

Журнал неорганической химии, 2022, T. 67, № 10, стр. 1425-1440
Квантово-химическое моделирование отщепления молекулярного водорода от триаммиаката боргидрида магния
А. С. Зюбин a, *, Т. С. Зюбина a, О. В. Кравченко a, М. В. Соловьев a, В. П. Васильев a, А. А. Зайцев a, А. В. Шиховцев a, Ю. А. Добровольский a
a Институт проблем химической физики РАН
142432 Московская обл., Черноголовка, пр-т Академика Семенова, 1, Россия
* E-mail: zyubin@icp.ac.ru
Поступила в редакцию 17.02.2022
После доработки 30.03.2022
Принята к публикации 04.04.2022
- EDN: VUVOQD
- DOI: 10.31857/S0044457X22100221
Аннотация
В рамках кластерного подхода с использованием базиса 6-31G* и гибридного функционала плотности (B3LYP) выполнено моделирование последовательного отрыва Н2 от комплексов Mg(BH4)2 · nNН3 (n = 2, 3, 4) и (Mg(BH4)2 · 3NН3)2. Установлено, что последовательное дегидрирование мономеров Mg(BH4)2 · nNН3 приводит к значительному росту потенциальных барьеров уже на третьем шаге, тогда как в димере (Mg(BH4)2·3NН3)2 такое увеличение не наблюдается почти до полного извлечения водорода. Начальный этап дегидрирования (примерно до 70–75%) должен быть экзотермическим, но для более высокой степени конверсии необходимы дополнительные затраты энергии.
ВВЕДЕНИЕ
Задача создания автономных источников водорода является ключевой для многих отраслей техники, прежде всего для электротранспорта [1, 2]. Большой интерес представляют боргидриды легких металлов, в первую очередь магния и натрия, и их комплексные соединения с аммиаком или водой, содержащие значительное количество водорода. Использование водорода для питания топливных элементов обусловливает дополнительные требования к его чистоте из-за высокой чувствительности электрохимических генераторов к отравляющим мембраны примесям, в частности к аммиаку [3].
Известно, что при термическом разложении боргидрида магния выделяется чистый водород, однако в силу высокого теплообразования для его разложения необходимо преодоление высоких потенциальных барьеров и требуются значительные энергозатраты [4]. При использовании комплексов с аммиаком или водой можно ожидать их снижения, поскольку формирование молекул Н2 будет происходить с участием отрицательно заряженных атомов Н из анионов ${\text{ВН}}_{4}^{ - }$ и протонов из молекул воды или аммиака. В работах [5–7] рассмотрено отщепление водорода от гидратированных комплексов боргидридов натрия и магния, дегидрирование которых проходит при умеренных температурах, но выход водорода на единицу массы не слишком велик.
В аммиакатах боргидридов легких металлов содержание водорода является более высоким, поэтому возможности его извлечения из данных соединений давно изучаются как экспериментально [8–22], так и с помощью компьютерного моделирования [23–27].
В системе боргидрид магния–аммиак образуются четыре устойчивые структуры Mg(BH4)2 · XNH3, где Х = 1, 2, 3 и 6 [18, 19, 21, 22]. Однако термолиз наиболее богатого водородом гексааммиаката при атмосферном давлении или в вакууме приводит к потере от трех до четырех молей аммиака и формированию три- или диаммиаката [9, 10, 19]. Триаммиакат имеет оптимальное соотношение противоположно заряженных атомов Н, входящих в анионы ${\text{ВН}}_{4}^{ - }$ и молекулы NH3, но при его термолизе при атмосферном давлении на первом этапе выделяется 1 моль аммиака, а затем 3 моля водорода [19]. При термолизе диаммиаката боргидрида магния аммиак в газообразных продуктах фиксируется в качестве примеси, а при разложении моноаммиаката боргидрида магния вовсе не фиксируется [18].
Кроме того, как это следует из результатов ДТА, для всех комплексов в процессе термолиза фиксируется экзотермический эффект, лежащий около 220°С, что снижает общие энергозатраты для их разложения. Можно предположить, что выделение аммиака при термолизе гекса- и триаммиакатов боргидрида магния связано с высоким равновесным давлением пара аммиака при температурах начала термораспада ~140–160°C и его отводом из зоны реакции.
Состав твердых продуктов термолиза элементно примерно соответствует составу Mg + 2BN и может образовываться в зависимости от скорости нагревания в аморфном состоянии или содержать кристаллическую фазу металлического магния. Можно ожидать, что при термолизе триаммиаката магний как активный металл будет образовывать продукты, содержащие нитрид магния Mg3N2 и (или) смешанные борнитриды [16], и образовывать дополнительное количество водорода, однако это не было подтверждено.
В [24] с помощью квантово-химического моделирования рассчитаны энтальпии и энергии Гиббса для шести реакций полного дегидрирования диаммиаката с формированием различных продуктов, находящихся в кристаллическом состоянии (BN, MgNB9, Mg3N2, MgB2, MgB4, MgB7, B). В рамках такого подхода выявлено, что для всех рассмотренных реакций значения энтальпий лежат в узком диапазоне (~0.2–0.15 эВ на Н2), а энергии Гиббса близки и отрицательны даже при комнатной температуре, т.е. реакции дегидрирования должны идти самопроизвольно. Однако такой подход является слишком формальным, поскольку каждый акт отрыва Н2 связан с существенным преобразованием структуры и требует преодоления значительных энергетических барьеров. Переход к наиболее стабильным конечным продуктам также связан с глубокой перестройкой строения, переключением системы связей и преодолением потенциальных барьеров. В большинстве работ, посвященных этой тематике, предполагается, что отщепление молекулы Н2 осуществляется при участии наиболее близко расположенных атомов Н из фрагментов ВН4 и NH3 без подробного изучения этого варианта. В [26] такая попытка была сделана для первого шага дегидрирования Mg(BH4)2·2NH3, и полученный барьер оказался достаточно высоким (~2.2 эВ). Однако наиболее простой вариант формирования Н2 –сближение атомов из ВН4 и NH3 – не является оптимальным. Согласно [5, 6], в процессе формирования Н2 в гидратированных комплексах образуется отрицательно заряженный фрагмент, стабилизируемый либо молекулами H2O, либо катионами металла, причем на каждом этапе реализуются разные варианты.
Очевидно, что процесс извлечения водорода из аммиакатов боргидрида магния является сложным и многоступенчатым, и для понимания его основных механизмов требуется детальное изучение отдельных этапов отрыва водорода от соответствующих комплексов. Одно из возможных направлений таких исследований – квантово-химическое моделирование, поэтому данная работа посвящена изучению с помощью такого моделирования путей отрыва молекулы водорода и величин соответствующих барьеров от аммиачных комплексов боргидрида магния, на первом этапе – триаммиаката.
МЕТОДИКА РАСЧЕТОВ
Для изучаемых систем моделирование выполнено в рамках того же подхода, что и в [5, 6], с использованием гибридного функционала плотности B3LYP и валентно-двухэкспонентного базиса 6-31G*, включающего поляризационные функции [28, 29], с помощью программного комплекса Gaussian [30]. Полученные в рамках самосогласованного поля энергии корректировали путем введения поправок на энергии нулевых колебаний (ЭНК, или ZPE) и энтропийных вкладов при нормальных условиях (1 атм., 20°С). Поиск переходных состояний (TS) между локальными минимумами проводили как с помощью процедуры QST3, так и путем сканирования перспективных участков потенциальной поверхности с дальнейшим уточнением структуры переходного состояния в рамках opt = TS. Соответствие найденного переходного состояния начальной и конечной конфигураций проверяли с помощью процедуры IRC. Согласно [5], использование более точных подходов B3LYP/6-311+G* и MP2/6-311++G** меняет величины рассчитанных барьеров в пределах 0.05–0.1 эВ, а при использовании достаточно большого количества молекул воды, обеспечивающего выход результатов на насыщение, в тех же пределах они согласуются с экспериментальными данными по определению энергии активации отрыва Н2 от боргидрида натрия в растворе.
Для оценки адекватности кластерного приближения для изучаемых систем выполнено моделирование кристаллических структур Mg(BH4)2·2NH3 и Mg(BH4)2·3NH3 в рамках периодических граничных условий с помощью программы VASP [31, 32] с использованием функционала PBE и базиса проектированных плоских волн (PAW) с пределом по энергии 600 эВ и с оптимизацией как координат атомов, так и параметров ячейки без ограничений по симметрии. Рассчитанные в рамках данного приближения параметры кристаллических решеток a, b, c составляют для диаммиаката 17.70, 9.40, 8.70 Å, а для триаммиаката 11.29, 9.40, 7.91 Å соответственно, что согласуется с экспериментальными данными [16, 22] в пределах 1%. При олигомеризации отдельных комплексов Mg(BH4)2·nNH3 (n = 2, 3) и объединении их в кристаллы строение первой координационной сферы сохраняется и расположение отдельных комплексов в олигомерах и кристаллах оказывается сходным (рис. 1), достаточно близкими в рассмотренных системах оказываются межатомные расстояния (табл. 1). Отсюда следует, что для данных систем взаимное влияние структурных блоков Mg(BH4)2·nNH3 не приводит к их существенным изменениям, и кластерное приближение адекватно воспроизводит основные характеристики конденсированной фазы.
Рис. 1.
Комплексы ди- и триаммиаката боргидрида магния и размножаемые ячейки кристаллов Mg(BH4)2·2NH3 и Mg(BH4)2·3NH3.
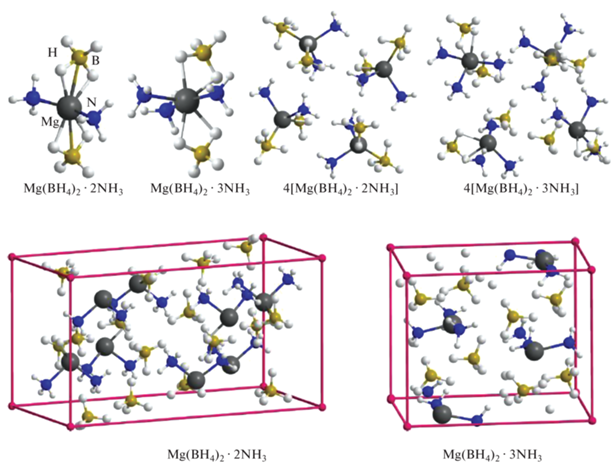
Таблица 1.
Равновесные расстояния (R, Å) в комплексах Mg(BH4)2·nNH3
| Параметр | Mg(BH4)2·2NH3 | Mg(BH4)2·3NH3 | ||||
|---|---|---|---|---|---|---|
| R(MgB) | R(MgN) | R(MgMg) | R(MgB) | R(MgN) | R(MgMg) | |
| Мономер | 2.24 | 2.19 | – | 2.46 | 2.20, 2.23 | – |
| Тетрамер | 2.21, 2.46 | 2.16 | 5.52 | 2.46, 2.56 | 2.18, 2.21 | 5.59, 6.12 |
| Кристалл, расчет | 2.26, 2.40 | 2.14 | 6.00, 6.19 | 2.51, 2.54 | 2.16, 2.18 | 6.08, 6.25 |
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
При моделировании отрыва молекулы Н2 от комплекса Mg(BH4)2 · 3NH3 найдено, что на этапе отделения от одной до четырех молекул Н2 величины наиболее низких потенциальных барьеров составляют 1.6–1.9 эВ, а далее резко возрастают, т.е. отщепление более четырех молекул Н2 от мономера весьма затруднительно. Для систем с другим количеством молекул аммиака (n = 2–6) качественная картина оказывается такой же, изменения носят лишь количественный характер. В наших предыдущих работах по моделированию отщепления водорода от комплексов боргидридов натрия и магния с водой отмечено, что олигомеризация таких комплексов ведет к снижению барьеров отрыва Н2 вследствие стабилизации промежуточных структур парой катионов [5, 6]. Поэтому в данной работе рассмотрена такая возможность на примере димера [Mg(BH4)2·3NH3]2.
При объединении молекул триаммиаката боргидрида магния в олигомеры их строение меняется не слишком существенно: вокруг катиона магния сохраняется искаженная бипирамида, сформированная двумя анионами ${\text{BH}}_{4}^{ - }$ и тремя молекулами аммиака, образующими вместе с Mg искаженную букву Т. При слипании в олигомер между катионами формируются цепочки Mg–${\text{BH}}_{4}^{ - }$–2H3N–Mg (например, в D0, рис. 2). При отщеплении молекулы водорода наиболее низким барьерам соответствуют конфигурации, в одной из которых объединяются атомы Н из ${\text{BH}}_{4}^{ - }$ и H3N в такой цепочке (D0ts1 ≥ D1b), а в другой (D0ts2) образуется группа из трех NH3, в которой молекула, максимально сближенная с катионами, формирует ослабленный мостик Mg–NH2–Mg и почти отщепляет протон, который отбирает Н– у ${\text{BH}}_{4}^{ - },$ образуя Н2, а ВН3 объединяется с NH3, формируя структуру D1a, которая через D1ts1 переходит в более стабильную D1b (рис. 2, 3). При дегидрировании этой конфигурации наиболее выгоден канал, в котором Н2 образуется через объединение атомов Н из фрагмента Н3В–NH2 и ближайшей молекулы аммиака (конфигурация D1ts2). У возникшей в результате структуры D2a есть возможность трансформироваться в D2b (через D2ts1) или отщепить водород через D2ts3 с образованием конфигурации D3a с анионом ${{{\text{B}}}_{2}}{\text{H}}_{7}^{ - },$ которая способна трансформироваться в более выгодную D3c через умеренный барьер D3ts1. Отщепление Н2 от D2b возможно через D2ts2 и ведет к D3b. Эти результаты показывают, что при дегидрировании не всегда реализуется наиболее прямой вариант – объединение атомов Н из NH3 и ${\text{BH}}_{4}^{ - }$, часто этот путь оказывается более сложным, с формированием промежуточных комплексов, объединенных в метастабильные структуры, которые в результате внутримолекулярных перегруппировок меняют свое строение.
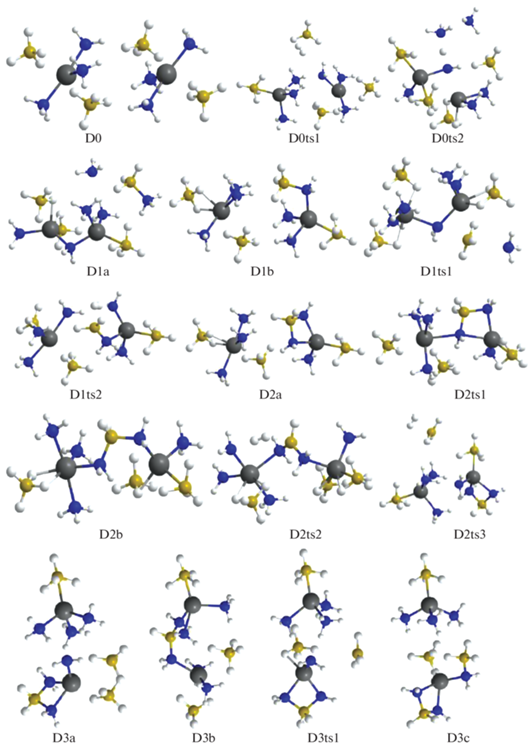
Рис. 3.
Энергии Гиббса для конфигураций системы [Mg(BH4)2·3NH3]2, возникающих при удалении до трех (D0–D3) и до шести (D3–D6) молекул Н2.
При дальнейшем дегидрировании отрыв Н2 от D3b реализуется в связывающей катионы цепочке NH3–${\text{BH}}_{4}^{ - }$ через конфигурацию D3ts3 и ведет к образованию D4a, а в D3c объединяются атомы Н из фрагмента H3B–NH2 и ближайшей к нему молекулы аммиака через D3ts2с формированием D4b. Для D4a отщепление Н2 реализуется через D4ts1, где объединяются атомы Н из NH3 и ${\text{BH}}_{4}^{ - }$ с формированием второго мостика Mg–NH2–Mg и слабосвязанного комплекса Н2·ВН3. Молекула ВН3 объединяется с NH2, H2 удаляется, а молекула H2B–NH2 включается во второй мостик Mg–NH2–Mg с формированием структуры D5a. У D4b Н2 отщепляется в мостике Mg–NH2–BH3–NH3–Mg (конфигурация D4ts2) с образованием D5b, при этом фрагмент H2B–NH2 отдает протон аниону ${\text{NH}}_{2}^{ - },$ восстанавливая NH3 (рис. 3, 4). Отщепление Н2 от D5a и D5b требует преодоления высоких барьеров, но после их преобразования в менее выгодные структуры D5c и D5d (через D5ts1 и D5ts2) отрыв молекулы водорода требует преодоления сравнительно умеренных барьеров D5ts3 и D5ts4 и перехода в D6a и D6b. Следует отметить, что оба варианта реализуются с помощью не встречавшегося ранее переноса Н в промежуточных структурах от атома бора к магнию. Рассмотрим этот механизм более подробно на примере дегидрирования кластера D5a (рис. 4). В конфигурации D5a расположенный между катионами фрагмент H2N–${\text{BH}}_{3}^{ - }$ отдает Н– на атомы магния, формируя мостик Mg–H–Mg (D5a ≥ D5ts1 ≥ D5c), при этом малликеновский заряд на Н меняется от –0.09 до –0.2 е, что облегчает его объединение с положительно заряженным атомом Н (0.39 е) из NH3-группы (D5с ≥ D5ts4 ≥ D6b). Для отрыва Н2 от D6b необходимо преобразовать ее в D6c через D6ts1 путем переноса Н из группы H2B(NH2)2 на Mg (рис. 5) с последующим объединением его с атомом Н из NH2 (конфигурация D6ts3) и переходом в D7b (рис. 5, 6).
Рис. 4.
Конфигурации системы [Mg(BH4)2·3NH3]2, возникающие при удалении от трех до шести молекул Н2.
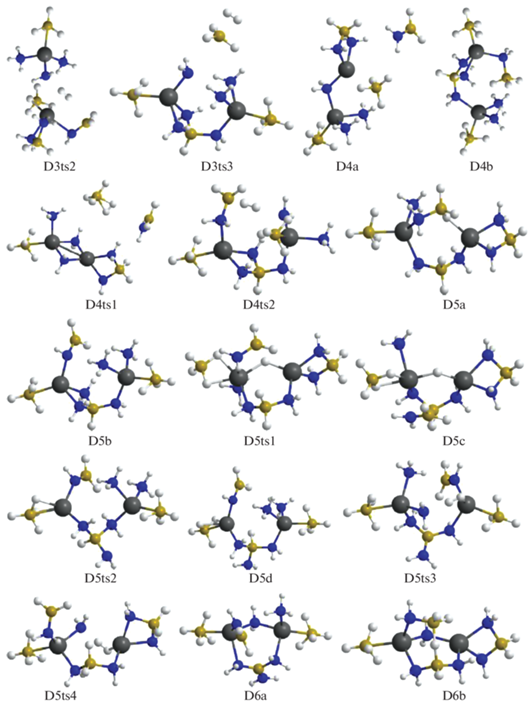
Рис. 5.
Конфигурации системы [Mg(BH4)2·3NH3]2, возникающие при удалении от шести до девяти молекул Н2.
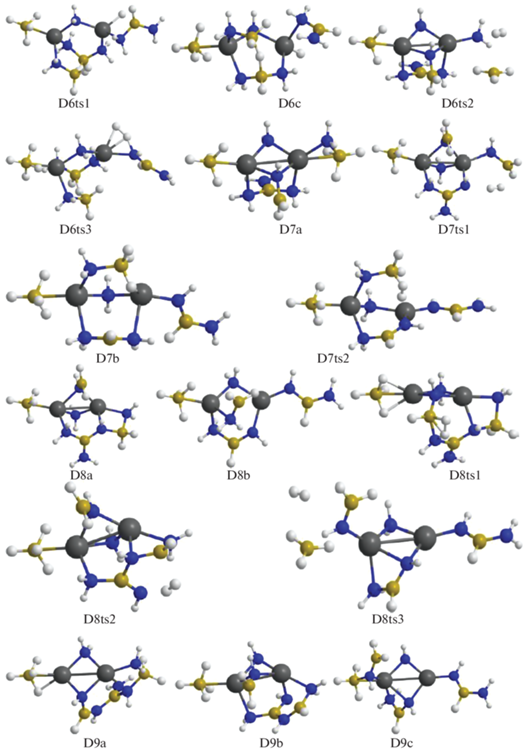
Рис. 6.
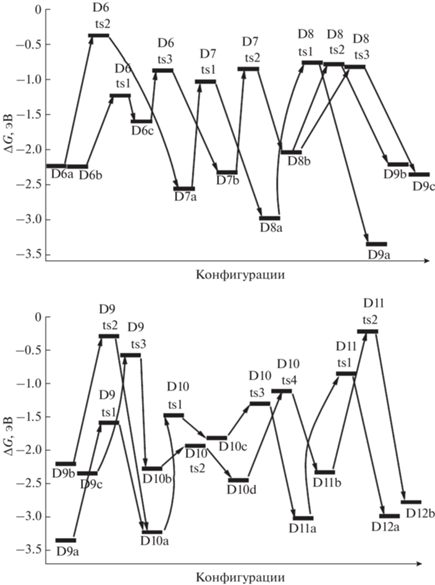
Энергии Гиббса для конфигураций системы [Mg(BH4)2·3NH3]2, возникающих при удалении от шести до девяти (6D–D9) и от девяти до двенадцати (D9–D12) молекул Н2.
Конфигурация D6a отщепляет Н2 при объединении атомов Н из ${\text{BH}}_{4}^{ - }$ и аммиака после преодоления сравнительно высокого барьера (D6ts2) с формированием структуры D7a. Молекула водорода отщепляется от D7a при взаимодействии фрагментов ВН3 из H3B–NH2 и H2N из H2N–B(NH2)2 через D7ts1, формируя D8a, а от D7b – через D7ts2 из фрагментов ВН3 и NH2 с участием атома Mg и образованием D8b (рис. 5, 6).
В структуре D8a молекула Н2 формируется через D8ts1 из атомов, входящих в группировки H2B–NH и цепочку H2N–BNH2–NH–BH2–NH2, что ведет к образованию еще более длинной цепочки, обвивающей катион магния (конфигурация D9a, рис. 5). Для D8b имеются два варианта, один из которых реализуется через объединение атомов Н из фрагментов H2B–NH2 и HN–BHNH2 (через D8ts2) с формированием структуры D9b, а другой – при взаимодействии H2B–NH2 с ${\text{BH}}_{4}^{ - }$ (через D8ts3) с формированием структуры D9c (рис. 5, 6).
D9a отщепляет Н2 через переходное состояние D9ts1 путем объединения атомов Н из фрагментов ВН2 и NH2 длинной цепочки и образует структуру D10a (рис. 6, 7). У D9b реализуется похожее взаимодействие, но между фрагментами H2NH и H2N–BNH–NH–BH–NH2 через D9ts2 с формированием той же структуры D10a. D9c отщепляет Н2 путем объединения атомов Н из групп –ВН3 и H2N– (фрагменты H3B–NH–BH2 и H2N–BH–NH, переходное состояние D9ts3) с образованием D10b (рис. 6, 7). Для дегидрирования D10a и D10b необходимо преобразовать их в менее выгодные D10c и D10d с помощью перевода отрицательно заряженного атома Н из фрагмента НВ< (через D10ts1) или H2B< (через D10ts2) на катион магния. Затем водород из фрагмента H–Mg объединяется с Н из фрагментов H2N< (через D10ts3) или HN< (через D10ts4) с формированием структур D11a или D11b.
Рис. 7.
Конфигурации системы [Mg(BH4)2·3NH3]2, возникающие при удалении от девяти до одиннадцати молекул Н2.
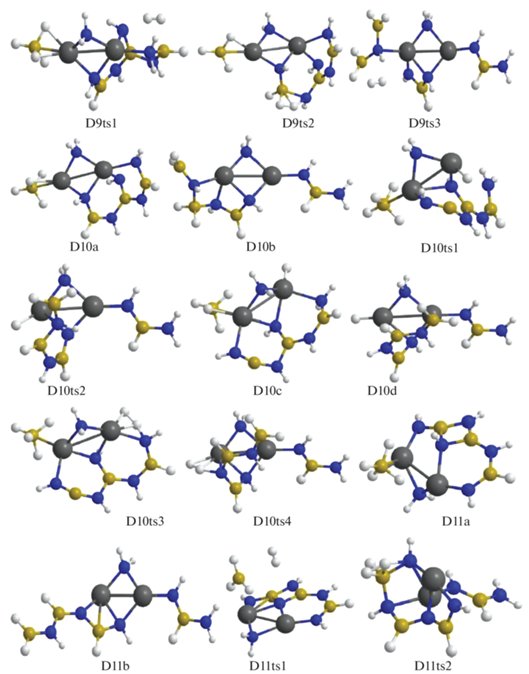
В конфигурации D11a отщепление Н2 идет через взаимодействие ${\text{BH}}_{4}^{ - }$ и фрагмента NH, в котором атом азота координирован к катиону магния и двум атомам бора (конфигурация D11ts1) с переходом в D12a. В структуре D11b при дегидрировании объединяются атомы Н из мостика Mg–NH2–Mg и цепочки H2B–NHBHNBHNH, которая при этом существенно изгибается (конфигурация D11ts2), в результате формируется структура D12b (рис. 6–8). В D12a отрыв Н2 реализуется через сближение фрагментов ВН3 и NH2 (конфигурация D12ts1) с переходом в D13b, а для D12b требуется преобразование в D12c (через D12ts2) с передачей Н от атома бора из фрагмента HN–BH–NH2 на Mg и дальнейшим объединением его с атомом Н из фрагмента H2N–BNH (конфигурация D12ts3) с формированием D13a. Для D13b требуется дальнейшая трансформация в D13c (через D13ts1) путем перемещения Н из фрагмента Н2В– на катион магния. Для отделения Н2 от D13a происходит сближение атомов Н из фрагмента H–NBNH и цепочки HN–BH–NBHNHBHNH (конфигурация D13ts2) с формированием D14a, а от D13c – атомов Н, связанных с магнием и азотом (конфигурация D13ts3), с переходом в D14b (рис. 8, 9).
Рис. 8.
Конфигурации системы [Mg(BH4)2·3NH3]2, возникающие при удалении от двенадцати до четырнадцати молекул Н2.
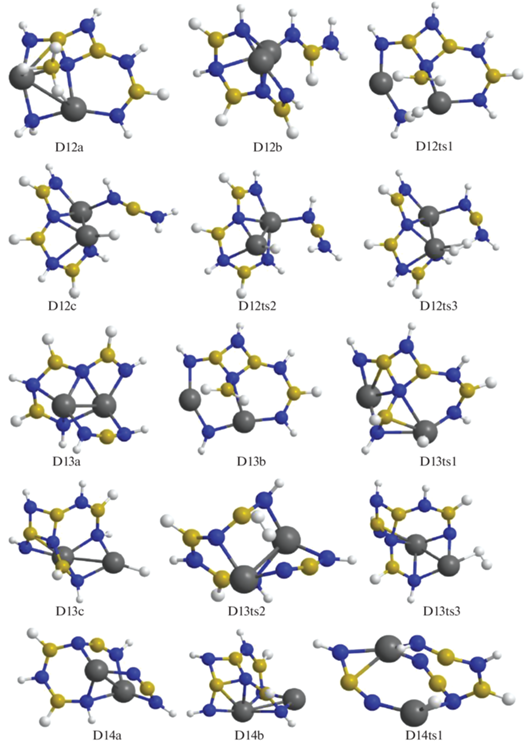
Рис. 9.
Энергии Гиббса для конфигураций системы [Mg(BH4)2·3NH3]2, возникающих при удалении от двенадцати до четырнадцати (D12–D14) и от четырнадцати до шестнадцати (D14–D16) молекул Н2.
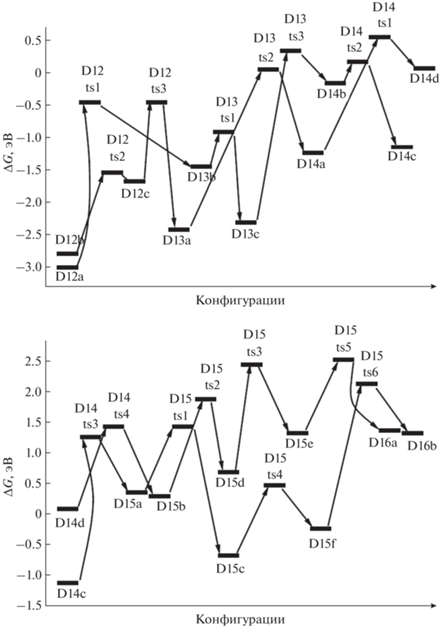
Для следующего шага дегидрирования требуется преобразование структур D14a и D14b в более активные D14c и D14d с помощью перемещения атома Н от бора на магний через конфигурации D14ts1 и D14ts2 (рис. 8–10). В D14c при формировании Н2 объединяются атомы из фрагментов Mg–H и ближайшего N–H (через D14ts3 с переходом в D15a), а в D14d – из более удаленного NH, входящего в цепочку N4B3H4 (через D14ts4 в D15b, рис. 9, 10).
Рис. 10.
Конфигурации системы [Mg(BH4)2·3NH3]2, возникающие при удалении от четырнадцати до шестнадцати молекул Н2.
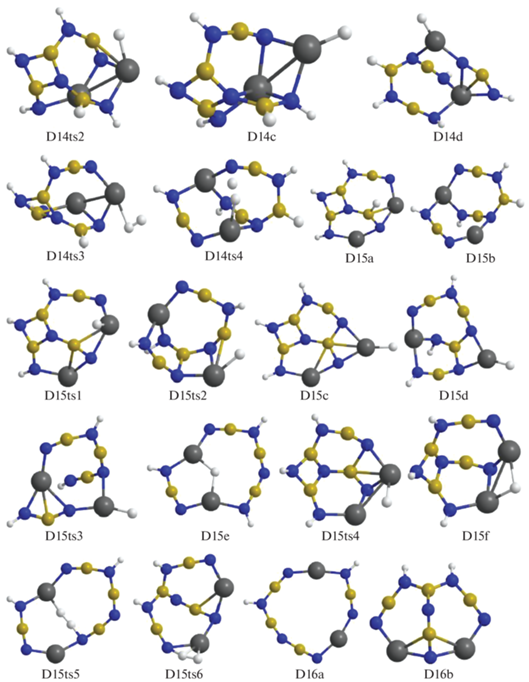
Для отделения Н2 от D15a и D15b необходимо перевести последний связанный с бором атом Н на магний (через D15ts1 и D15ts2), преобразовав их в D15c и D15d, но при этом расстояния от фрагмента H–Mg до ближайших H–N оказываются слишком большими, поэтому необходимо дальнейшее преобразование для сближения активных фрагментов. Рассмотрим отрыв Н2 от D15a более подробно. В исходной конфигурации связь Н–В направлена почти перпендикулярно к поверхности, сформированной остальными атомами (D15a, рис. 10). Атом Н из этого фрагмента переходит на ближайший катион магния (D15a ≥ D15ts1 ≥ D15c), затем на другой Mg+, контактирующий с фрагментом N–H (D15c ≥ D15ts4 ≥ D15f), и далее через D15ts6 в D16b (рис. 9, 10).
У D15d для отрыва Н2 требуется преобразование каркаса, в результате которого центральный фрагмент HN–B< отделяется от Mg, на его место приходит атом N из соседней группировки, причем связь этого атома с бором разрушается (конфигурация D15ts3), а связь H–Mg разворачивается ко второму атому Mg, формируя мостик Mg–H–Mg (структура D15e). Через D15ts5 от этой структуры отщепляется Н2 и формируется D16a (рис. 9, 10).
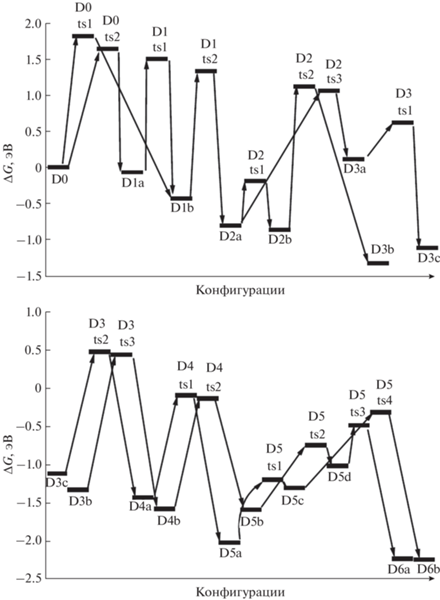
Рассмотрим тенденции изменений энергий Гиббса для наиболее низких энергетических барьеров при последовательном отщеплении молекул водорода и возникающих при этом конфигураций. Согласно полученным данным, при отделении до 50% водорода (от конфигураций D0 до D8, i = 1–8) соответствующие энергии последовательно снижаются (табл. 2), затем в интервале D8–D12 (i = 9–13, 50–75% водорода) наступает стабилизация, при дальнейшем извлечении водорода эти энергии начинают резко возрастать, причем почти одинаково растут энергии как переходных конфигураций (TS_i), так и промежуточных локальных минимумов (D_i). Для энтальпии соответствующая тенденция оказывается иной: в интервале i = 1–6 (~40% водорода) она слабо снижается, т.е. отщепление водорода ведет к небольшому выделению энергии, но дальнейшее извлечение Н2 требует энергетических затрат, наиболее существенных после удаления ~80% водорода (i = 13–16, табл. 2). Эти различия можно объяснить тем, что в энергию Гиббса существенный вклад дает энтропийный член (TΔS, где Т – абсолютная температура, а ΔS – увеличение энтропии при отрыве водорода), т.е. базой такого поведения является изменение энтальпии (ΔH).
Таблица 2.
Энергии Гиббса ΔG и теплоты образования ΔH (эВ) для двух наиболее низких переходных конфигураций (TS_i', TS_i") и локальных минимумов (D_i', D_i"), возникающих при удалении от одной (i = 1) до шестнадцати (i = 16) молекул Н2 из системы [Mg(BH4)2·3NH3]2. В качестве начала отсчета принята энергия димера D0
| i | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 |
|---|---|---|---|---|---|---|---|---|
| ΔG ts_i" | 1.82 | 1.50 | 1.12 | 0.47 | –0.10 | –0.32 | –0.36 | –0.84 |
| ΔG ts_i' | 1.64 | 1.33 | 1.06 | 0.43 | –0.14 | –0.49 | –0.86 | –1.02 |
| ΔG D_i" | –0.07 | –0.81 | –1.12 | –1.43 | –1.59 | –2.23 | –2.32 | –2.03 |
| ΔG D_i' | –0.43 | –0.87 | –1.33 | –1.58 | –2.02 | –2.24 | –2.55 | –2.98 |
| ΔH D_i" | –0.19 | –0.22 | –0.11 | –0.11 | –0.44 | –0.15 | 0.58 | |
| ΔH D_i' | –0.12 | –0.31 | –0.50 | –0.48 | –0.58 | –0.50 | –0.45 | –0.58 |
| i | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 |
| ΔG ts_i" | –0.75 | –0.58 | –1.12 | –0.23 | –0.45 | 0.34 | 1.42 | 2.51 |
| ΔG ts_i' | –0.81 | –1.59 | –1.31 | –0.86 | –0.45 | 0.05 | 1.24 | 2.11 |
| ΔG D_i" | –2.35 | –2.45 | –2.33 | –2.78 | –2.30 | –1.14 | 0.34 | 1.31 |
| ΔG D_i' | –3.35 | –3.23 | –3.02 | –2.99 | –2.41 | –1.23 | –0.69 | 1.35 |
| ΔH D_i" | 0.52 | 0.82 | 1.32 | 1.12 | 1.77 | 3.31 | 5.12 | 6.82 |
| ΔH D_i' | –0.53 | –0.11 | 0.38 | 0.72 | 1.70 | 3.30 | 4.19 | 6.57 |
Попытаемся на основе полученных структурных данных понять причины таких изменений. В исходном комплексе D0 отрицательные заряды рассредоточены в анионах ${\text{BH}}_{4}^{ - }.$ После удаления от одной до семи молекул Н2 (комплексы D1–D8, рис. 2, 3, 5) формируются как объемные (H3BNH–, ${{H}_{2}}B(N{{H}_{2}})_{2}^{--}HB(N{{H}_{2}})_{3}^{--}$), так и более компактные анионы с более высокой локализацией заряда преимущественно на атоме азота с точки зрения формальной валентности (N–H2, HN––BH2, HN––BH–NH2). Конечно, такая картина является сугубо качественной, и при образовании комплекса заряды будут существенно перераспределяться. Увеличение локализации зарядов должно приводить к повышению энергии вследствие увеличения кулоновского отталкивания, но более компактные анионы могут подходить к катионам на более короткие расстояния, понижая энергию, так что в целом она меняется слабо. Основным следствием появления компактных анионов является то, что они занимают позиции между катионами, выталкивая более объемных конкурентов на периферию.
На следующих шагах удаления Н2 остающиеся фрагменты вынуждены объединяться для замыкания освобождающихся связей во все более сложные структуры, в некоторых случаях несущие два и даже три формальных отрицательных заряда. Так, в D9a возникает разветвленная цепочка HN––BH–NH–BNH2–NH–B–H2–NH2 с зарядами на атомах азота и бора, а в D9b – НN––ВNH2–NH–BH–NH2 с зарядом на азоте, в D10a и D10b – цепочки HN––BH–NH–BN–H–NH–BH–NH2 и HN––BH–NH–B–H2–NH–BH2, в D11a и D11b – HN––BH–NH–B(NH)2B–N–H и HN––BH–N––BH–NH–BH2 (рис. 5, 7), в D12a и D12b, D13b и D13c, D14a и D14c, D15a и D15c – трехзарядные анионы HN––BH–NH–B(NH)(N–BH3)B–N–H и HN––BH–N––BH–NH–BH–N–H, HN––BH–N––BH–NH–BH–N–H и (B4N3H4)(N–H)3, HN––B–N––BH–NH–BH–N–H и N–=B–NH–(B3N2H2)(N–H)2, N–=B–NH–BH–N–=B–N–H и HN––(B4N4H2)–N2– (рис. 8, 10). Вследствие слишком большого объема информации не приводим данные по распределению электронной плотности и геометрическому строению рассмотренных конфигураций. Эти данные могут быть предоставлены всем желающим с помощью электронной почты.
По мере удаления атомов водорода и замыкания освобождающихся связей структура анионного окружения становится все менее гибкой и хуже экранирует катионы, что приводит к повышению полной энергии системы. Однако у таких кластеров из-за менее полного экранирования заряженных участков должна увеличиваться энергия димеризации, и их агломерация будет снижать полную энергию системы. Чтобы проверить, насколько существенным может быть такой вклад в энергию, было выполнено моделирование формирования димеров из кластеров D8–D16. Согласно полученным результатам, для D8 энтальпия димеризации (ΔH) невелика, немного возрастает для D9, более существенно увеличивается для D10–D13 и наиболее резко растет для D14–D16 (табл. 3). Если учесть влияние этого эффекта на величины, приведенные в табл. 2 (при этом приведенные в табл. 3 значения следует делить пополам, так как они приходятся на две частицы), то значительный рост энергетических затрат при дегидрировании можно ожидать лишь после D12 (75% Н2). Если ввести аналогичные поправки для энергий Гиббса (ΔG), то энергии переходных конфигураций и промежуточных структур, возникающих при отщеплении водорода, могут остаться на прежнем уровне и даже слегка понизиться почти до полного извлечения водорода.
Таблица 3.
Энергии димеризации (эВ) промежуточных структур, возникающих при последовательном отщеплении Н2 от комплекса [Mg(BH4)2 · 3NH3]2
| Параметр | D8a | D9a | D10a | D11a | D12a | D13b | D14a | D15c | D16b |
|---|---|---|---|---|---|---|---|---|---|
| ΔH | 0.50 | 0.95 | 2.25 | 1.99 | 2.11 | 2.69 | 4.10 | 5.09 | 5.33 |
| ΔG | –0.07 | 0.15 | 1.77 | 1.60 | 1.61 | 1.97 | 3.53 | 4.41 | 4.74 |
Приведенные в настоящей работе результаты позволяют предположить, что проведение начальной стадии термического разложения триаммиаката боргидрида магния при повышенном давлении будет препятствовать отрыву аммиака с образованием диаммиаката. При достижении уровня конверсии D6–D7 (отщепление более трех молей Н2 на моль триаммиаката) весь аммиак преобразуется в более тяжелые фрагменты, после чего давление можно будет снизить. Не исключено, что такой вариант способен привести к увеличению выхода водорода.
ЗАКЛЮЧЕНИЕ
Для извлечения водорода из триаммиаката боргидрида магния требуется существенный нагрев, связанный с необходимостью преодоления энергетических барьеров величиной до 1.5–2.0 эВ. Первый этап дегидрирования (примерно до 70–75%) должен идти с выделением энергии, но для более высокой степени конверсии потребуются значительные энергозатраты, поэтому полное извлечение Н2 из этого соединения может оказаться нецелесообразным и диаммиакат в плане извлечения водорода при умеренных энергозатратах может оказаться более перспективным. Моделирование этого процесса планируется в следующей работе.
Необходимо добавить, что в настоящей работе рассмотрен только мономолекулярный вариант дегидрирования, который не требует столкновения молекул при определенной ориентации и поэтому является наиболее быстрым, если накопленной энергии достаточно для преодоления очередного барьера. Однако при медленном нагреве система может находиться на очередном этапе достаточно долго для того, чтобы мог реализоваться вариант столкновения уже имеющихся фрагментов, приводящий к дегидрированию с такой величиной барьера, который может быть преодолен при достигнутом уровне энергии. Из этого следует, что в зависимости от скорости нагрева могут возникать разные каналы дегидрирования. К сожалению, поиск таких вариантов является трудоемкой задачей, поскольку связан с перебором большого количества разных вариантов столкновения, возникающих в процессе дегидрирования фрагментов, и выходит за рамки данной работы.
Список литературы
Graetz J. // Chem. Soc. Rev. 2009. V. 38. P. 73. https://doi.org/10.1039/B718842K
Makaryan I.A., Sedov I.V. // Russ. J. Gen. Chem. 2021. V. 91. P. 1912. https://doi.org/10.1134/S1070363221090371
Llerena F.I., Jiménez A.D.H., González E.L. et al. // Fuel. Cells. 2019. V. 19. P. 651. https://doi.org/10.1002/fuce.201900031
Zavorotynska O., El-Kharbachi A., Deledda S. et al. // Int. J. Hydrogen Energy. 2016. V. 41. P. 14387. https://doi.org/10.1016/j.ijhydene.2016.02.015
Zyubin A.S., Zyubina T.S., Kravchenko O.V. et al. // Russ. J. Inorg. Chem. 2016. V. 61. P. 731. [Зюбин А.С., Зюбина Т.С., Кравченко О.В. и др. // Журн. неорган. химии. 2016. Т. 61. С. 767.] https://doi.org/10.1134/S0036023616060231
Zyubin A.S., Zyubina T.S., Kravchenko O.V. et al. // Russ. J. Inorg. Chem. 2018. V. 63. P. 201. [Зюбин А.С., Зюбина Т.С., Кравченко О.В. и др. // Журн. неорган. химии. 2018. Т. 63. С. 190.] https://doi.org/10.1134/S0036023618020237
Solovev M.V., Chashchikhin O.V., Dorovatovskii P.V. et al. // J. Power Sources. 2018. V. 377. P. 93. https://doi.org/10.1016/j.jpowsour.2017.11.090
Семененко К.Н., Шилкин С.П., Полякова В.Б. // Изв. АН СССР. Сер. хим. 1975. № 4. С. 735.
Коноплев В.Н., Силина Т.А. // Журн. неорган. химии. 1985. Т. 30. С. 1125.
Кравченко О.В., Кравченко С.Е., Семененко К.Н. // Журн. общ. химии. 1990. Т. 60. С. 2641.
Кравченко О.В., Хафизова Г.М., Бурдина К.П. и др. // Журн. общ. химии. 1994. Т. 64. С. 6.
Schlapbach L., Zuttel A. // Nature. 2001. V. 414. P. 353. https://doi.org/10.1038/35104634
Züttel A. // Mater. Today. 2003. V. 6. P. 24. https://doi.org/10.1016/S1369-7021(03)00922-2
Orimo S.-I., Nakamori Y., Eliseo J.R. et al. // Chem. Rev. 2007. V. 107. P. 4111. https://doi.org/:10.1021/cr0501846
Sakintuna B., Lamari-Darkrim F., Hirscher M. // Int. J. Hydrogen Energy. 2007. V. 32. P. 1121. https://doi.org/10.1016/j.ijhydene.2006.11.022
Soloveichik G., Her J.-H., Stephens P.W. et al. // Inorg. Chem. 2008. V. 47. P. 4290. https://doi.org/10.1021/ic7023633
Guo Y., Wu H., Zhou W., Yu X. // J. Am. Chem. Soc. 2011. V. 133. P. 4690. https://doi.org/10.1021/ja1105893
Yang Y., Liu Y., Li Y. et al. // Chem. Asian J. 2013. V. 8. P. 476. https://doi.org/10.1002/asia.201200970
Yang Y., Liu Y., Li Y. et al. // J. Phys. Chem. C. 2013. V. 117. P. 16326. https://doi.org/10.1021/jp404424m
Jepsen L.H., Ley M.B., Filinchuk Y. et al. // ChemSusChem. 2015. V. 8. P. 1452.
Paskevicius M., Jepsen L.H., Schouwink P. et al. // Chem. Soc. Rev. 2017. V. 46. P. 1565. https://doi.org/10.1039/c6cs00705h
Yan Y., Dononelli W., Jorgensen M. et al. // Phys. Chem. Chem. Phys. 2020. V. 22. P. 9204. https://doi.org/10.1039/d0cp00158a
Chen X., Yu X. // J. Phys. Chem. C. 2012. V. 116. P. 11900. https://doi.org/10.1021/jp301986k
Yuan P.-F., Wang F., Sun Q. et al. // Int. J. Hydrogen Energy. 2013. V. 38. P. 2836. https://doi.org/10.1016/j.ijhydene.2012.12.075
Wang K., Zhang J.-G., Lang X.-Q. // Phys. Chem. Chem. Phys. 2016. V. 18. P. 7015. https://doi.org/10.1039/C5CP06808H
Chen X., Li R., Xia G. et al. // RSC Adv. 2017. V. 7. P. 31027. https://doi.org/10.1039/c7ra05322c
Chen X., Zou W., Li R. et al. // J. Phys. Chem. C. 2018. V. 122. P. 4241. https://doi.org/10.1021/acs.jpcc.8b00455
Becke A.D. // J. Chem. Phys. 1993. V. 98. P. 5648. https://doi.org/10.1063/1.464913
Johnson B.J., Gill P.M.W., Pople J.A. // J. Chem. Phys. 1993. V. 98. P. 5612. https://doi.org/10.1063/1.464906
Gaussian 09, Revision B.01. Gaussian, Inc., Wallingford CT, 2010.
Kresse G., Furthmüller J. // Phys. Rev. B. 1996. V. 54. P. 11169. https://doi.org/10.1103/physrevb.54.11169
Kresse G., Joubert D. // Phys. Rev. B. 1999. V. 59. P. 1758. https://doi.org/10.1103/physrevb.59.1758
Дополнительные материалы отсутствуют.
Инструменты
Журнал неорганической химии