

Журнал неорганической химии, 2022, T. 67, № 9, стр. 1283-1291
Синтез и экстракционные свойства дифенилфосфорилмочевин с ω-(алкокси/тетрагидрофурил)алкильными заместителями у терминального атома азота
А. М. Сафиулина a, *, Н. Е. Борисова b, А. В. Лизунов a, Т. В. Баулина c, Е. И. Горюнов c, А. С. Перегудов c, В. К. Брель c, И. Г. Тананаев d
a Высокотехнологический научно-исследовательский институт неорганических материалов им. А.А. Бочвара
123098 Москва, ул. Рогова, 5а, Россия
b Московский государственный университет им. М.В. Ломоносова
119991 Москва, Ленинские горы, 1, Россия
c Институт элементоорганических соединений им. А.Н. Несмеянова РАН
119991 Москва, ул. Вавилова, 28, Россия
d Дальневосточный федеральный университет
690922 Владивосток, о. Русский, п. Аякс, 10, Россия
* E-mail: AMSafiulina@bochvar.ru
Поступила в редакцию 17.01.2022
После доработки 21.03.2022
Принята к публикации 30.03.2022
- EDN: MJMPAQ
- DOI: 10.31857/S0044457X22090100
Аннотация
На основе высокоэффективного технологичного каталитического “one-pot”-процесса синтезированы N-дифенилфосфорил-N'-[ω-(алкокси/тетрагидрофурил)алкил]мочевины и исследованы их экстракционные свойства по отношению к f-элементам в азотнокислых средах. Выявлено влияние на экстракционные характеристики наличия алкоксильной или тетрагидрофурильной группировки, находящейся в ω-положении нормального N'-алкильного радикала фосфорилмочевины. Показано, что наилучшими экстракционными свойствами по отношению к исследуемым f-элементам обладает N-(дифенилфосфорил)мочевина, содержащая N'-[2-(тетрагидрофур-2-ил)этильный] радикал, особенно этот эффект выражен для урана(VI). Данная зависимость получила теоретическое обоснование при моделировании комплексообразования методом теории функционала плотности (DFT, PBE, cc-pVDZ), поскольку координирование иона f-элемента к атому кислорода фурильного радикала, присоединенному к этиленовой углеродной цепочке, оказывается предпочтительным.
ВВЕДЕНИЕ
С развитием уровня современной техники потребление редких и редкоземельных металлов (РМ и РЗМ) имеет тенденцию к неуклонному росту. Это происходит из-за предпочтительного или безальтернативного применения РМ и РЗМ в качестве функциональных и конструкционных материалов в ряде технологий и технических устройств: в оптике, электронике, системах хранения энергии, реакторостроении различного назначения, средствах транспорта и т.п. [1–3].
Вследствие высокого потребления РМ и РЗМ, а также геополитического взаимодействия ряда стран возник дефицит редких металлов, что привело к росту цен на них [4, 5]. Эти обстоятельства вызвали необходимость разработки новых подходов к производству редкометалльных концентратов из низкорентабельных руд и техногенных отходов [6–10]. Традиционно технологические схемы производства концентратов РМ и РЗМ включают экстракционные переделы из водных кислотных растворов. При переработке сырья с низким содержанием ценных компонентов важен выбор органического экстрагента с высокой координирующей способностью к f-элементам. Коммерчески доступные на рынке фосфорорганические экстрагенты обладают низкой эффективностью и селективностью в отношении к f-элементам [11, 12], поэтому остается актуальным поиск новых эффективных и селективных экстрагентов для извлечения и разделения РЗМ.
Ранее установлено, что N-(диорганилфосфорил)мочевины R2P(O)NHC(O)NHR'(I), преимущественно N-дифенилфосфорилмочевины (Ia, R = Ph), обладают высокой экстракционной способностью к f-элементам из азотнокислых растворов в широком диапазоне концентраций [13–15]. На экстракционную способность фосфорилмочевин оказывает влияние природа заместителей у терминального атома азота. Были исследованы N-(дифенилфосфорил)-N'-н-алкил(С6-С10)мочевины Ph2P(O)NHC(O)NHCnH2n+ 1 (n = 6–10) и показано, что наибольшей экстракционной способностью к f-элементам обладает N'-н-октильное производное (Ib) [13].

Недавно исследовано влияние имидазолильного, диэтиламино, пирид-2-ильного и 2-оксопирролидинового радикалов у терминального углеродного атома пропильного фрагмента дифенилфосфорилмочевин [Ia, R' = RN(CH2)3] на эффективность при экстракции f-элементов [16]. Показано, что наилучшими экстракционными свойствами обладает N-(дифенилфосфорил)мочевина, содержащая ω-(2-оксопирролидино)пропильный радикал в N’-положении. Этот эффект обусловлен наличием дополнительного центра координации амидного атома кислорода ω-(2-оксопирролидино)пропильного радикала. Следует предположить, что введение алкоксильной или тетрагидрофурильной группировки, также содержащих потенциальный дополнительный центр координации – атом кислорода, в ω-положение алкильного радикала у терминального атома азота N-дифенилфосфорилмочевины, будет способствовать увеличению эффективности и селективности соответствующих лигандов при экстракции f-элементов.
В настоящей работе исследовано влияние алкоксиалкильного и тетрагидрофурилалкильных радикалов у N'-атома азота дифенилфосфорилмочевин (Ia) на их экстракционную способность по отношению к f-элементам.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
N-(Дифенилфосфорил)-N'-алкилмочевины, содержащие в ω-положении алкильного радикала алкоксильные или тетрагидрофур-2-ильные заместители (II–V), синтезировали с использованием оригинальных “one-pot”-процессов, в которых в качестве исходного фосфорорганического реагента применяли коммерчески доступный дифенилхлорфосфин (схема). Эти процессы включают в себя три стадии: окисление дифенилхлорфосфина до дифенилхлорфосфината, каталитическое превращение последнего в дифенилфосфорилизоцианат и взаимодействие этого изоцианата с кислородсодержащими первичными аминами RО(CH2)nNH2 (RO = н-BuO, 2-метилтетрагидрофур-2-ил, тетрагидрофур-2-ил; n = 1, 2). Все три стадии могут быть проведены при комнатной температуре, общее время процесса не превышает 6 ч, а выход аналитически и спектрально чистых целевых продуктов находится на уровне 90%.
Дифенилхлорфосфин (Aldrich, 98%) непосредственно перед реакцией перегоняли в вакууме. Безводный MgCl2 (Aldrich, ≥98%) использовали без дополнительной очистки. Циановокислый натрий (Aldrich, 96%) сушили 4 ч при 120°С в вакууме (1 Торр) над P2O5. Хлористый сульфурил (Acros, 98.5%) перегоняли непосредственно перед реакцией. Четыреххлористый углерод и ацетонитрил абсолютировали перегонкой над P2O5. Все операции проводили в атмосфере аргона.
Общая методика синтеза N-дифенилфосфорил-N'-(ω-RО-алкил)мочевин (II–V). К раствору 1.82 г (8.25 ммоль) дифенилхлорфосфина в 4 мл абсолютированного CCl4 при перемешивании на магнитной мешалке при комнатной температуре в течение 20 мин добавляли по каплям раствор 1.35 г (10 ммоль) SO2Cl2 в 4 мл абсолютированного CCl4, перемешивали еще 1 ч при этой температуре, растворитель и другие летучие компоненты реакционной смеси удаляли в вакууме. Остаток растворяли в 20 мл абсолютированного MeCN; к полученному раствору добавляли 20 мг (0.21 ммоль) мелко растертого безводного MgCl2, перемешивали до полного растворения последнего, прибавляли 1.08 г (16.5 ммоль) NaOCN и перемешивали 1 ч при комнатной температуре; к полученной суспензии в течение 15 мин добавляли по каплям раствор 8.25 ммоль RO(CH2)nNH2 в 5 мл абсолютированного MeCN, перемешивали 1 ч при комнатной температуре. Удаляли растворитель, и к сухому остатку добавляли 30 мл дистиллированной воды, перемешивали 1 ч при комнатной температуре. Осадок отфильтровывали, последовательно промывали смесью 8 мл дистиллированной воды и 2 мл MeCN, дистиллированной водой (2 × 10 мл); сушили на воздухе.
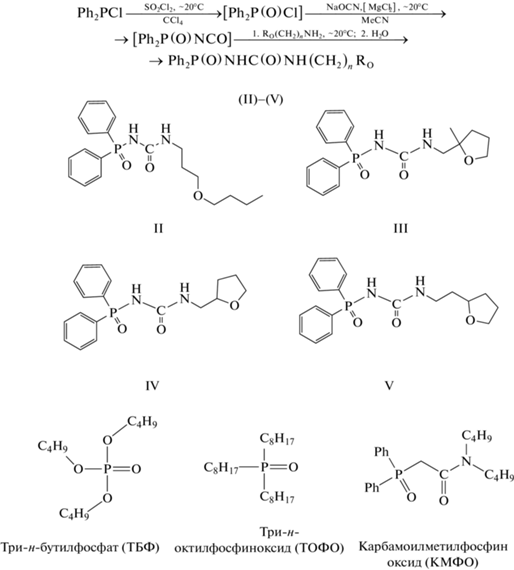
N-Дифенилфосфорил-N'-(3-н-бутоксипропил)мочевина (II). Выход 91.8%. tпл = 159.5–160.5°С (хлороформ-гексан).
Спектр ЯМР 1Н (δН, м.д.): 0.87 т (3H, CH3, 3JH–H = = 7.3 Гц); 1.30 секстет (2Н, CH3CH2, 3JH–H = 7.4 Гц); 1.46 квинт (2H, СН3СН2CH2, 3JH–H = 7.0 Гц); 1.58 квинт (2H, ОCH2CH2CH2NН, 3JH–H = 6.7 Гц); 3.04 дт (2H, CH2NH, 3JH–СH = 6.4 Гц, 3JH–NH = 6.2 Гц); 3.32 т (4H, CH2O, 3JH–H = 6.5 Гц); 6.52 т (1Н, CH2NH, 3JH–H = 5.7 Гц); 7.47–7.62 м (6Н, м- + р-С6Н5); 7.75 дд (4Н, о-С6Н5, 3JH–Н = 7.9 Гц, 3JH–Р = = 12.4 Гц); 8.34 д [1Н, NHP(O), 2JH–P = 10.5 Гц].
Спектр ЯМР 31P{1H} (δP, м.д.): 16.80 с.
N-Дифенилфосфорил-N’-[(2-метилтетрагидрофур-2-ил)метил]мочевина (III). Выход 92.9%. tпл = = 161.5–162.0°С.
Спектр ЯМР 1Н (δН, м.д.): 1.06 с (3Н, СН3); 1.48–1.56 м (1Н, 3СНАНв–тетрагидрофур-2-ил); 1.61–1.70 м (1Н, 3СНАНв-тетрагидрофур-2-ил); 1.76–1.89 м (2Н, 4СН2-тетрагидрофур-2-ил); 3.01 дд (1Н, NHCHАНВ, 3JH–H = 5.5 Гц, 2JHА–HВ = = 13.3 Гц); 3.07 дд (1Н, NHCHАНВ, 3JH–H = 6.0 Гц, 2JHВ–HА = 13.3 Гц); 3.66–3.75 м (2Н, 5СН2-тетрагидрофур-2-ил); 6.66 т (1Н, NHCH2, 3JH–H = 5.5 Гц); 7.51 дт (4Н, m-C6H5, 3JH–H = 7.3 Гц, 4JH–Р = 2.6 Гц); 7.57 т (2Н, p-C6H5, 3JH–H = 7.3 Гц); 7.75 дд (4Н, o-C6H5, 3JH–H = 7.8 Гц, 3JH–Р = 12.4 Гц); 8.43 д [1Н, NHP(O), 2JH–P = 11.0 Гц].
Спектр ЯМР 13С{1H} (δC, м.д.): 24.64 с (СН3); 26.24 с (4С-тетрагидрофур-2-ил); 34.50 с (3С-тетрагидрофур-2-ил); 47.50 с (NHCH2); 67.41 с (5С-тетрагидрофур-2-ил); 82.15 с (2С-тетрагидрофур-2-ил); 128.99 д (m-C6H5, 3JC–P = 12.2 Гц); 131.52 д (o-C6H5, 2JC–P = 9.9 Гц); 132.26 д (p-C6H5, 4JC–P = = 1.7 Гц); 133.41 д (ипсо-C6H5, 1JC–P = 128.8 Гц); 133.43 д (ипсо-C6H5, 1JC–P = 129.4 Гц); 155.60 c (C=O).
Спектр ЯМР 31Р{1H} (δР, м.д.): 15.37 с.
N-Дифенилфосфорил-N'-[(тетрагидрофур-2-ил)метил]мочевина (IV). Выход 91.2%. tпл = 196–197°С.
Спектр ЯМР 1Н (δН, м.д.): 1.39–1.48 м (1Н, 3СНАНв-тетрагидрофур-2-ил); 1.72–1.88 м (3Н, 3СНАНв- + 4СН2-тетрагидрофур-2-ил); 3.02 дт (1Н, NHСНAHB, 3JH–H = 6.0 Гц, 2JHA–HB = 13.5 Гц); 3.15 ддд (1Н, NHСНAHB, 3JH–СH = 4.5 Гц, 3JH–NH = = 5.6 Гц, 2JHB–HA = 13.5 Гц); 3.58–3.65 м (1Н, 5СНАНв-тетрагидрофур-2-ил); 3.71–3.77 м (1Н, 5СНАНв-тетрагидрофур-2-ил); 3.77–3.83 м (1Н, 2СН-тетрагидрофур-2-ил); 6.67 т (1Н, NHCH2, 3JH–H = 5.7 Гц); 7.51 дт (4Н, m-C6H5, 3JH–H = 7.3 Гц, 4JH–Р = 3.2 Гц); 7.57 дт (2Н, p-C6H5, 3JH–H = 7.4 Гц, 5JH–Р = 1.2 Гц); 7.75 дд (4Н, o-C6H5, 3JH–H = 7.2 Гц, 3JH–P = 12.4 Гц); 8.41 шс [1Н, NHP(O)].
Спектр ЯМР 13С{1H} (δC, м.д.): 25.81 с (4С-тетрагидрофур-2-ил); 28.49 с (3С-тетрагидрофур-2-ил); 43.47 с (NHCH2); 67.73 c (5С-тетрагидрофур-2-ил); 77.73 c (2С-тетрагидрофур-2-ил); 128.99 д (m-C6H5, 3JC–P = 12.7 Гц); 131.53 д (o-C6H5, 2JC–P = = 10.0 Гц); 132.24 c (p-C6H5); 133.41 д (ипсо-C6H5, 1JC–P = 129.0 Гц); 133.46 д (ипсо-C6H5, 1JC–P = = 128.1 Гц); 155.47 c (C=O).
Спектр ЯМР 31P{1H} (δP, м.д.): 15.32 с.
N-Дифенилфосфорил-N'-[2-(тетрагидрофур-2-ил)этил]мочевина (V). Выход 89.6%. tпл = 187–188°С.
Спектр ЯМР 1Н (δН, м.д.): 1.32–1.42 м (1Н, 3СНАНв-тетрагидрофур-2-ил); 1.48–1.59 м (2Н, NHCH2CH2); 1.71–1.85 м (2Н, 4СН2-тетрагидрофур-2-ил); 1.86–1.95 м (1Н, 3СНАНв-тетрагидрофур-2-ил); 3.00–3.13 м (2Н, NHCH2); 3.53–3.60 м (1Н, 5СНАНв-тетрагидрофур-2-ил); 3.65–3.76 м (2Н, 2СН- + 5СНАНв-тетрагидрофур-2-ил); 6.58 т (1Н, NHCH2, 3JH–H = 5.5 Гц); 7.51 дт (4Н, m-C6H5, 3JH–H = 7.4 Гц, 4JH–P = 2.9 Гц); 7.57 т (2Н, p-C6H5, 3JH–H = 7.3 Гц); 7.75 дд (4Н, o-C6H5, 3JH–H = 7.6 Гц, 3JH–P = 12.1 Гц); 8.41 шс [1Н, NHP(O)].
Спектр ЯМР 13С{1H} (δC, м.д.): 25.64 с (4С-тетрагидрофур-2-ил); 31.39 с (3С-тетрагидрофур-2-ил); 35.70 с (NHCH2CH2); 37.34 с (NHСН2); 67.27 c (5С-тетрагидрофур-2-ил); 76.98 с (2С-тетрагидрофур-2-ил); 128.97 д (m-C6H5, 3JC-P = 12.7 Гц); 131.55 д (o-C6H5, 2JC-P = 10.0 Гц); 132.21 д (p-C6H5, 4JC-P = 1.8 Гц); 133.48 д (ипсо-C6H5, 1JC-P = 129.0 Гц); 155.33 с (C=O).
Спектр ЯМР 31Р{1H} (δР, м.д.): 15.36 c.
Спектры ЯМР 1H и 31P{1H} мочевин (II–V), а также спектры ЯМР 13С{1H} мочевин (III–V) регистрировали на приборе Bruker AV-500, рабочая частота 500.13 MHz (1H), 125.77 MHz (13C{1H}) и 202.46 MHz (31P{1H}), в растворе (CD3)2SO (с = = 0.1 М). Внутренний эталон для спектров ЯМР 1H-сигналы остаточных протонов дейтерированного растворителя, а для спектров ЯМР 13С{1H} – сигналы ядер атомов углерода дейтерированного растворителя; внешний эталон для спектров ЯМР-31Р{1H} – 85%-ная H3PO4. В случае мочевин (III–V), содержащих тетрагидрофурильный фрагмент, включающий асимметрический атом углерода в 2-положении гетероциклической матрицы, корректное отнесение сигналов в спектрах ЯМР 1H и 13С{1H} проводили с использованием корреляций COSY, HMQC и HMBC.
Исследование экстракции металлов. В работе использовали хлороформ (х. ч.), арсеназо III (ч. д. а.), HNO3 (ос. ч.), ГСО 8363-2003 закись-окись урана (содержание урана 84.784 ± 0.016%), La(NO3)3·6H2O (х. ч.), Nd(NO3)3 · 6H2O (х. ч.), Ho(NO3)3·6H2O (х. ч.). Растворы готовили объемно-весовым методом. Водные растворы готовили в бидистиллированной воде. Растворы нитратов исследуемых элементов готовили растворением навески соответствующего нитрата в 0.01 моль/л растворе HNO3. Концентрацию растворов нитратов металлов (0.1 ммоль/л) уточняли спектрофотометрически по методике [17] с использованием спектрофотометра Cary 5000 Scan (Varian). Концентрацию растворов HNO3 определяли потенциометрическим титрованием 0.1 моль/л NaOH с использованием рН-метр/кондуктометра S470 SevenExcellence™ (MettlerToledo) с точностью ±0.01 ед. рН. Электродную пару калибровали по стандартным буферным растворам с рН 1.68, 4.01 и 9.21 (MettlerToledo) (значения при 20°С). Концентрацию раствора NaOH уточняли потенциометрическим титрованием с 0.1 моль/л HCl (фиксанал).
Исследование экстракции катионов металлов выполняли по следующей методике. В пробирку с притертой пробкой вносили 1.5 мл раствора азотной кислоты, концентрация которой варьировалась от 0.052 до 5.0 моль/л; 0.5 мл 0.1 ммоль/л раствора нитрата металла, 2 мл 0.01 моль/л раствора лиганда в хлороформе. Фазы перемешивали в течение 20 мин в ротаторе. Время установления равновесия экстракции проверяли, увеличивая время контакта фаз до 120 мин, коэффициенты распределения при этом не изменялись. Расслаивание фаз осуществляли центрифугированием. После разделения фаз концентрацию катионов металлов в водной фазе определяли спектрофотометрическим методом [17]. Для каждой концентрации проводили не менее трех независимых опытов. Суммарная погрешность результатов составила ~20%, учитывая неисключенную и случайную составляющие. Доверительный интервал определяемых концентраций металлов в эксперименте составляет 0.002 ммоль/л.
Все эксперименты проводили при температуре 20 ± 1°С.
Коэффициенты распределения при экстракции (D = [M]org/[M]aq) определяли при постоянных концентрациях экстрагента (0.01 моль/л в хлороформе) и исходных концентрациях металла в эксперименте (0.025 ммоль/л в водной фазе).
Расчеты выполняли на суперкомпьютере MVS-50K Межведомственного суперкомпьютерного центра РАН (www.jscc.ru). Все расчеты вели с использованием программы Природа [18, 19] (функционал PBE0 [20, 21]). Для всех систем проводили разложение электронной плотности во вспомогательном базисе. Геометрии всех соединений оптимизировали без ограничения на симметрию. Анализ колебательных спектров использовали для идентификации стационарных точек.
Комплексы с f-элементами изучали с использованием базиса cc-pVDZ [18]. Геометрии лиганда и его комплексов с f-элементами оптимизировали без ограничений по спину системы.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Известно, что экстракционная способность органического лиганда во многом зависит от прочности образуемого комплекса в соответствии с донорноакцепторными свойствами координирующих центров, а также и от гидрофобно-липофильного баланса соединения [11, 22, 23]. Варьирование строения углеводородных радикалов у терминального атома азота фосфорилмочевины, а именно введение алкоксиалкильного и тетрагидрофурилалкильного радикалов, значительно меняет липофильность как самого лиганда, так и экстрагируемых комплексов.
Результаты жидкостной экстракции f-элементов при концентрации 0.01 моль/л в хлороформе и нитрата соответствующего лантанида в водной фазе (2.5 × 10–4 моль/л) представлены на рис. 1а, 1б.
Рис. 1.
Экстракция лантана(III) (а) и неодима(III) (б) растворами лигандов II–V (0.01 моль/л в хлороформе).
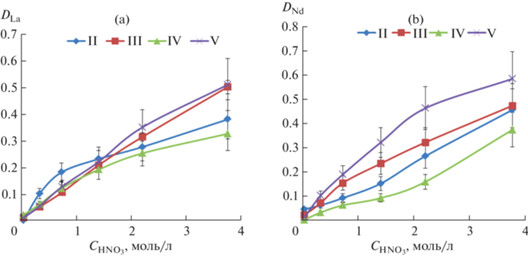
При экстракции лантана(III) и неодима(III) экстракционные зависимости для фосфорилмочевин II–V имеют практически одинаковый вид, возрастая с увеличением концентрации азотной кислоты в водной фазе (рис. 1а, 1б). Согласно исследованиям комплексообразования с родственными бидентатными фосфиноксидами R2P(O)CH2C(O)R [24–27], R2P(O)CH2C(O)NR2 [28–30], а также фосфорилмочевинами R2P(O)NHC(O)NHR [31, 32], можно допустить, что лантаниды экстрагируется в виде катионных и нейтральных комплексов [Ln(L)n(NO3)2]+(NO3)– и [Ln(L)n(NO3)3]0 (n ≥ 3). Поскольку с ростом концентрации азотной кислоты содержание нейтральных комплексов должно увеличиваться, соответственно возрастают и коэффициенты распределения при экстракции, как лантана, так и неодима. Коэффициенты распределения лантана(III) и неодима(III) при 3.8 моль/л HNO3 находятся в диапазоне от 0.32 до 0.58. Необходимо отметить, что наилучшие экстракционные свойства для лантана(III) и неодима(III) выявлены при использовании фосфорилмочивины V, содержащей [2-(тетрагидрофур-2-ил)этильный] радикал в N'-положении. При использовании N-(дифенилфосфорил)-N'-[2-(тетрагидрофур-2-ил)этил]мочевины в качестве экстрагента лантана(III) и неодима(III) степени извлечения достигают 34 и 37% соответственно в одну стадию.
Экстракционная способность фосфорилмочевин II–V по отношению к гольмию(III) возрастает (рис. 2а) по сравнению с данными по экстракции, полученными в одних экспериментальных условиях для лантана(III) и неодима(III). Коэффиценты распределения гольмия(III) при экстракции из 3.8 моль/л HNO3 находятся в пределах от 0.46 до 1.2. Можно предположить, что экстрагируемые комплексы, как и в случае с лантаном и неодимом, будут представлять нейтральные частицы [Ho(L)n(NO3)3]0, а также ионные пары [Ho(L)n(NO3)2]+(NO3)–. Степень извлечения при экстракции Ho(III) раствором V в хлороформе из 3.8 моль/л HNO3 составляет 54% в одну стадию. Увеличение экстракционной способности у лиганда V связано, возможно, с дополнительным вкладом в координирование металла [2-(тетрагидрофур-2-ил)этильной] группировки у N’-атома азота фосфорилмочевины. Введение этиленового фрагмента, связывающего тетрагидрофурильную группировку с остовом фосфорилмочевины увеличивает не только липофильность лиганда и комплексов, но и создает благоприятные условия для образования полости тридентатной координации к металлу. В то время как лиганды III и IV, также содержащие тетрагидрофурильный фрагмент, который соединен с остовом фосфорилмочевины более коротким метиленовым радикалом, по-видимому, не обладают дополнительными координирующими возможностями, что наглядно демонстрируют рис. 2а и 2б.
Рис. 2.
Экстракция гольмия(III) (а) и урана(VI) (б) растворами лигандов II–V (0.01 моль/л в хлороформе).

В случае извлечения урана(VI) коэффициенты распределения в целом выше по сравнению с экстракцией лантанидов и находятся в диапазоне от 1.5 до 7.5 при 3.8 моль/л HNO3 (рис. 2б). При этом степени извлечения U(VI) варьируются в интервале от ~60 до ~90% в одну стадию. Экстракционное поведение лигандов II–V по отношению к урану(VI) схоже с лантанидами(III), за исключением более высоких значений коэффициентов распределения. Необходимо заметить, что значения количественных характеристик (DU) экстракции урана(VI) при использовании лиганда V значительно больше по сравнению с другими лигандами, а тем более при экстракции лантанидов(III). Возможно причиной этому эффекту стали благоприятные условия возникновения тридентатной координации к иону уранила фосфорилмочевины, содержащей в структуре [2-(тетрагидрофур-2-ил)этильную] группировку в N'-положении, которые наиболее выражены в случае с ураном(VI) (рис. 2б).
Для объяснения причин значительного различия в экстракционной способности реагентов II–V по отношению к урану провели квантово-химическое моделирование строения возможных комплексов, образующихся в процессе экстракции, в предположении образовании комплексов стехиометрии UO2(L)2. Все исследованные реагенты образуют хелатные комплексы с ионом уранила. Для всех лигандов характерна координация UO2 с атомами кислорода фосфорильной и карбонильной группы обоих координированных лигандов (рис. 4). Формирующийся при такой координации шестичленный хелатный металлоцикл находится в конформации ванны для всех исследуемых лигандов. Важнейшие межатомные расстояния и углы в координационной сфере иона металла приведены в табл. 1.
Рис. 3.
Сравнение коэффициентов распределения La(III), Nd(III), Ho(III) и U(VI) при экстракции лигандами II–V, а также коммерчески доступными фосфорорганическими экстрагентами – ТБФ, ТОФО и КМФО (0.01 моль/л растворы в хлороформе) из 3.8 моль/л HNO3.

Рис. 4.
Структура комплексов стехиометрии UO2(L)2 для трех типов реагентов линейного эфира II (а), тетрагидрофуранового лиганда с короткой цепью III (б) и тетрагидрофуранового лиганда с длинной цепью V (в).
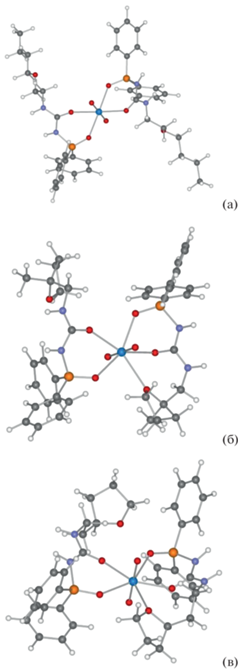
Таблица 1.
Важнейшие межатомные расстояния (Å) и углы (град) в ближайшей координационной сфере иона уранила для комплексов состава UO2(L)2 с лигандами II–V и в свободных молекулах (L) и свободные энергии Гиббса по реакции (1)
| Лиганд | ΔG0, ккал/моль | U–O(P) | U–O(C) | U–O(CH2) | Угол O–CHR–CH2 | |
|---|---|---|---|---|---|---|
| L | UO2(L)2 | |||||
| II | –103.8 | 2.332/2.338 | 2.346/2.356 | – | 107.899 | 108.028/106.162 |
| III | –104.3 | 2.424*/2.373 | 2.371*/2.367 | 2.676 | 106.117 | 111.899/108.033 |
| IV | –104.9 | 2.413*/2.366 | 2.354*/2.368 | 2.794 | 107.570 | 113.457*/109.101 |
| V | –110.3 | 2.424*/2.386 | 2.342*/2.374 | 2.579 | 108.814 | 112.848*/108.557 |
Производные тетрагидрофурана III–V, в которых положение атома кислорода вблизи карбамидного фрагмента закреплено, образуют дополнительную координационную связь между атомом кислорода бокового заместителя и ионом урана, в то время как линейный эфир II не способен к этому из-за большей гибкости линейного алифатического фрагмента. Расстояния U–O сокращаются при переходе от незамещенного тетрагидрофуранового кольца (IV) к 2-метилированному (III) и далее при увеличении расстояния до фрагмента мочевины(V), свидетельствуя в пользу повышения устойчивости этой связи. При этом координация с ионом уранила приводит к увеличению угла O–CHR–CH2 в мостике, связывающем тетрагидрофурильный фрагмент с карбамидной группой (112°–113°), по сравнению c геометрией свободного лиганда (106°–109°), что свидетельствует о стерическом напряжении в sp3-гибридизованном мостиковом атоме углерода. Максимального значения искажение достигает в комплексах лиганда IV. Такие напряжения объясняются формированием вокруг иона урана пентагональнобипирамидального окружения, требующего расположения всех донорных атомов кислорода в одной плоскости. Дополнительная координация атома кислорода бокового заместителя приводит к разрыхлению связи иона уранила с атомами кислорода фосфорильной и карбамидной групп обоих органических фрагментов, причем максимально этот эффект сказывается на тридентатно координированном лиганде.
Свободные энергии Гиббса реакций комплексообразования по модельному уравнению (1) приведены в табл. 1. Все рассчитанные величины отрицательны, а их значения находятся в соответствии с наблюдаемой экстракционной эффективностью для извлечения тетрагидрофурильными производными фосфорилмочевин (III–V). Наибольший энергетический выигрыш наблюдается для реакции реагента V, а наименьший – для реагента III, что и подтверждается экспериментально.
(1)
$~{\text{U}}{{{\text{O}}}_{2}}\left( {{{{\text{Н}}}_{{\text{2}}}}{\text{О}}} \right)_{5}^{{2 + }} + 2{\text{L}} = {\text{U}}{{{\text{O}}}_{{\text{2}}}}{\text{(L}})_{2}^{{2 + }} + 5{{{\text{Н}}}_{{\text{2}}}}{\text{О}}.~~$Для реагента II модельная реакция должна выглядеть сложнее, в связи с координационной ненасыщенностью модельного комплекса UO2(II)2, который, скорее всего, существует в виде гидрата с дополнительной координацией молекулы воды или сольвата с дополнительной координацией еще одной молекулы реагента II. Этим объясняется его бóльшая экстракционная способность по сравнению с реагентами III и IV.
Проведено сравнение в одинаковых экспериментальных условиях с коммерчески доступными нейтральными фосфорилсодержащими экстрагентами. Как видно из рис. 3, экстракционные способности фосфорилмочевин II–V в отношении к лантанидам несколько больше по сравнению с ТБФ, ТОФО и КМФО. При этом экстракционная способность исследуемых фосфорилмочевин II–V в отношении урана также больше, особенно для лигандов II и V. Показано, что N-дифенилфосфорил-N'-[2-(тетрагидрофур-2-ил)этил]мочевина обладает более высокой экстракционной способностью по отношению к f-элементам в сравнении с ТБФ, ТОФО и КМФО.
Список литературы
Zepf V. Rare Earth Elements. Springer Theses. 2013. https://doi.org/10.1007/978-3-642-35458-8
Moss R., Tzimas E., Willis P. et al. Critical metals in the path towards the decarbonisation of the EU energy sector: assessing rare metals as supply-chain bottlenecks in low-carbon energy technologies. Luxembourg: Publication Office of the European Union, 2013. 242 p.
Schulz K.J., De Young J.H. Jr., Seal R.R.II, Bradly D.C. Critical mineral resources of the United States-Economic and environmental geology and prospects for future supply. 2017. 797 p. https://doi.org/10.3133/pp1802
Romanova O.A., Sirotin D.V. // KnE Materials Sci. 2019. V. 5. № 1. P. 15. https://doi.org/10.18502/kms.v5i1.3949
Samsonov N.Yu. // Prostranstvennaya Ekonomika – Spatial Economics. 2018. № 3. P. 43. https://doi.org/10.14530/se.2018.3.043-066
Крюков В.А., Яценко В.А., Крюков Я.В. // Горная промышленность. 2020. № 5. С. 68. https://doi.org/10.30686/1609-9192-2020-5-68-84
Yahorava V., Lakay E., Clark W., Strauss J. // Extraction. 2018. P. 2415. https://doi.org/10.1007/978-3-319-95022-8_204
Genkin M., Evtushenko A., Komkov A. et al. // Pat. US 9657371 B2. 5 March 2013.
Moalla R., Gargouri M., Khmiri F. et al. // Environ. Engin. Res. 2018. V. 23. № 1. P. 36. https://doi.org/10.4491/eer.2017.055
Soukeur A., Szymczyk A., Berbar Y., Amara M. // Separat. Purificat. Technol. 2021. V. 256. 117857. https://doi.org/10.1016/j.seppur.2020.117857
Розен А.М., Крупнов Б.В. // Успехи химии. 1996. Т. 65. № 11. С. 1052. [Rozen A.M., Krupnov B.V. // Russ. Chem. Rev. 1996. V. 65. № 11. P. 973.] https://doi.org/10.1070/RC1996v065n11ABEH000241
Шарова Е.В., Артюшин О.И., Одинец И.Л. // Успехи химии. 2014. Т. 83. № 2. С. 95. [Sharova E.V., Artyushin O.I., Odinets I.L. // Russ. Chem. Rev. 2014. V. 83. № 2. P. 95.] https://doi.org/10.1070/RC2014v083n02ABEH004384
Тананаев И.Г., Летюшов А.А., Сафиулина А.М. и др. // Докл. АН. 2008. Т. 422. № 6. С. 762. [Tananaev I.G., Letyushov A.A., Safiulina A.M. et al. // Dokl. Chem. 2008. V. 422. № 2. P. 260.] https://doi.org/10.1134/S0012500808100054
Горюнов Е.И., Шипов А.Э., Горюнова И.Б. и др. // Докл. АН. 2011. Т. 438. № 4. С. 480. [Goryunov E.I., Shipov A.E., Goryunova I.B. et al. // Dokl. Chem. 2011. V. 438. № 2. Р. 151.] https://doi.org/10.1134/S0012500811060012
Safiulina A.M., Goryunov E.I., Letyushov A.A. et al. // Mendeleev Commun. 2009. V. 19. № 5. P. 263. https://doi.org/10.1016/j.mencom.2009.09.010
Сафиулина А.М., Лизунов А.В., Борисова Н.Е. и др. // Журн. неорган. химии. 2021. Т. 66. № 5 С. 731. [Safiulina A.M., Lizunov A.V., Borisova N.E. et al. // Russ. J. Inorg. Chem. 2021. V. 66. № 5. P. 731.] https://doi.org/10.1134/S0036023621050156
Саввин С.Б. Органические реагенты группы арсеназо III. М.: Атомиздат, 1971. 352 с.
Laikov D.N. // Chem. Phys. Lett. 2005. V. 416. № 1–3. P. 116. https://doi.org/10.1016/j.cplett.2005.09.046
Laikov D.N. // Chem. Phys. Lett. 1997. V. 281. № 1–2. P. 151. https://doi.org/10.1016/S0009-2614(97)01206-2
Perdew J.P., Burke K., Ernzerhof M. // Phys. Rev. Lett. 1996. V. 77. P. 3865. https://doi.org/10.1103/PhysRevLett.77.3865
Perdew J.P., Burke K., Ernzerhof M. // Phys. Rev. Lett. 1997. V. 78. P. 1396. https://doi.org/10.1103/PhysRevLett.78.1396
Рейхардт К. Растворители и эффекты среды в органической химии. М.: Мир, 1991. С. 763.
Кабачник М.И., Медведь Т.Я., Дятлова Н.М. и др. // Успехи химии. 1968. Т. 37. № 7. С. 1161. [Kabachnik M.I., Medved’ T.Ya., Dyatlova N.M. et al. // Russ Chem Rev. 1968. V. 37. № 7. P. 503.] https://doi.org/10.1070/RC1968v037n07ABEH001662
Bowen S.M., Duesler E.N., Paine R.T. // Inorg. Chim. Acta. 1982. V. 61. P. 155. https://doi.org/10.1016/S0020-1693(00)89134-3
Babecki R., Platt A.W.G., Russell D.R. // Inorg. Chim. Acta. 1990. V. 171. № 1. P. 25. https://doi.org/10.1016/S0020-1693(00)84658-7
Сафиулина А.М., Матвеева А.Г., Дворянчикова Т.К. и др. // Изв. АН. Сер. хим. 2012. № 2. С. 390. [Safiulina A.M., Matveeva A.G., Dvoryanchikova T.K. et al. // Russ. Chem. Bull. 2012. V. 61. № 2. P. 392.] https://doi.org/10.1007/s11172-012-0055-0
Матвеева А.Г., Григорьев М.С., Дворянчикова Т.К. и др. // Изв. АН. Сер. хим. 2012. № 2. С. 397. [Matveeva A.G., Grigoriev M.S., Dvoryanchikova T.K. et al. // Russ. Chem. Bull. V. 61. № 2. P. 399.] https://doi.org/10.1007/s11172-012-0056-z
McCabe D.J., Duesler E.N., Paine R.T. // Inorg. Chim. Acta. 1988. V. 147. №. 2. P. 265. https://doi.org/10.1016/S0020-1693(00)83383-6
Шарова Е.В., Артюшин О.И., Нелюбина Ю.В. и др. // Изв. АН. Сер. хим. 2008. № 9. С. 1856. [Sharova E.V., Artyushin O.I., Nelyubina Yu.V. et. al. // Russ. Chem. Bull. 2008. V. 57. № 9. P. 1890.] https://doi.org/10.1007/s11172-008-0255-9
Wang C.-Zhi, Shi W.-Q., Lan J.-H. et al. // Inorg. Chem. 2013. V. 52. № 19. P. 10904. https://doi.org/10.1021/ic400895d
Матросов Е.И., Горюнов Е.И., Баулина Т.В. и др. // Докл. АН. 2010. Т. 432. № 2. С. 191. [Matrosov E.I., Goryunov E.I., Baulina T.V. et al. // Dokl. Chem. 2010. V. 432. № 1. P. 136.] https://doi.org/10.1134/S0012500810050058
Матросов Е.И., Горюнова И.Б., Лысенко К.А. и др. // Журн. неорган. химии. 2011. Т. 56. № 4. С. 579. [Matrosov E.I., Goryunova I.B., Lysenko K.A. et. al. // Russ. J. Inorg. Chem. 2011. V. 56. № 4. P. 539.] https://doi.org/10.1134/S003602361104019X
Дополнительные материалы отсутствуют.
Инструменты
Журнал неорганической химии