугин М.А., Багаев Т.А., Горлачук П.В., Яроцкая И.В. Модель расчета состава твердых растворов GaAsP в условиях МОС-гидридной эпитаксии // Неорган. материалы. 2017. Т. 53. № 4. С. 362–368. https://doi.org/10.7868/S0002337X17040121
Samuelson L., Omling P., Grimmeiss H.G. Alloying Mechanisms in MOVPE GaAs1 – xPx // J. Cryst. Growth. 1983. V. 61. P. 425–426.
Kangawa Y., Akiyama T., Ito T., Shiraishi K., Nakayama T. Surface Stability and Growth Kinetics of Compound Semiconductors: An Ab Initio-Based Approach // Materials. 2013. V. 6. P. 3309–3360.
Blinder S.M. Chapter 14 – Density Functional Theory. Introduction to Quantum Mechanics (Second edition). Amsterdam: Elsevier, 2021. P. 235–244.
Hasnip P.J., Refson K., Prober M.I.J., Yates J.R., Clark S.J., Pickard C.J. Density Functional Theory in the Solid State // Philos. Trans. R. Soc. A. 2014. V. 372. P. 20130270.
Andersen M., Panosetti C., Reuter K. A Practical Guide to Surface Kinetic Monte Carlo Simulations. // Front. Chem. 2019. V. 7. P. 202–223.
Laidler K.J. Chemical Kinetics. N.Y.: Harper & Row, 1987. P. 272.
Nørskov J.K., Bligaard T., Logadottir A., Bahn S., Hansen L.B. et al. Universality in Heterogeneous Catalysis // J. Catal. 2002. V. 209. № 2. P. 275–278.
Michaelides A., Liu Z.-P., Zhang C.J., Alavi A., King D.A., Hu P. Identification of General Linear Relationships between Activation Energies and Enthalpy Changes for Dissociation Reactions at Surfaces // J. Am. Chem. Soc. 2003. V. 125. P. 3704–3705.
Kühne1 T.D., Iannuzzi M., Del Ben M., Rybkin V.V., Seewald P. CP2K: An Electronic Structure and Molecular Dynamics Software Package – Quickstep: Efficient and Accurate Electronic Structure Calculations // J. Chem. Phys. 2020. V. 152. № 19. P. 194103-1-48.
Светогоров В.Н., Акчурин Р.Х., Мармалюк А.А., Ладугин М.А., Яроцкая И.В. Расчет упругонапряженной гетероструктуры AlxGayIn1 –x–yAs/InP с квантовыми ямами для эффективных лазерных излучателей // Рос. технол. журн. 2018. Т. 6. № 2. С. 46–54.
Jordan A.S., Robertson A. Copyrolysis of AsH3 and PH3 in the Epitaxial Growth of Ternary and Quaternary III–V Alloys // J. Cryst. Growth. 1994. V. 137. P. 224–230.
Kobayashi Y., Kobayashi N. Chemical-Bonding Structure of InP Surface in MOVPE Studied by Surface Photo-Absorption // J. Electron. Mater. 1996. V. 25. P. 691–624.
Smeets E.T.J.M., Cox A.M.W. Influence of Alkyl Substituents of Oms and Operating Pressure on the Quality of InxGa1 – xAs/InP Heterostructures Grown by OMVPE // J. Cryst. Growth. 1986. V. 77. № 1–3. P. 347–353.
Biefeld R.M. The Preparation of InAs1 –xSbx alloys and strained-layer superlattices by MOCVD // J. Cryst. Growth. 1986. V. 77. № 1–3. P. 392–399.
Дополнительные материалы отсутствуют.
Инструменты
Неорганические материалы