

Журнал неорганической химии, 2022, T. 67, № 9, стр. 1292-1300
Синтез и спектроскопия замещенных бензоилацетонатов дифторида бора
А. Г. Мирочник a, З. Н. Пузырьков a, b, Е. В. Федоренко a, *, И. В. Свистунова b
a Институт химии ДВО РАН
690022 Владивосток, пр-т 100-летия Владивостока, 159, Россия
b Дальневосточный федеральный университет
690922 Владивосток, о. Русский, п. Аякс, 10,Россия
* E-mail: gev@ich.dvo.ru
Поступила в редакцию 27.02.2022
После доработки 21.03.2022
Принята к публикации 30.03.2022
- EDN: QQCVZU
- DOI: 10.31857/S0044457X22090082
Аннотация
Предложен простой метод препаративного синтеза замещенных бензоилацетонатов дифторида бора в одну стадию из донорно-замещенных аренов путем ацетоацетилирования под действием трифторида бора. Проанализировано влияние природы алкильных и арильных заместителей фенильного кольца бензоилацетонатов дифторида бора на их спектрально-люминесцентные свойства. Показано, что алкильные заместители незначительно увеличивают квантовый выход люминесценции. Существенное увеличение квантового выхода люминесценции до 0.75 и значительный батохромный сдвиг до 536 нм достигаются при увеличении π-системы молекулы: переход от фенила к нафтилу, дифенилу, антрацилу.
ВВЕДЕНИЕ
За последнее время хелаты дифторида бора приобрели статус перспективных люминофоров [1–5] с широким спектром практического применения в качестве лазерных красителей, биомаркеров, хемосенсоров, компонентов светочувствительных покрытий для фотогальванических устройств и др. [6–8].
β-Дикетонаты дифторида бора представляют собой хелаты, в которых в качестве лиганда выступает енольная форма β-дикетона, в качестве комплексообразователя – катион дифторида бора. β-Дикетонаты дифторида бора обладают интенсивной люминесценцией как в растворах, так и в кристаллах. Люминесцентные свойства кристаллов и растворов одного и того же β-дикетоната дифторида бора, в отличие от большинства известных люминофоров, значительно различаются и существенно зависят от концентрации раствора, что представляет уникальную возможность для управления оптическими свойствами функциональных материалов. Ароилацетоны, как и 1,3-дикетоны, являются универсальными прекурсорами для получения различных типов гетероциклических соединений. Из подобных ароилацетонов, β-дикетонов и их борных комплексов легко осуществляется синтез пиразолов [9, 10] и полиметиновых красителей [11, 12]. Кроме того, такие комплексы обладают биологической активностью и могут быть ингибиторами различных гидролаз и оксигеназ, например, циклооксигеназы-2, гемеоксигеназы-1, фосфолипазы D [13–17].
Поэтому особый интерес представляют препаративные методы синтеза β-дикетонатов дифторида бора. Как правило используется двухстадийный синтез: 1) конденсация Кляйзена ацетофенонов со сложными эфирами [18]; 2) хелатирование полученного β-дикетона эфиратом трехфтористого бора [19]. Значительно реже применяют конденсацию Меервейна под действием трифторида бора [20]. Ацилирование ароматических соединений уксусным ангидридом и трифторидом бора c образованием β-дикетонатов дифторида бора впервые было проведено Меервейном при исследовании способности кислот Льюиса к ацилированию по Фриделю–Крафтсу [20]. В ряде работ эта реакция упоминается как конденсация Меервейна. Впоследствии в серии работ [21, 22] установлен механизм реакции. Метод Меервейна для получения β-дикетонатов дифторида бора получил развитие в работах [23–25].
В настоящем сообщении проведено сравнение методик получения замещенных бензоилцетонатов дифторида бора конденсацией Меервейна с использованием газообразного BF3 и его аддукта с уксусной кислотой (BF3‧2AcOH). Исследованы люминесцентные свойства полученных комплексов дифторида бора.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Температуру плавления определяли на приборе Buchi Melting Point B-540. ИК-спектры записывали на ИК-Фурье-спектрометре Spectrum BX 400 (Perkin Elmer, США) в бромиде калия. Спектры ЯМР регистрировали на спектрометре высокого разрешения Avance II 400 МГц (Bruker, Германия) на ядрах 1H и 13C при различных рабочих частотах. В качестве растворителя использовали дейтерированный хлороформ. Данные ЯМР приведены в табл. 1 и 2. Спектры поглощения регистрировали на спектрофотометре Shimadzu UV 2550 (Shimadzu, Япония). Спектры люминесценции и возбуждения получены на спектрофлуориметре Shimadzu RF 5301 (Shimadzu, Япония). Спектры флуоресценции с временным разрешением измерены по технологии время-коррелированного счета одиночных фотонов (TCSPC) на лазерном пикосекундном спектрофлуориметре FluoTime 200 (PicoQuant, Германия), источник PDL 800-B (λex = 370 нм). Для определения квантового выхода люминесценции в качестве стандарта использовали раствор нафталина в этаноле (φ = 0.12) [26].
Таблица 1.
Данные 1H ЯМР замещенных бензоилацетонатов дифторида бора I–XII в CDCl3 (δ, м.д.)
| № | 6-CH3 | 5-CH | 4-Ar | Другие сигналы |
|---|---|---|---|---|
| I | 2.42 | 6.61 | 7.51–7.55 (2Н), 7.67–7.68 (1Н), 8.04–8.06 (2Н) | – |
| II | 2.43 | 6.59 | 7.35–7.37 (2H), 7.98–8.00 (2H) | 2.49 (с, 3H, CH3) |
| III | 2.40 | 6.55 | 7.34–7.36 (2H), 7.97–8.00 (2H) | 1.25–1.29 (с, 3H, CH3), 2.72–2,77 (2H, CH2) |
| IV | 2.39 | 6.56 | 7.26–7.28 (1H), 7.77–7.80 (1H), 7.85 (1H) | 2.33 (с, 3H, CH3), 2.36 (с, 3H, CH3) |
| V | 2.40 | 6.34 | 7.18–7.20 (1Н), 7.28–7.31 (1Н), 7.46 (1Н) | 2.36 (с, 3H, CH3), 2.54 (с, 3H, CH3) |
| VI | 2.37 | 6.49 | 6.98–7.01 (2H), 8.04–8.06 (2H) | 3.92 (с, 3H, OCH3) |
| VII | 2.43 | 6.48 | 7.50–7.63 (3H), 7.90–7.92 (2H), 8.07–8.09 (1H), 8.47–8.49 (1H) | – |
| VIII | 2.46 | 6.73 | 7.61–7.68 (2H), 7.90–8.01 (4H), 8.70–8.71 (1H) | – |
| IX | 2.43 | 6.62 | 7.44 (1H), 7.61 (1H), 7.88–7.90 (2H), 8.09–8.11 (1H), 8.28 (1H) | – |
| X | 2.25 | 5.96 | 7.46–7.58 (5H), 8.00–8.13 (5H), 8.51 (1H) | – |
| XI | 2.43 | 6.61 | 7.44–7.52 (3H), 7.64–7.66 (2H), 7.74–7.76 (2H), 8.13–8.15 (2H) | – |
| XII | 2.42 | 6.62 | 7.12–7.17 (1H), 7.29–7.42 (4H), 7.55–7.57 (2H), 7,63–7.66 (2H), 8.05–8.07 (2H) | – |
Таблица 2.
Данные 13С ЯМР замещенных бензоилацетонатов дифторида бора I–XII в CDCl3 (δ, м.д.)
| № | C1′* | C2′ | C3′ | C4′ | C5′ | C6′ | C4 | C5 | C6 | 6-CH3 | Остальные сигналы |
|---|---|---|---|---|---|---|---|---|---|---|---|
| I | 131.′10 | 129.21 | 129.01 | 135.51 | 129.01 | 129.21 | 182.83 | 97.52 | 192.70 | 24.77 | – |
| II | 147.20 | 129.94 | 129.14 | 128.36 | 129.14 | 129.94 | 182.72 | 97.01 | 191.60 | 24.60 | 21.94 (р-CH3) |
| III | 153.24 | 128.75 | 128.75 | 128.59 | 128.75 | 129.27 | 182.76 | 97.01 | 191.56 | 24.60 | 14.89 (р-CH3), 29.13 (-CH2-) |
| IV | 146.05 | 130.03 | 126.82 | 128.64 | 130.43 | 137.83 | 182.92 | 97.01 | 191.25 | 24.54 | 19.72 (м-CH3), 20.95 (р-CH3) |
| V | 131.57 | 135.92 | 130.35 | 136.84 | 134.46 | 132.37 | 181.47 | 100.98 | 191.88 | 24.62 | 20.73 (CH3), 20.95 (CH3) |
| VI | 165.83 | 114.24 | 131.69 | 123.24 | 131.69 | 114.24 | 181.82 | 96.35 | 190.17 | 24.46 | 55.76 (OCH3) |
| VII | 130.20 | 133.84 | 125.11 | 134.97 | 124.56 | 127.02 | 186.96 | 102.07 | 192.45 | 24.75 | 128.56, 128.90 |
| VIII | 131.81 | 136.56 | 123.36 | 132.42 | 129.07 | 129.96 | 182.62 | 97.66 | 192.16 | 24.79 | 127.46, 127.89 |
| IX | 143.83 | 149.24 | 125.72 | 139.89 | 127.31 | 121.36 | 182.70 | 97.17 | 191.11 | 24.63 | 36.79 (-CH2-), 120.23, 121.36, 125.39, 128.54, 129.00 |
| X | 128.92 | 130.75 | 127.91 | 131.56 | 127.91 | 130.75 | 188.60 | 105.54 | 193.55 | 24.83 | 124.35, 125.77, 128.86 |
| XI | 148.21 | 129.09 | 127.29 | 139.02 | 127.29 | 129.09 | 182.31 | 97.30 | 192.08 | 24.74 | 127.66, 128.89, 129.63 |
| XII | 136.22 | 126.93 | 127.01 | 144.63 | 127.01 | 126.93 | 181.91 | 97.13 | 191.62 | 24.70 | 126.83, 128.79, 128.86, 129.62, 133.20, 160.74 |

В качестве растворителей и реагентов в работе применяли: толуол, этилбензол, о-ксилол, п-ксилол, нафталин, антрацен, флуорен (Реахим, Россия), бензоилацетон (Alfa Aesar, США), этанол, хлороформ, уксусную кислоту, ацетонитрил (ЭКОС-1, Россия), бортрифторид уксусный кислотный комплекс, анизол (Sigma Aldrich, США). Очистку растворителей проводили по известным методикам [27].
Соединение I получали из бензоилацетона и эфирата трифторида бора по методике [28]. Выход 95.4%; Rf = 0.57 (хлороформ); tпл 155–156°C; ИК (KBr), ν, см–1: 3138 (OHass), 2928 (C–HPh), 1597 (C=O), 1547 (C=C), 1439 (CH3), 1367, 1358 (δs B–O), 1156, 1138, 1113 (δ B–F), 1090, 1057 (δass B–O).
Соединение II. К смеси 0.11моль BF3‧2AcOH и 0.33 моль уксусного ангидрида добавляли по каплям 0.054 моль толуола при 45°C в течение 6 ч. После добавления толуола смесь перемешивали еще 3 ч при 45°C. Оставили на ночь в холодильнике, осадок отфильтровали. Промыли уксусной кислотой. Продукт перекристаллизовали из ацетонитрила. Выход 25.3%; Rf = 0.59 (хлороформ); tпл 158–159°C; ИК (KBr), ν, см–1: 3153, 3225 (OHass), 2933 (C–HPh), 1612 (C=O), 1544 (C=C), 1444 (CH3), 1373, 1353 (δs B–O), 1149, 1130 (δ B–F), 1085, 1051 (δass B–O).
Соединение IV получали из орто-ксилола аналогично II. Выход 68.5%; Rf = 0.55 (хлороформ); tпл 137–138°C; ИК (KBr), ν, см–1: 3165 (OHass), 3028 (C–HPh), 1608 (C=O), 1548 (C=C), 1434 (CH3), 1384 (δs B–O), 1168, 1103, (δ B–F), 1047, 1018 (δass B–O).
Соединение VI получали из анизола аналогично II. Выход 71.8%; Rf = 0.4 (хлороформ); tпл 163–165°C; ИК (KBr), ν, см–1: 3152 (OHass), 2981 (C–HPh), 1602 (C=O), 1569 (C=C), 1427 (CH3), 1352, 1352 (δs B–O), 1141, 1130 (δ B–F), 1087, 1056 (δass B–O).
Соединение III (модифицированная методика [24]). К смеси 0.15 моля BF3·2AcOH и 0.45 моля уксусного ангидрида добавляют по каплям 0.075 моля этилбензола в течение 6 ч при 45°C. После добавления этилбензола смесь перемешивали еще 3 ч при 45°C. После охлаждения реакционную смесь вылили в воду и экстрагировали хлороформом. Органический слой сушили сульфатом натрия, растворитель удаляли на роторном испарителе. Продукт перекристаллизовали из ацетонитрила. Выход 7.6%; Rf = 0.62 (хлороформ); tпл 163–165°C; ИК (KBr), ν, см–1: 3142 (OHass), 2972 (C–HPh), 1610 (C=O), 1551, 1543 (C=C), 1442 (CH3), 1377, 1352 (δs B–O), 1153, 1130, 1109 (δ B–F), 1086, 1057 (δass B–O).
Соединение V получали из пара-ксилола аналогично III. Выход 5.0%; Rf = 0.64 (хлороформ); tпл 65–67°C; ИК (KBr), ν, см–1: 3142 (OHass), 2990, 3076 (C–HPh), 1609 (C=O), 1535 (C=C), 1412 (CH3), 1365 (δs B–O), 1159, 1145 (δ B–F), 1087, 1055 (δass B–O).
Соединение VII получали из α-ацетонафтона по методике [29]. Выход 53.5%; Rf = 0.59 (хлороформ); tпл 153–155°C; ИК (KBr), ν, см–1: 3147 (OHass), 3061 (C–HPh), 1599 (C=O), 1539 (C=C), 1433 (CH3), 1371 (δs B–O), 1151, 1142 (δ B–F), 1076, 1047 (δass B–O).
Соединение VIII получали из β-ацетонафтона по методике [29]. Выход 81.2%; Rf = 0.57 (хлороформ); tпл 182–183°C; ИК (KBr), ν, см–1: 3147 (OHass), 3061 (C–HPh), 1599 (C=O), 1539 (C=C), 1433 (CH3), 1371 (δs B–O), 1151, 1142 (δ B–F), 1076, 1047 (δass B–O).
Соединение IX получали из 9H-флуорена по методике [30]. Выход 61.0%; Rf = 0.54 (хлороформ); tпл 261–262°C; ИК (KBr), ν, см–1: 3141 (OHass), 3057 (C–HPh), 1614 (C=O), 1537 (C=C), 1429 (CH3), 1377 (δs B–O), 1182, 1153 (δ B–F), 1076, 1055 (δass B–O).
Соединение X получали из антрацена по методике [31]. Выход 43.2%; Rf = 0.5 (хлороформ); tпл 228°C; ИК (KBr), ν, см–1: 3141 (OHass), 3057 (C–HPh), 1614 (C=O), 1537 (C=C), 1429 (CH3), 1377 (δs B–O), 1182, 1153 (δ B–F), 1076, 1055 (δass B–O).
Соединение XI получали из бифенила по методике [32]. Выход 71.0%; Rf = 0.59 (хлороформ); tпл 211–212°C; ИК (KBr), ν, см–1: 3147 (OHass), 3035 (C–HPh), 1606 (C=O), 1541 (C=C), 1452 (CH3), 1355 (δs B–O), 1182, 1139 (δ B–F), 1091, 1060 (δass B–O).
Соединение XII получали по реакции с газообразным трифторидом бора. 18.1 г (0.1 моль) (E)-стильбена добавили к 60 мл уксусного ангидрида и систему насыщали газообразным трехфтористым бором, поддерживая температуру реакционной смеси ниже 60°C. Реакционную смесь перемешивали до начала интенсивной кристаллизации. Осадок отфильтровали, промывали уксусной кислотой. Продукт перекристаллизовали из ацетонитрила. Выход 65.0%; Rf = 0.59 (хлороформ). tпл = 210–212°C; ИК (KBr), ν, см–1: 3151 (OHass), 3031 (C–HPh), 1600 (C=O), 1556 (C=C), 1442 (CH3), 1377 (δs B–O), 1188, 1153 (δ B–F), 11080, 1053 (δass B–O)
.РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Для получения замещенных бензоилацетонатов дифторида бора I–XII из ароматических углеводородов в качестве исходных соединений использовали бензол, нафталин, антрацен и соответствующие замещенные производные бензола. Например, при взаимодействии с орто- и пара-ксилолами, анизолом, флуореном и дифенилом соединения IV–VI, IX–XI получены с удовлетворительными выходами (рис. 1).
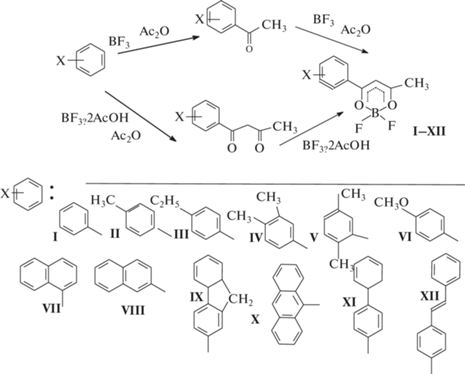
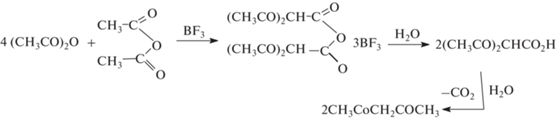
В случае бензола из-за его малой активности реакция ацилирования не идет даже под действием трифторида бора, исходный бензол был выделен в индивидуальном виде. При этом наблюдалось поглощение BF3 реакционной смесью и образование белого осадка хорошо растворимого в воде. Согласно работе [33], самоконденсация уксусного ангидрида (рис. 2) проходит через образование интермедиата, который гидролизом и последующим декарбоксилированием можно перевести в ацетилацетон.
Бензоилацетонат дифторида бора (I) получен классическим методом – комплексованием бензоилацетона с эфиратом трехфтористого бора в присутствии трибутилбората. При использовании в качестве исходных аренов, активированных σ-донорами: толуола, этилбензола, орто- и пара-ксилолов, реакция идет практически без побочных процессов и с BF3·2AcOH и с газообразным BF3. При использовании BF3·2AcOH некоторые комплексы дифторида бора оказались растворимы в реакционной смеси. Поэтому процесс выделения целевых продуктов модифицировали: реакционную смесь выливали в воду и выпавший осадок отфильтровали (комплексы II, IV, VI), в противном случае, продукт экстрагировали хлороформом (III и V).
1-(β-Нафтил)бутандионат-1,3 дифторида бора (VIII) получали с выходом 73% ацилированием нафталина при использовании газообразного трифторида бора [34]. Замена сильной кислоты Льюиса – трифторида бора на более слабую BF3 · ⋅ 2AcOH приводит к получению смеси изомерных продуктов VII и VIII [29], которую трудно разделить методами фракционной кристаллизации и колоночной хроматографией.
Молекула нафталина имеет два реакционных центра: α- и β-положения. α-Положение является более выгодным и при кинетическом контроле реакции замещения идут по α-положению, при термодинамическом возможна реакция в β-положение [35]. Обе возможности могут реализоваться при ацилировании нафталина под действием хлорида алюминия: в дихлорэтане образуется α- изомер, а в нитробензоле β-изомер [36]. Следовательно, при использовании газообразного BF3 ацилирование нафталина идет по термодинамическому контролю, а при использовании BF3·2AcOH по кинетическому. Единственная возможность получить 1-(α-нафтил)бутандионат-1,3 дифторида (7) – это использовать α-ацетилнафталин. Также неоднозначно ацилируется антрацен: при использовании BF3·2AcOH даже при длительном нагревании реакционной смеси остается сложная смесь продуктов. С газообразным BF3 ацилирование проходит строго по положению 9 молекулы антрацена.
Для анизола, содержащего сильный π-донор метокси-группу, ацилирование с газообразным BF3 проходит очень интенсивно, сопровождается выделением тепла и значительным осмолением реакционной смеси. При использовании BF3·2AcOH осмоления не происходит, получается достаточно чистый продукт анизоилацетонат дифторида бора (VI). Для аренов, содержащих π-доноры средней силы (дифенил, флуорен, стильбен) реакция удовлетворительно проходит и с газообразным BF3 и с BF3·2AcOH.
β-Дикетонаты дифторида бора можно получить не только непосредственным С-ацилированием, но и косвенно – О-ацилированием кетона с последующим С‑ацилированием полученного енола кетона (рис. 3). Некоторые эфиры енолов кетонов удалось выделить из реакционной смеси и превратить, обработав ангидридом кислоты и трифторидом бора, в эфир енола β-дикетона и, затем, в β-дикетонат дифторида бора [22].

В начальный период реакции, пока не образовалось большое количество карбоновой кислоты, преобладает С-ацилирование (рис. 1, верхний путь реакции). Кислота, образующаяся в качестве побочного продукта, является катализатором О-ацилирования (рис. 3), следовательно возможно протекание реакции по косвенному пути (рис. 1, нижний путь реакции). С течением времени косвенный путь может даже преобладать. Прямой путь преобладает при быстром насыщении реакционной смеси трифторидом бора, а косвенный при медленном. Особенно быстро накопление кислоты происходит при использовании BF3·2AcOH, высвобождающаяся в ходе реакции уксусная кислота способствует реакции по пути О-ацилирования.
Таким образом, выбор между газообразным трифторидом бора и его аддуктом с уксусной кислотой – это выбор между механизмами С- и О-ацилирования, что объясняет образование смеси продуктов реакции при ацилировании антрацена и нафталина BF3·2AcOH и образование индивидуальных веществ при использовании газообразного BF3. Как показано на примере ацилирования анизола (синтез комплекса VI), использование BF3 · 2AcOH хорошо подходит для ацилирования активных соединений.
Строение всех полученных бензоилацетонатов дифторида бора подтверждено данными элементного анализа, ИК- и ЯМР-спектроскопии. В инфракрасных спектрах ряда соединений I–XII появляются специфические для этого класса соединений полосы: в диапазоне 1380–1360 см–1 сигналы валентно-симметричных колебаний связей В–О, полоса расщепляется из-за наличия изотопов 10В и 11В; в области 1150–1000 см–1 сигналы валентных колебаний образованных связью B–F и валентных асимметричных колебаний B–O. Стоит отметить наличие интенсивных сигналов связей С=О и С=С в хелатном цикле при 1560–1540 см–1, в области 1450–1410 см–1 появляется полоса деформационных асимметричных колебаний метильной группы.
Строение всех полученных β-дикетонатов дифторида бора подтверждено характерными сигналами в спектрах ЯМР 1H: при 6.4–6.7 м.д. (синглетный протон в гамма-положении хелатного цикла) и 2.3–2.5 м.д. (синглетная метильная группа) (табл. 1). В спектрах ЯМР 13С (табл. 2) отчетливо прослеживаются три группы сигналов: в области 24–25 м.д. сигнал метильной группы, в области 97–100 м.д. сигнал углерода в γ-положении хелатного цикла, сигналы 182–184 и 190–193 м.д. С–О и С=O групп, соответственно. Когда в α-положении хелатного цикла сопряженная π-система увеличивается, идентификация всех фенильных сигналов становится проблематичной из-за стерических и ориентационных факторов.
В табл. 3 приведены спектральные характеристики растворов I–XII. Для комплексов I–IV максимумы спектров люминесценции находятся в голубой области спектра (390–395 нм), квантовый выход люминесценции I незначителен и составляет 0.001, для комплексов II–V, содержащих алкильные заместители (σ-доноры) в фенильном кольце, происходит увеличение квантового выхода до 0.12, что, по-видимому, связано с замедлением вращения фенильного кольца. Введение сильного π-донора метоксигруппы в VI не вызывает заметных спектральных изменений. Увеличение размера π-системы молекулы путем замены фенила на нафтил, антрацил, дифенил, стильбенил (комплексы VII–XII) приводит к значительному батохромному смещению спектров поглощения, возбуждения люминесценции и люминесценции и заметному увеличению квантового выхода люминесценции до 0.53–0.75 (табл. 3). Максимальное батохромное смещение спектров наблюдается для IX (λabs = 443 нм, λlum = 536 нм), для которого из-за стерических затруднений и неплоского строения молекулы реализуется состояние с переносом заряда [31].
Таблица 3.
Спектральные характеристики растворов замещенных бензоилацетонатов дифторида бора I–XII в хлороформе
| № | λabs, нм | λex, нм | λlum, нм | ΔνST, см–1 | φ | τ, нс |
|---|---|---|---|---|---|---|
| I | 328 | 345 | 395 | 5171 | 0.001 | 0.54 |
| II | 334 | 340 | 392 | 4585 | 0.01 | 1.40 |
| III | 335 | 360 | 390 | 2264 | 0.12 | 1.11 |
| IV | 323 | 325 | 419 | 2386 | 0.01 | 1.86 |
| V | 347 | 406 | 470 | 7542 | 0.001 | 1,66 |
| VI | 360 | 366 | 386 | 2403 | 0.04 | 2.91 |
| VII | 367 | 373 | 444 | 4725 | 0.61 | 9.2 |
| VIII | 343, 379 | 380 | 444 | 3862 | 0.53 | 7.1 |
| IX | 383 | 382 | 436 | 4270 | 0.86 | 1.85 |
| X | 443 | 447 | 536 | 3917 | 0.09 | 0.85 |
| XI | 358 | 360 | 426 | 4459 | 0.75 | 1.13 |
| XII | 394 | 390 | 470 | 4104 | 0.70 | 2.00 |
Для V наблюдается батохромный сдвиг спектров поглощения и люминесценции и значительно увеличивается величина стоксова сдвига по сравнению с остальными алкилбензоилацетонатами дифторида бора I–IV. Наличие метильной группы в орто-положении фенильного кольца в V не позволяет фенильному и дикетонатному кольцам лежать в одной плоскости и приводит образованию ротамеров (рис. 4).
Рис. 4.
Схема образования ротамеров V, стрелками показаны стерические затруднения в молекуле, препятствующие свободному вращению фенильного кольца.

Геометрия молекулы V оптимизирована методом TD DFT (6-311G(d, p)) (рис. 5). Обнаружено, что угол междуплоскостями фенильного и хелатного колец составляет 30°. При этом для V вероятно образование ротамеров, различающихся положением метильной группы относительно дикетонатного кольца (рис. 3). Батохромное смещение спектра люминесценции и увеличение стоксова сдвига V относительно комплексов I–IV (табл. 3), связано с переносом заряда в молекуле V при переходе в возбужденное состояние (ВЗМО–НСМО) (рис. 5а).
Рис. 5.
Строение граничных орбиталей молекулы V (а), оптимизированная геометрия молекулы IV (б), оптимизированная геометрия молекулы XII (в).

Как показал анализ данных РСА, для соединений I–III, VI, VIII, X, XI фенильное и хелатное кольца лежат в одной плоскости [36]. Расчет геометрии IV и XII методом TD DFT (6-311G(d, p)) также подтвердил плоское строение молекул (рис. 5б, 5в). Среди исследованных двенадцати комплексов три (V, VII и IX) имеют значительный угол между плоскостями фенильного и дикетонатного колец (30°, 34° [29] и 77° [31] соответственно). Для этих комплексов переход ВЗМО–НСМО является переходом с переносом заряда, что приводит к значительному батохромному сдвигу спектров поглощения люминесценции относительно спектров других соединений и существенному снижению квантового выхода люминесценции.
ЗАКЛЮЧЕНИЕ
Ряд замещенных бензоилацетонатов дифторида бора получили конденсацией Меервейна (ацилирование ароматических соединений уксусным ангидридом под действием трифторида бора или его аддуктов). Показано, что реакция успешно проходит с ароматическими соединениями, имеющими донорные заместители в фенильном кольце, и с конденсированными ароматическими соединениями (нафталин или антрацен). При попытке ацилирования бензола преимущественно проходит самоконденсация уксусного ангидрида. Большое значение имеет выбор кислоты Льюиса нужной активности (трифторид бора и его аддукты). Для конденсированных ароматических соединений необходимо использовать газообразный трифторид бора для направления реакции по термодинамическому пути, а для соединений с активными донорными группами – его аддукт с уксусной кислотой для уменьшения количества побочных процессов, приводящих к осмолению смеси. Конденсацию Меервейна можно рекомендовать для получения замещенных бензоилацетонатов дифторида бора в препаративных количествах, с целью дальнейшего их использования, например, как прекурсоров при получении красителей.
Систематизированы данные по люминесцентным свойствам бензоилацетонатов I–XII. По сравнению с незамещенным бензоилацетонатом дифторида бора I введение алкильных заместителей в фенильное кольцо (комплексы II–V) не приводит к заметным спектральным изменим, при этом увеличивается квантовый выход с 0.001 (I) до 0.12 (III). Существенное увеличение квантового выхода люминесценции до 0.75 (X) и значительный батохромный сдвиг до 536 нм (IX) достигаются при увеличении π-системы молекулы: переход от фенила к нафтилу, антрацилу, дифенилу.
Список литературы
Bumagina N.A., Kritskaya A.Y., Antina E.V. et al. // Russ. J. Inorg. Chem. 2018. V. 63. № 10. P. 1326. https://doi.org/10.1134/S0036023618100030
Антина Е.В., Березин М.Б., Вьюгин А.И. и др. // Журн. неорган. химии 2022. Т. 67. № 3. С. 342. https://doi.org/10.31857/S0044457X22030035
Wang F., Song D.L., Dickie D.A. // J. Fluores. 2021. V. 31. P. 39. https://doi.org/10.1007/s10895-020-02626-8
Lee S., Kwak S., Wan K.-K. et al. // ECS J. Solid State Sci. Technol. 2020. V. 110. P. 086007. https://doi.org/10.1149/2162-8777/ac1d60
Zhang Q.-Z.H., Chen P.-Z., Niu L.-Y. et al. // Mater. Chem. Front. 2020. V. 4. P. 285. https://doi.org/10gbk.1039/C9QM00672A
Loudet A., Burgess K. // Chem. Rev. 2007. V. 107. P. 4891. https://doi.org/10.1021/cr078381n
Collot M. // Mater. Horiz. 2021. V. 8. P. 50. https://doi.org/10.1039/d0mh01186j
Kamkaew A., Lim S.H., Lee H.B. et al. // Chem. Soc. Rev. 2013. V. 42. P. 77. https://doi.org/10.1039/C2CS35216H
Nieto C.I., Cabildo M.P., Cornago M.P. et al. // Molecules. 2015. V. 20. P. 15643. https://doi.org/10.3390/molecules200915643
Svistunova I.V., Shapkin N.P., Nikolaeva O.V. // Russ. J. Gen. Chem. 2002. V. 72. P. 899. https://doi.org/10.1023/A:1020426105849
Halik M., Hartmann H. // Chem. Eur. J. 1999. V. 5. P. 2511.
Traven V.F., Chibisova T.A., Manaev A.V. // Dyes Pigments. 2003. V. 58. № 1. P. 41 https://doi.org/10.1016/S0143-7208(03)00022-6
Goel A., Kunnumakkara A.B., Aggarwal B.B. // Biochem. Pharmacol. 2008. V. 75. № 4. P. 787. https://doi.org/10.1016/j.bcp.2007.08.016
Mayadevi M., Sherin D.R., Keerthi V.S et al. // Bioorg. Med. Chem. 2012. V. 20. P. 6040. https://doi.org/10.1016/j.bmc.2012.08.029
Hatchera H., Planalp R., Chob J. et al. // Cell. Mol. Life Sci. 2008. V. 65. P. 1631. https://doi.org/10.1007/s00018-008-7452-4
Benoit I., Asther M., Sulzenbacher G. et al. // FEBS Lett. 2006. V. 580. P. 5815. https://doi.org/10.1016/j.febslet.2006.09.039
Hermoso J.A., Sanz-Aparicio J., Molina R. et al. // J. Mol. Biol. 2004. V. 338. P. 495. https://doi.org/10.1016/j.jmb.2004.03.003
Liu K., Chen J., Chojnacki J. et al. // Tetrahedron Lett. 2013. V. 54. P. 2070. https://doi.org/10.1016/j.tetlet.2013.02.015
Brown N.M.D., Bladon P. // J. Chem. Soc. A. 1969. P. 526. https://doi.org/10.1039/J19690000526
Meerwein H., Vossen D. // J. Prakt. Chem. 1934. V. 141. P. 149. https://doi.org/10.1002/prac.19341410503
Hauser C.R., Swamer F.W., Adams J.T. // The Acylation of Ketones to Form β-Diketones or β-Keto Aldehydes. Organic Reactions. 2011. https://doi.org/10.1002/0471264180.or008.03
Hauser C.R., Frostik F.C., Man E.H. // J. Am. Chem. Soc. 1952. V. 74. P. 3231. https://doi.org/10.1021/ja01133a0082
Gorlitz G., Hartmann H., Kossanyi J. et al. // Ber. Bunsenges. Phys. Chem. 1998. V. 102. № 10. P. 1449. https://doi.org/10.1002/(SICI)1521-3897(199902)34-1:23.0.CO;2-A
Gorlitz G., Hartmann H., Nuber B. // J. Pract Chem. 1999. V. 341. № 2. P. 167/ https://doi.org/10.1002/(SICI)1521-3897(199902)34-1:23.0.CO;2-A
Dromzee Y., Kossanyi J., Wintgens V. et al. // Z. Kristallographie. 1997. V. 212. P. 372. https://doi.org/10.1524/zkri.1997.212.5.372
Demas J.N., Crosby G.A. // J. Phys. Chem. J. Phys. Chem. 1971. V. 75. № 8. P. 991. https://doi.org/10.1021/j100678a001
Weissberger A., Proskauer E.S., Riddick J.A., Toops E.E. Organic Solvents. Physical Properties and Methods of Purifiation. N.Y.: Interscience Publ., 1955. 552 p.
Karasev V.E., Korotkich O.A. // Russ. J. Inorg. Chem. 1986. V. 31. № 4. P. 869.
Fedorenko E.V., Mirochnik A.G., Gerasimenko A.V. et al. // J. Photochem. Photobiol. A. 2021. V. 412. P. 113220. https://doi.org/10.1016/j.jphotochem.2021.113220
Bukvetskii B.V., Fedorenko E.V., Mirochnik A.G. // J. Struct. Chem. 2011. V. 52. № 1. P. 221. https://doi.org/10.1134/s0022476611010331
Fedorenko E.V., Bukvetskii B.V., Mirochnik A.G. et al. // J. Lumines. 2010. V. 130. № 5. P. 756. https://doi.org/10.1016/j.jlumin.2009.11.027
Bukvetskii B.V., Fedorenko E.V., Mirochnik A.G. // J. Struct. Chem. 2010. V. 51. № 4. P. 785. https://doi.org/10.1007/s10947-010-0118-8
Bukvetskii B.V., Fedorenko E.V., Mirochnik A.G. // Struct. Chem. 2010. V. 51. № 3. P. 545. https://doi.org/10.1007/s10947-010-0079-y
Марч Дж. Органическая химия. Реакции, механизмы и структура. Т. 2. / Пер. с англ. под ред. Белецкой И.П. и др. M.: Мир, 1987. 504 с.
Donaldson N. // The Chemistry and Technology of Naphthalene Compounds / Ed. Arnold E. London, 1958.
Fedorenko E.V., Mirochnik A.G., Beloliptsev A.Yu. // J. Lumines. 2018. V. 196. P. 316. https://doi.org/10.1016/j.jlumin.2017.12.071
Дополнительные материалы отсутствуют.
Инструменты
Журнал неорганической химии