

Журнал неорганической химии, 2019, T. 64, № 2, стр. 181-189
1,8-бис[2-(дифенилфосфорил)фенокси-4-фенилдиазенил)]-3,6-диоксаоктан (L): синтез, комплексообразующие и ионоселективные свойства. Кристаллическая и молекулярная структура L ⋅ 0.25H2O и [LiL]I3 ⋅ MePh
И. С. Иванова 1, *, А. Б. Илюхин 1, И. Н. Полякова 1, Ю. И. Рогачева 2, Е. Н. Пятова 1, Г. С. Цебрикова 3, В. Е. Баулин 2, 3, А. Ю. Цивадзе 3
1 Институт общей и неорганической химии им. Н.C. Курнакова РАН
119991 Москва, Ленинский пр-т, 31, Россия
2 Институт физиологически активных веществ РАН
142432 Московской обл, Черноголовка, Северный пр-д, 1, Россия
3 Институт физической химии и электрохимии им. А.Н. Фрумкина РАН
119991 Москва, Ленинский пр-т, 31, Россия
* E-mail: isivanova@mail.ru
Поступила в редакцию 07.03.2018
После доработки 04.07.2018
Принята к публикации 09.04.2018
Аннотация
Описаны синтез, ИК- и электронные спектры поглощения и ионоселективные свойства нового фосфорилподанда 1,8-бис[2-(дифенилфосфорил)фенокси-4-фенилдиазенил]-3,6-диоксаоктана (L). Методом РСА установлены кристаллические структуры L ⋅ 0.25H2O и синтезированного комплекса [LiL]I3 · MePh. Определен состав пластифицированной полимерной мембраны литийселективного электрода на основе L.
ВВЕДЕНИЕ
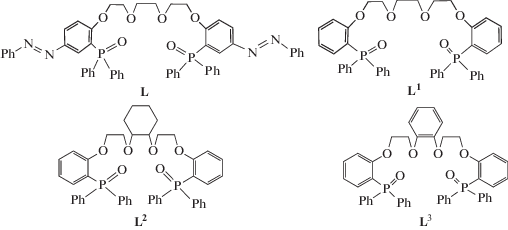
Несмотря на большие успехи в области разработки оптических химических сенсоров на основе различных макроциклических соединений [1, 2], поиск новых хромогенных соединений, позволяющих проводить мониторинг различных ионов в окружающей среде и биологических объектах, остается актуальным направлением исследований. Интерес к фосфорсодержащим подандам, концевые группы которых представляют собой фрагменты орто-фосфорилированных фенолов, связан с простотой синтеза и довольно широкими возможностями их практического применения [3–6]. Комплексообразующие и другие свойства подандов зависят от многих факторов, в частности, от количества полиэфирных фрагментов и различных функциональных групп [7–14]. Установлено, что гексадентатные фосфорилподанды L1−L3 обладают ионофорными свойствами по отношению к катионам щелочных и щелочноземельных металлов, проявляя селективность по отношению к катиону лития [15–17].
Известно [1, 2, 18], что присутствие хромофорных фрагментов позволяет получать соединения, способные изменять фотофизические свойства при селективном связывании катиона металла. Поэтому весьма перспективно с точки зрения получения оптически активных и ионоселективных фосфорилподандов объединение в одной молекуле комплексообразующих и хромофорных фрагментов.
Цель настоящей работы – синтез и исследование ИК- и электронных спектров поглощения и ионоселективных свойств нового фосфорилподанда – 1,8-бис[2-(дифенилфосфорил)фенокси-4-фенилдиазенил]-3,6-диоксаоктана (L) с двумя π-электронными диазофенильными фрагментами, которые являются распространенным типом хромофоров и характеризуются полосой поглощения в области УФ/видимого диапазона, а также установление кристаллических структур L ⋅ 0.25H2O (I) и [LiL]I3 ⋅ MePh (II).
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Спектры ЯМР регистрировали на приборах Bruker-СPX-200 и Bruker-DXР-200 (200 МГц) относительно тетраметилсилана (1Н) или 85%-ной Н3РО4 (31P). ТСХ-анализ выполняли на пластинах Silufol с проявлением парами иода. Температуру плавления определяли с помощью укороченного термометра Аншутца. Элементный анализ проводили на С, Н, N-анализаторе (Carlo Erba Strumentazione, Italy) и атомно-эмиссионном спектрометре с индуктивно-связанной плазмой IRIS Advantage (“Thermo Jarrell Ash”, США).
Синтез
2-(Дифенилфосфорил)-4-(фенилдиазенил)фенол. Смесь 3.98 г (16.9 ммоль) 2-(дифенилфосфорил)фенола [19] и 0.67 г (16.9 ммоль) NaOH в 10 мл воды и 20 мл диоксана перемешивали при 100°C. Через 0.5 ч в эту смесь добавляли раствор 26.0 г СН3СООNa в 50 мл воды и охлаждали до 0°С. Затем туда же по каплям прибавляли раствор фенилдиазония, полученный из 4.7 г (50.0 ммоль) свежеперегнанного анилина, 3.5 г (50.0 ммоль) NaNO2 и 10 мл 35%-ной HCl. Температуру повышали до комнатной и перемешивали в течение 2 ч. Осадок отфильтровывали, промывали 40 мл разбавленной (1 : 1) HCl, водой (50 мл) и высушивали в вакууме. Выход 5.0 г, 74%, tпл = 201–202°С (бензол).
Спектр ПМР (DMSO-d6), δ, м. д.: 8.80–8.30 м (18Н, Ar–H), 11.25 c (1Н, Ar–OH). Спектр ЯМР 31Р (DMSO-d6), δ, м. д.: 18.87.
1,8-Бис[2-(дифенилфосфорил)фенокси-4-фенилдиазенил]-3,6-диоксаоктан (L). К суспензии 3.98 г (10.0 ммоль) 2-(дифенилфосфорил)-4-(фенилдиазенил)фенола в 50 мл безводного диоксана добавляли 0.43 г (10.0 ммоль) 55%-ной суспензии NaH в вазелиновом масле и перемешивали в течение 0.5 ч при 100°С. Затем в реакционную смесь добавляли 2.29 г (5.0 ммоль) дитозилата триэтиленгликоля в виде порошка и смесь кипятили 8 ч. После удаления растворителя к остатку добавляли 45 мл разбавленной HCl (1 : 1) и экстрагировали CHCl3 (3 × 35 мл). Экстракт последовательно промывали 10%-ным раствором KOH (2 × 30 мл), разбавленной HCl (2 × 30 мл), водой (3 × 35 мл) и упаривали в вакууме. Остаток хроматографировали на колонке с силикагелем марки L (100–160 µ), элюент СHCl3 и СHCl3 : i-PrOH = 100 : 3. Выход L составил 3.36 г (74%), tпл = 201–202°С.
Спектр ПМР (CDCl3), δ, м. д.: 3.21, т (4Н, 3Jн–н = 4.89 Гц, ОСН2СН2), 3.32, с (4Н, ОСН2СН2ОСН2СН2ОСН2СН2О), 3.90, т (4Н, 3Jн–н = = 4.89 Гц, ОСН2СН2).
Кристаллы L ⋅ 0.25H2O (I) получены перекристаллизацией из ацетонитрила.
Комплекс [LiL]I3⋅ MePh (II) в виде очень мелких и тонких кристаллов получен взаимодействием фосфорилподанда с LiI в ацетонитриле (М : L = = 3 : 1) при комнатной температуре. Кристаллы, пригодные для рентгеноструктурного анализа (РСА), получены в результате перекристаллизации комплекса из смеси MePh + MeOH (1 : 1).
Попытки получить комплексы L с катионами не только лития, но и натрия с другими анионами в виде монокристаллов не увенчались успехом.
Спектральные исследования. ИК-спектры поглощения записывали на спектрометре VERTEX 70 фирмы Bruker в диапазоне 4000–400 см–1 (суспензия в вазелиновом масле и гексахлорбутадиене) и методом НПВО на спектрометре Nexsus, Nicolete. Электронные спектры поглощения (ЭСП) записывали на UV-VIS спектрофотометре Carry 100 Scan.
РСА. Экспериментальные данные для соединений I и II получены на дифрактометре Bruker SMART APEX2 (λ(MoKα), графитовый монохроматор) [20] (табл. 1). Поглощение учтено полуэмпирическим методом по эквивалентам по программе SADABS [21]. Структуры определены комбинацией прямого метода и Фурье-синтеза. Величины заселенностей разупорядоченных фрагментов получены при изотропном уточнении структур с фиксированными тепловыми параметрами разупорядоченных атомов и в последующих расчетах не уточнялись. Структуры уточнены полноматричным анизотропно-изотропным (часть разупорядоченных атомов) методом наименьших квадратов. Атомы водорода рассчитаны из геометрических соображений. Все расчеты выполнены по программам SHELXS и SHELXL [22].
Таблица 1.
Основные кристаллографические данные и результаты уточнения структур I и II
| Параметр | I | II |
|---|---|---|
| T, K | 100(2) | 150(2) |
| Сингония | Триклинная | Ромбическая |
| Пр. гр. | P1 | Pna21 |
| a, Å | 12.3507(14) | 18.6038(13) |
| b, Å | 13.3063(15) | 15.5201(11) |
| c, Å | 15.8366(17) | 20.5045(15) |
| α, град | 110.138(2) | 90 |
| β, град | 95.856(2) | 90 |
| γ, град | 103.536(2) | 90 |
| V, Å3 | 2327.6(4) | 5920.3(7) |
| Z | 2 | 4 |
| ρвыч, г/см3 | 1.306 | 1.560 |
| µ, мм–1 | 0.151 | 1.691 |
| Размер кристалла, мм | 0.20 × 0.20 × 0.12 | 0.20 × 0.16 × 0.06 |
| Интервал θ, град | 2.048, 27.526 | 2.189, 25.393 |
| Интервал индексов | –16 ≤ h ≤ 16 –17 ≤ k ≤ 17 –20 ≤ l ≤ 20 |
–22 ≤ h ≤ 20 –18 ≤ k ≤ 18 –24 ≤ l ≤ 24 |
| Собранных отражений | 23043 | 38471 |
| Независимых отражений (Rint) | 10652, 0.0490 | 10725, 0.0573 |
| Полнота до θ = 25.242°, % | 99.8 | 99.7 |
| Max, min пропускание | 0.7456, 0.5801 | 0.259, 0.1963 |
| Ограничения/параметры | 30/670 | 103/787 |
| GООF | 0.952 | 1.179 |
| R1, wR2 (I > 2σ(I)) | 0.0529, 0.1225 | 0.0712, 0.1327 |
| R1, wR2 (весь массив) | 0.1078, 0.1506 | 0.0993, 0.1426 |
| Δρmax/Δρmin, е Å–3 | 0.489, –0.310 | 0.796, –0.767 |
Экспериментальные данные для структур I и II депонированы в Кембриджском банке структурных данных (CCDC № 1826436, 1826437).
ЭДС измеряли pH-ионометром ОР-300 (Radelkis). В качестве электрода сравнения использовали хлорсеребряный электрод ОР-0820Р “Раделкис”. Электроаналитические параметры мембраны ионоселективного электрода (ИСЭ) определяли согласно рекомендациям IUPAC [23] при рН 5−7. Коэффициенты потенциометрической селективности определяли методами как смешанных, так и отдельных растворов [24].
Пластифицированные полимерные мембраны для ИСЭ, содержащие L в качестве активного компонента, готовили по известной методике [25]. Мембраны имели следующий состав (мас. %): активный компонент − 1−2%, поливинилхлорид − 30−33%, пластификатор − 65−69%, липофильная добавка (тетракис(4-хлорфенил)борат калия) – 0–2%. Содержание липофильной добавки варьировали от 0 до 100% относительно содержания электродоактивного компонента (ЭАК). В качестве пластификатора использовали дибутилфталат (ДБФ) или ортонитрофенилоктиловый эфир (ОНФОЭ).
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
1,8-Бис[2-(дифенилфосфорил)фенокси-4-фенилдиазенил]-3,6-диоксаоктан (L) построен по принципу объединения комплексообразующей платформы фосфорилподанда 1,8-бис[2-(дифенилфосфорил)фенокси]-3,6-диоксаоктана (L1) [15], обладающего литиевой селективностью, и двух диазофенильных π-электронных “хвостиков” в качестве хромофорных фрагментов. Фосфорилподанд L получен взаимодействием 2-(дифенилфосфорил)-4-(фенилдиазенил)фенола с дитозилатом триэтиленгликоля в присутствии гидрида натрия в качестве основания в кипящем диоксане. Исходный 2-(дифенилфосфорил)-4-(фенилдиазенил)фенол впервые получен реакцией азосочетания 2-(дифенилфосфорил)фенола с фенилдиазонием (схема).
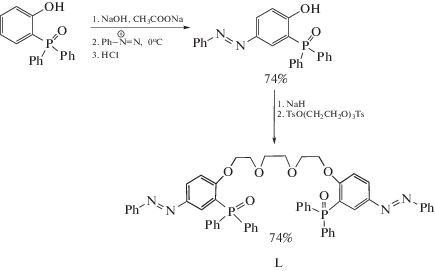
Схема получения 1,8-бис[2-(дифенилфосфосфорил)фенокси-4-фенилдиазенил]-3,6-диоксаоктана (L).
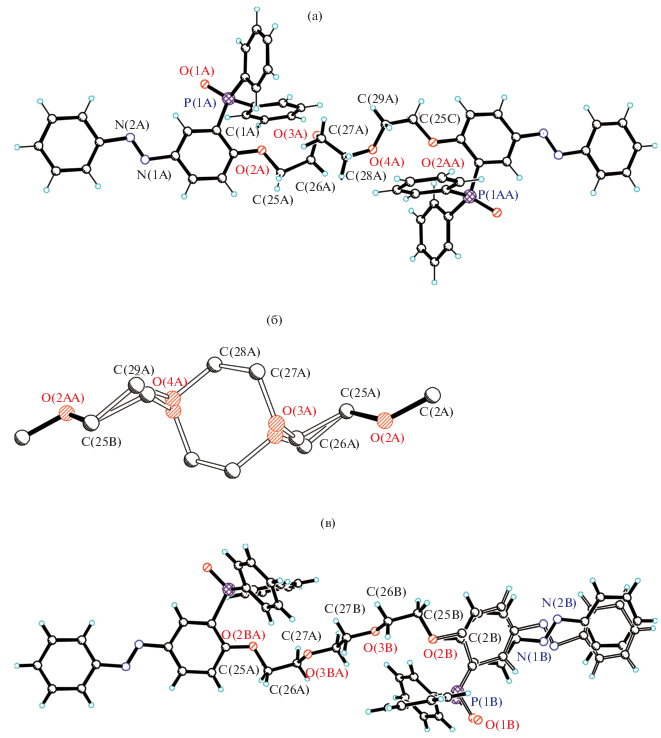
Структура I образована двумя кристаллографически независимыми центросимметричными лигандами L и кристаллизационной молекулой H2O. Обе молекулы L разупорядочены. В молекуле P(1A) (рис. 1а) разупорядочена триэтиленгликолевая цепочка (рис. 1б), в молекуле P(1B) − дифенилазеновый фрагмент (рис. 1в). Уточнение структуры в пр. гр. P1 не приводит к снятию разупорядоченности. Правильность выбора пр. р. P1 подтверждается и соотношением заселенностей разупорядоченных фрагментов – 1 : 1 для молекулы P(1A) и 1 : 4 для молекулы P(1B). Конформация триэтиленгликолевой цепочки в двух молекулах различна – t-g-g-g-g-g-t-g-t и t-t-g-t-t-t-g-t-t.
Рис. 1.
Строение свободного лиганда L в структуре I, молекула P(1A) (а), разупорядочение триэтиленгликолевой цепочки в молекуле P(1A) (б), молекула P(1B) (в).
Структура II образована комплексами (рис. 2а), анионами I3– и сольватными молекулами толуола. Молекула толуола и анион разупорядочены в соотношении 0.55 : 0.45. КЧ(Li) = 4 + 2 (табл. 2), полиэдр нерегулярный. Атомы O(2)–О(5) с точностью 0.32 Å лежат в одной плоскости. Аналогичное строение комплекса найдено нами в [Li(1,8-бис[2-(дифенилфосфорил)фенокси]-3,6-диоксаоктане)]ClO4 ⋅ 2C6H6 (III) [15] (рис. 2б).
Рис. 2.
Строение комплекса в структуре II (а), сравнение строения комплексов из соединений II и III (б), атомы H для ясности опущены.
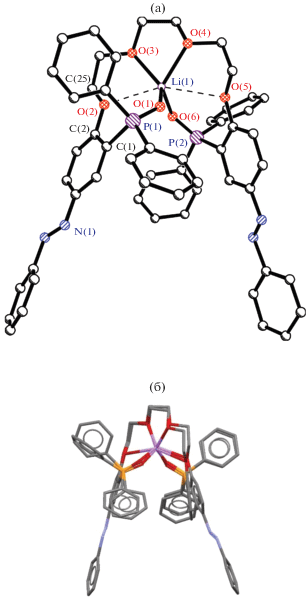
Таблица 2.
Длины связей Li–O в структуре II
| Связь | d, Å | Связь | d, Å |
|---|---|---|---|
| Li(1)–O(1) | 1.92(2) | Li(1)–O(4) | 2.16(2) |
| Li(1)–O(2) | 2.58(2) | Li(1)–O(5) | 2.76(2) |
| Li(1)–O(3) | 2.13(2) | Li(1)–O(6) | 1.92(2) |
Отнесение основных колебательных частот в ИК-спектре L и комплекса II проведено на основании выполненных ранее спектральных и структурных исследований близкого аналога 1,8-бис[2-(дифенилфосфорил)фенокси]-3,6-диоксаоктана (L1) и его комплекса лития (III) (табл. 3) [15]. Спектры L и L1 аналогичны практически во всем спектральном диапазоне, и полосы поглощения, обусловленные колебаниями электронодонорных групп, проявляются в одних и тех же интервалах волновых чисел. Что касается колебаний, обусловленных связью –N=N–, то к ним, согласно [26], можно отнести очень малоинтенсивную полосу при 1528 см–1, отсутствующую в спектре L1. Валентному колебанию связи Р=О в ИК-спектре L соответствует интенсивная полоса при 1178 см–1 (1179 см–1 в спектре L1). Полоса ν(Рh–О) лежит около 1258 см–1, что на 13 см–1 выше, чем в спектре L1(1245 см–1).
Таблица 3.
Отнесение некоторых колебательных частот (см–1) в ИК-спектрах L и [LiL]I3 ⋅ MePh
| Отнесение | L | [LiL]I3 ⋅ MePh |
|---|---|---|
| ν(Ph=O) | 1178 | 1182 |
| δ(Ph) | 1164 1145 1121 |
1139 1122 |
| νas(COC) | 1103 | 1102 |
| δ(Ph) | 1073 | |
| νs(COC) + ν(CC) | 1061 1043 | 1069 1045 1025 |
| δ(Ph) | 999 | 999 |
| ν(CO) + ν(CC) + ρ(CH2) |
932 922 875 839 |
955 930 870 843 824 807 |
| δ(Ph) | 765 755 740 721 701 688 650 |
769 752 737 706 683 663 |
| δ(СОС) | 617 597 |
617 591 |
| δ(PhРО) | 559 | 557 |
В ИК-спектре II, в отличие от L, около 1684 см–1 появляется новая интенсивная полоса, связанная, по-видимому, с присутствием в составе комплекса молекулы толуола. Полоса ν(Р=О) лежит при 1182 см–1, что немного выше по сравнению с ее положением в спектре L (1178 см–1), а полоса ν(Рh–О) не меняет своего положения. Аналогичная ситуация наблюдалась для комплексов лития с L1: ν(Р=О) повышалась на 5−7 см–1, а ν(Рh–О) оставалась неизменной.
Основные различия спектров L и комплекса II, обусловленные конформационной перестройкой поданда, наблюдаются только в конформационно-чувствительной области 1000–800 см–1 (табл. 3).
В работе [16] о комплексообразовании фосфорилподандов в растворе судили на основании данных кондуктометрических исследований, которые являются очень трудоемкими и проводились в других средах (смешанный растворитель ТГФ–СНCl3 (4 : 1)). Введение хромофорных фрагментов в молекулу L позволило исследовать комплексообразование в растворе с помощью простого метода электронной спектроскопии в том же растворителе, из которого комплекс получен в твердом виде.
ЭСП L в ацетонитриле состоит из двух полос: интенсивной полосы при 208 нм (lg ε = 5.5), соответствующей π−π*-электронным переходам бензольных колец, и полосы при 342 нм (lg ε = 4.6) с плечом при 437 нм (lg ε = 3.3), обусловленной наличием в молекуле L хромофорной группы –N=N–. В ЭСП L1 – L3 такой группы нет и полоса при 342 нм отсутствует.
При титровании раствора иодида лития раствором L обнаружен небольшой ионохромный эффект, который наблюдался и при обратном титровании. В результате комплексообразования при добавлении соли в ацетонитриле к L в ацетонитриле происходит уменьшение интенсивности полосы при 342 нм и небольшой гипсохромный сдвиг ее максимума до 338 нм, что, вероятно, связано с перераспределением электронной плотности в молекуле лиганда по сопряжению в сторону групп P=O (рис. 3а). На кривой титрования наблюдается излом при соотношении сL : сМ ≈ 1 (сL и сМ – аналитические концентрации реагентов), что указывает на преимущественное образование комплекса состава M : L = 1 : 1. Уменьшение интенсивности после точки излома M : L = 1 : 1, вероятно, связано с небольшим разбавлением системы при дальнейшем добавлении соли. Кроме того, в процессе титрования происходит увеличение интенсивности новой полосы при 245 нм (рис. 3б). Спектр исходной соли содержит полосы при 247 нм (lg ε = 4.1) и 209 нм (lg ε = 4.2). Однако рост полосы 245 нм при добавлении иодида лития неравномерен: на кривой титрования имеется излом также при соотношении сL : сМ ≈ 1. Результаты обратного титрования (добавление раствора L к раствору иодида лития) также свидетельствуют об образовании комплекса состава M : L = 1 : 1, что находится в согласии с данными РСА.
Рис. 3.
Спектрофотометрическое титрование раствора L в ацетонитриле раствором иодида лития (I) в ацетонитриле: а − cL = 1.13 × 10–5 моль/л, б − cL = 5.70 × 10–6 моль/л.
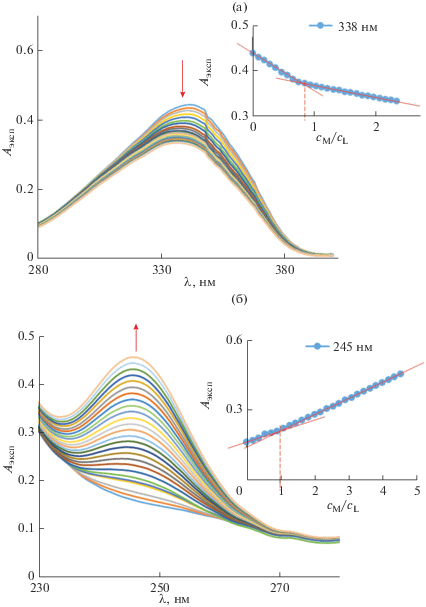
На основе данных ЭСП с помощью компьютерной программы CHEMEQUI [27, 28] выполнен расчет константы равновесия для Li + L = LiL в ацетонитриле: lg K = 2.8 ± 0.2.
Известно [16, 29], что аналитические свойства ИСЭ (предел обнаружения, потенциометрическая селективность и др.) с пластифицированными полимерными мембранами на основе моноподандов c дифенилфосфорильными группами зависят от особенностей их строения. Ранее нами было установлено, что гексадентатные фосфорилподанды с дифенилфосфорильными концевыми группами в той или иной степени обладают литиевой селективностью [15, 17]. Тестирование аналитических свойств потенциально гексадентатных фосфорилподандов показало [17], что введение жестких заместителей в этиленгликолевую цепочку подандов и закрепление С-образной формы 1,2-бис[2-((2-дифенилфосфорил)фенокси)этокси]-циклогексана (L2) и 1,2-бис[2-((2-дифенилфосфорил)фенокси)этокси]-бензола (L3) и их структурных аналогов приводят к некоторому изменению мембрано-активных свойств ИСЭ по отношению к катиону лития.
В настоящей работе протестированы ионоселективные свойства полимерных пластифицированных мембран на основе L по отношению к катионам щелочных, щелочноземельных и некоторых переходных металлов.
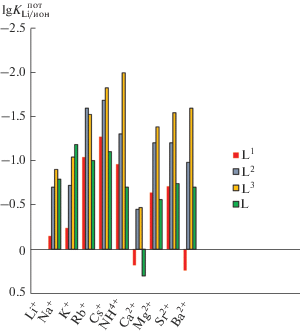
Электродная функция для Li+-селективного электрода на основе L прямолинейна в пределах концентраций 5 × 10–4–10–1 моль/л, угловой наклон электродной характеристики составляет 48.0 мВ/рLi+, предел обнаружения равен 1.3 × × 10–5 моль/л. В табл. 4 представлены электродные характеристики литиевых электродов на основе L и сравнительных электродов на основе L1−L3, ДБФ и липофильной добавки – тетракис(4-хлорофенил)бората калия (содержание относительно ЭАК – 50%). На рис. 4 приведены коэффициенты потенциометрической селективности (Kпот) литиевых электродов на основе L и L1−L3 с мембранами одинакового состава по отношению к катионам щелочных, щелочноземельных металлов. Из рис. 4 видно, что значения Kпот литий/натриевой селективности для L значительно выше, чем у L1, и примерно равны таковым для L2 и L3.
Таблица 4.
Элeктродные характеристики мембран литийселективных электродов на основе L и L1−L3 (ДБФ и тетракис(4-хлорофенил)борат калия – 50%-ное содержание от ЭАК)
| Фосфорилподанд | Li+-ИСЭ | ||
|---|---|---|---|
| линейный диапазон электродной функции, моль/л | наклон электродной функции, мВ/pLi+ | предел обнаружения, моль/л | |
| L1 | 5 × 10–4–10–1 | 55.0 | 4. 1 × 10–4 |
| L2 | 10–4–10–1 | 53.0 | 7. 5 × 10–5 |
| L3 | 10–4–10–1 | 55.0 | 4. 2 × 10–5 |
| L | 5 × 10–5–10–1 | 48.0 | 1. 3 × 10–5 |
Рис. 4.
Коэффициенты потенциометрической селективности (Kпот) литиевых электродов на основе L и L1−L3 (ДБФ и тетракис(4-хлорoфенил)борат калия − 50%-ное содержание от ЭАК).
На основании проверки влияния природы растворителя (ДБФ и ОНФОЭ) и количества липофильной добавки (содержание тетракис(4-хлорофенил)бората калия 0, 50, 100% относительно ЭАК) на электроаналитические параметры ИСЭ на основе L определена оптимальная композиция мембраны.
ВЫВОДЫ
Таким образом, синтезирован новый фосфорилподанд 1,8-бис[2-(дифенилфосфорил)фенокси-4-фенилдиазенил]-3,6-диоксаоктан (L), установлены кристаллические структуры L ⋅ 0.25H2O и комплекса [LiL]I3 ⋅ MePh. Показано, что введение хромофорного заместителя в пара-положение бензольного кольца позволяет исследовать комплексообразование в растворе с помощью простого метода электронной спектроскопии, не отражается на способе координации, не оказывает существенного влияния на потенциометрическую избирательность поданда, однако приводит к повышению литий/натриевой селективности.
БЛАГОДАРНОСТЬ
Исследования проводили с использованием оборудования ЦКП ФМИ ИОНХ РАН. Авторы статьи выражают благодарность д. х. н. В.П. Соловьеву за помощь в определении константы равновесия.
Работа выполнена в рамках государственного задания 2018 г. (темы № 0088-2014-0001, № 0081-2014-0015, № 0090-2017-0024). Синтез целевых соединений и спектрофотометрические исследования выполнены за счет гранта Российского научного фонда (проект №14-13-01286).
Список литературы
Панченко П.А., Федорова О.А., Федоров Ю.В. // Успехи химии. 2014. Т. 83. № 2. С. 155. [Panchenko P.A., Fedorova O.A., Fedorov Yu.V. // Russ. Chem. Rev. 2014. V. 83. № 2. P. 152. doi 10.1070/ RC2014v083n02ABEH004380].
Ушаков Е.Н., Алфимов М.В., Громов С.П. // Успехи химии. 2008. Т. 77. № 1. С. 39. [Ushakov E.N., Alfi-mov M.V., Gromov S.P. // Russ. Chem. Rev. 2008. V. 77. № 1. P. 39. doi 10.1070/RC2008v077n01ABEH003757].
Баулин В.Е., Баулин Д.В., Калашникова И.П. и др. Пат. РФ на изобр. № 2630695. 2017. 12. 09.
Баулин В.Е., Коваленко О.В., Туранов А.Н. и др. // Радиохимия. 2015. Т. 57. № 1. С. 53. [Baulin V.E., Kovalenko O.V., Turanov A.N. et al. // Radiochemistry. 2015. V. 57. № 1. P. 61. doi 10.1134/S1066362215010099].
Баулин В.Е., Калашникова И.П., Коваленко О.В. и др. // Физикохимия поверхности и защита материалов. 2016. Т. 52. № 6. С. 604. [Baulin V.E., Kalashnikova I.P., Kovalenko O.V. et al. // Protection Met. Phys. Chem. Surfaces. 2016. V. 52. № 6. P. 996. doi 10.7868/S0044185616060085].
Сафиулина А.М., Матвеева А.Г., Иванец Д.В. и др. // Изв. АН Сер. хим. 2015. № 1. С. 169. [Safiulina A.M., Matveeva A.G., Ivanets D.V. et al. // Russ. Chem. Bull. Int. Ed. 2015. V. 64. № 1. P. 169. doi 10.1007/s11172-015-0838-1].
Młynarz P., Ptak T., Czernicka A. et al. // J. Mol. Struct. 2011. № 991. P. 18.
Филякова В.И., Слепухин П.А., Болтачева Н.С. и др. // Журн. общ. химии. 2017. Т. 87. № 5. С. 766. [Filyakova V.I., Slepukhin P.A., Boltacheva N.S. et al. // Russ. J. Gen. Chem. 2017. V. 87. P. 957. doi 10.1134 S1070363217050115].
Гейн В.Л., Замараева Т.М., Дмитриев М.В. и др. // Журн. орг. химии. 2017. Т. 53. № 7. С. 1077. [Gein V.L., Zamaraeva T.M., Dmitriev M.V. et al. // Russ. J. Org. Chem. 2017. V. 53. P. 1090. doi 10.1134/S1070428017070223].
Anh Le.T., Nhung D.T., Tuyet Le.A. et al. // Macroheterocycles. 2017. V. 10. № 2. P. 243. doi 10.6060/mhc170289l
Чижов Д.Л., Слепухин П.А., Овчинникова И.Г. и др. // Изв. АН. Сер. хим. 2010. № 11. С. 2068. [Chizhov D.L., Slepukhin P.A., Ovchinnikova I.G. et al. // Russ. Chem. Bull. 2010. V. 59. № 11. P. 2122. doi 10.1007/s11172-010-0365-z]
Овчинникова И.Г., Федорова О.В., Слепухин П.А. и др. // Кристаллография. 2009. Т. 54. № 1. С. 37. [Ovchinnikova I.G., Fedorova O.V., Slepukhin P.A. et al. // Crystallogr. Rep. 2009. V. 54. P. 31. https://doi.org/ 10.1134/S1063774509010064.]
Князева И.Р., Бурилов А.Р., Пудовик М.А., Хабихер В.Д. // Успехи химии. 2013. Т. 82. № 2. С. 150. [Knyazeva I.R., Burilov A.R., Pudovik M.A., Habicher W.D. // Russ. Chem. Rev. 2013. V. 82. № 2. P. 150. 10.1070/RC2013v082n02ABEH004296].
Didarul A., Takashi Ogata, Satsuo Kamata // Anal. Chem. 1996. № 68. P. 366.
Баулин В.E., Иванова И.С., Полякова И.Н. и др. // Журн. общ. химии. 2015. Т. 85. № 4. С. 659. [Baulin V.E., Ivanova I.S., Polyakova I.N. et al. // Russ. J. Gen. Chem. 2015. V. 85 P. 899. doi 10.1134/ S1070363215040234].
Евреинов В.И., Вострокнутова З.Н., Баулин В.Е. и др. // Изв. АН СССР. Сер. хим. 1991. № 9. С. 1992.
Криворотько E.С., Полякова И.Н., Иванова И.С. и др. // Журн. неорган. химии. 2016. Т. 61. № 10. С. 1298. [Krivorot’ko E.S., Polyakova I.N., Ivanova I.S. et al. // Russ. J. Inorg. Chem. 2016. V. 61. № 10. P. 1241. doi 10.1134/S0036023616100132].
Oshchepkov A.S., Mittapalli R.R., Fedorova O.A., Kataev E.A. // Chem. Eur. J. 2017. V. 23. № 40. P. 9657. doi 10.1002/chem.201701515
Цветков Е.Н., Сюндюкова B.Х., Баулин В.Е. // Журн. общ. химии. 1987. Т. 57. № 11. С. 2456.
APEX II and SAINT. Madison (W, USA): Bruker AXS Inc., 2007.
Sheldrick G.M. SADABS. Göttingen (Germany): Univ. of Göttingen, 1997.
Sheldrick G.M. // Acta Crystallogr. Sect. C. 2015. V. 71. № 1. P. 3.
IUPAC. Recommendation for Nomenclature of Ion Selective Electrodes // Pure Appl. Chem. 1976. V. 48. № 1. P. 127.
Bakker E., Pretsch E., Buhlmann P. // Anal. Chem. 2000. V. 72. P. 1127.
Матерова Е.А., Грекович А.Л., Дидина С.У. // Электрохимия. 1972. Т. 8. № 8. С. 1829.
Белами Л. Инфракрасные спектры сложных молекул / Под ред. Пентина Ю.А. М.: Иностр. литер., 1963. 590 с.
Solov’ev V.P., Tsivadze A.Y. // Prot. Met. Phys. Chem. Surf. 2015. V. 51. № 1. P. 1. doi 10.1134/ S2070205115010153
Соловьев В.П. Программа ChemEqui для расчета констант химических равновесий и сопутствующих параметров, исходя из экспериментальных результатов физико-химических методов, таких как УФ, ИК и ЯМР спектроскопия, калориметрия, потенциометрия и кондуктометрия. http://vpsolovev.ru/programs/.
Петрухин О.М., Евсевлеева Л.Г., Урусов Ю.И. и др. // Журн. аналит. химии. 1995. Т. 50. № 1. С. 60.
Дополнительные материалы отсутствуют.
Инструменты
Журнал неорганической химии