

Журнал неорганической химии, 2023, T. 68, № 11, стр. 1656-1663
Влияние природы мостикового радикала на экстракционную способность дифенилфосфорилкетонов по отношению к актинидам и лантанидам
А. М. Сафиулина a, b, *, Н. Е. Борисова c, d, А. В. Лизунов a, А. В. Голубев c, Г. В. Бодрин d, Е. И. Горюнов d, И. Б. Горюнова d, В. К. Брель d, И. Г. Тананаев e, f, g
a АО “Высокотехнологический научно-исследовательский институт неорганических материалов
им. академика А.А. Бочвара”
123098 Москва, ул. Рогова, 5а, Россия
b Российский химико-технологический университет им. Д.И. Менделеева
125047 Москва, Миусская пл., 9, Россия
c Московский государственный университет им. М.В. Ломоносова
119991 Москва, Ленинские горы, 1, Россия
d Институт элементоорганических соединений им. А.Н. Несмеянова РАН
119334 Москва,
ул. Вавилова, 28, стр. 1, Россия
e Федеральный исследовательский центр “Кольский научный центр РАН”
184209 Апатиты, ул. Ферсмана, 14, Россия
f Институт химии и технологии редких элементов и минерального сырья
им. И.В. Тананаева КНЦ РАН
184209 Апатиты, ул. Академгородок, 26а, Россия
g Дальневосточный федеральный университет
690090 Владивосток, о. Русский, п. Аякс, 10, Россия
* E-mail: AMSafiulina@bochvar.ru
Поступила в редакцию 08.04.2023
После доработки 23.06.2023
Принята к публикации 01.07.2023
- EDN: DJFGHV
- DOI: 10.31857/S0044457X23600585
Аннотация
Исследовано влияние на экстракционную способность по отношению к актинидам и лантанидам соединений класса фосфорилкетонов, содержащих гибкие алкильные и конформационно жесткие фрагменты в линкере. Показано, что введение в структуру молекулы конформационно жестких циклоалкандиильных и алкендиильных радикалов мостика, разделяющего Р=О- и С=О-группировки, снижает эффективность экстрагентов как в случае актинидов, так и в случае лантанидов. В то же время введение в структуру линкера гибких алкильных радикалов повышает комплексообразующую и экстракционную способность. Данная зависимость получила теоретическое обоснование при моделировании комплексообразования (DFT, PBE, cc-pVDZ), наблюдалась монодентатная координация для всех исследуемых фосфорилалканонов.
ВВЕДЕНИЕ
Нейтральные фосфорорганические соединения (НФОС), в частности три-н-бутилфосфат, успешно используются при переработке отработавшего ядерного топлива с выделением целевых элементов: урана, плутония, нептуния и иногда сопутствующего технеция на разных стадиях технологии [1–4]. При развитии стратегии фракционирования высокоактивных отходов (ВАО) предусматривается экстракционное выделение минорных актинидов, особенно Am(III) и Cm(III) [4–7].
Для решения задачи фракционирования и кондиционирования ВАО ранее были разработаны и синтезированы соединения класса бидентатных НФОС: диоксиды дифосфинов и карбамоилметилфосфиноксиды (КМФО) [5, 8–11]. Несмотря на более низкую экстракционную способность КМФО по сравнению с диоксидами дифосфинов по отношению к целевым минорным актинидам, при практическом применении они более эффективны вследствие лучшей совместимости с растворителем как самого лиганда, так и экстрагируемых комплексов. Необходимо отметить более простой и технологичный синтез КМФО, чем диоксидов дифосфинов [8, 12]. При замещении карбамоильной группировки –С(О)NR2 на алканоильную –C(O)R (фосфорилкетоны) синтез таких соединений становится более доступным, по сравнению с КМФО. Кроме того, дифенилфосфорилкетоны выгодно отличаются от аналогов с алкильными заместителями, а также от КМФО тем, что они обладают селективностью к лантанидам на фоне актинидов [8, 13–15, 19].
Ранее в [16] было показано, что целенаправленная изомеризация фосфорилалканонов на примере 5-(дифенилфосфорил)гексан-3-она значительно увеличивает экстракционную способность по отношению к лантанидам. В работе [17] выявлено влияние дополнительных координационных центров на способность фосфорилкарбонильных соединений экстрагировать актиниды. На примере бис(дифенилфосфорил)бутан-2-она показано, что введение второй фосфорильной группы повышает экстракционную способность и селективность экстрагента по отношению к U(VI), в то время как дополнительная карбонильная группировка не сказывается на эффективности экстракции не только урана(VI), но и лантанидов [17].
На эффективность и селективность при экстракции f-элементов фосфорилкарбонильными соединениями влияет не только количество функциональных групп [17], но и строение радикалов у атома фосфора фосфорильной группы [18, 19], а также длина линкера и природа заместителя R фрагмента >Р(О)CHRCH2С(О)– [20]. Влияние на экстракционные свойства лиганда могут оказывать также геометрически более жесткие линкеры, соединяющие координирующие центры, при использовании циклоалкандиильных и алкендиильных радикалов в структуре мостика. В настоящей работе исследовано влияние гибких алкильных и конформационно жестких фрагментов в структуре мостикового радикала фосфорилкетонов на экстракционную и комплексообразующую способность по отношению к лантанидам и актинидам.
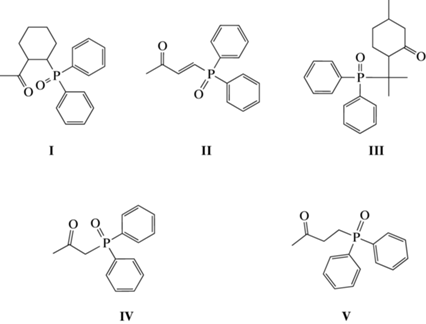
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Лиганды [4-(дифенилфосфорил)бут-3-ен-2-он] (II) [14], [1-(дифенилфосфорил)пропан-2-он] (IV) и [4-(дифенилфосфорил)бутан-2-он] (V) [21, 22] синтезировали по известным методикам.
Не описанные в литературе фосфорилкарбонильные лиганды I и III, в которых мостик, разделяющий Р=О- и С=О-группы, содержит алициклические фрагменты, синтезировали исходя из коммерчески доступного дифенилхлорфосфина (ДФХФ) с использованием двухстадийных “one-pot”-процессов. На первой стадии этих процессов ДФХФ взаимодействует с циклическим α,β-еноном (1-ацетилциклогексеном (при получении I) или пулегоном (при синтезе III)) в отсутствие растворителя при комнатной температуре с образованием твердых аддуктов, которые при обработке абсолютным этанолом превращаются в соответствующие целевые соединения.
ДФХФ (Aldrich, 98%) непосредственно перед реакцией перегоняли в вакууме. Все операции проводили в атмосфере аргона.
1-Ацетил-2-(дифенилфосфорил)циклогексан (I). К 3.7 г (0.0168 ммоль) ДФХФ добавляли 2.15 г (0.0173 моль) 1-ацетилциклогексена, тщательно перемешивали и выдерживали реакционную смесь при комнатной температуре в отсутствие света в течение 70 ч. Затвердевшую реакционную массу разлагали 3.9 г (0.085 моль) абсолютного EtOH, избыток спирта удаляли в вакууме, остаток последовательно экстрагировали кипящим гексаном (3 × 20 мл) и кипящим гептаном (3 × 20 мл), экстракты объединяли, охлаждали до комнатной температуры, выпавший осадок отделяли и сушили в вакууме (~10 Торр) 1 ч при 110°С. Получено 3.4 г соединения I.
Выход 62.0%. tпл = 143–144°С (циклогексан).
Спектр ЯМР 1Н (δН, м.д.): 1.25–1.44 м (3Н, 3СН2- + 4CH2- + 6СН2-циклогексан); 1.50–1.62 м (1Н, 3СН2-циклогексан); 1.62–1.85 м (3Н, 4CH2- + + 5СН2-циклогексан); 1.90 с (3Н, СН3); 2.00–2.09 м (1Н, 6СН2-циклогексан); 2.96–3.06 м (2Н, 1СН- + + 2СН-циклогексан); 7.44 дт (2Н, m-C6H5, 3JH–H = = 7.4 Гц, 4JH–Р = 2.8 Гц); 7.46–7.55 м (4Н, m- + + p-C6H5); 7.76–7.86 м (4Н, o-C6H5).
Спектр ЯМР 13С{1H} (δC, м.д.): 24.40 с (5С-циклогексан); 24.64 д (3С-циклогексан, 2JC–P = 1.8 Гц); 24.83 д (4С-циклогексан, 3JC–P = 10.9 Гц); 29.13 с (СН3); 29.63 д (6С-циклогексан, 3JC–P = 9.1 Гц); 36.12 д (2С-циклогексан, 1JC–P = 69.9 Гц); 48.33 д (1С-циклогексан, 2JC–P = 1.8 Гц); 128.41 д (m-C6H5, 3JC–P = 11.3 Гц); 128.60 д (м-C6H5, 3JC–P = 11.4 Гц); 131.18 д (o-C6H5, 2JC–P = 9.1 Гц); 131.60 д (o-C6H5, 2JC–P = 9.1 Гц); 131.64 д (p-C6H5, 4JC–P = 2.3 Гц); 131.78 д (ипсо-C6H5, 1JC–P = 95.8 Гц); 131.80 д (p-C6H5, 4JC–P = 2.7 Гц); 132.24 д (ипсо-C6H5, 1JC–P = 94.0 Гц); 210.87 д (C=O, 3JC–P = 3.6 Гц).
Спектр ЯМР 31P{1H} (δP, м.д.): 35.87 с.
2-[1-(Дифенилфосфорил)-1-метилэтил]-5-метилциклогексанон (III). Аналогичным образом из 3.70 г (0.0168 моль) ДФХФ и 2.63 г (0.173 моль) пулегона получено 5.3 г соединения III.
Выход 89.1%. tпл = 130–131°С (бензол–гептан).
Спектр ЯМР 1Н (δН, м.д.): 0.93 д (3H, 5CH3-циклогексанон, 3JH–H = 6.4 Гц); 1.04–1.14 м (1Н, 4CH2-циклогексанон); 1.41 д [3H, СН3(A)СН3(B)C, 3JH–P = 17.6 Гц]; 1.49–1.58 м (1H, 3CH2-циклогексанон); 1.52 д [3H, СН3(A)СН3(B)C, 3JH–P = 16.2 Гц]; 1.70–1.78 м (1H, 4CH2-циклогексанон); 1.78–1.86 м (1H, 5CH-циклогексанон); 1.93–2.00 м (1H, 6CH2-циклогексанон); 2.17–2.23 м (1H, 6CH2-циклогексанон); 2.45–2.54 м (1H, 3CH2-циклогексанон); 2.73–2.80 м (1H, 2CH-циклогексанон); 7.43–7.51 м (3Н, м- + р-С6Н5); 7.51–7.60 м (3Н, м- + + р-С6Н5); 7.92–7.98 м (2Н, о-С6Н5); 8.00–8.06 м (2Н, о-С6Н5).
Спектр ЯМР 13С{1H} (δC, м.д.): 17.70 д [СН3(A)СН3(B)C, 2JC–P = 1.6 Гц]; 22.21 c (5CH3-циклогексанон); 22.27 с [СН3(A)СН3(B)C]; 30.34 д (3СН2-циклогексанон, 3JC–P = 3.2 Гц); 34.35 с (4СН2-циклогексанон); 36.84 с (5СН-циклогексанон); 39.70 д [(СН3)2C 1JC–P = 67.2 Гц]; 52.43 д (6СН2-циклогексанон, 4JC–P = 2.8 Гц); 54.07 с (2СН-циклогексанон); 128.37 д (м-C6H5, 3JC–P = = 11.1 Гц); 128.49 д (м-C6H5, 3JC–P = 11.1 Гц); 131.23 д (ипсо-C6H5, 1JC–P = 89.4 Гц); 131.47 д (р-C6H5, 4JC–P = = 2.8 Гц); 131.65 д (p-C6H5, 4JC–P = 2.8 Гц); 131.89 д (ипсо-C6H5, 1JC–P = 89.8 Гц); 132.01 д (о-C6H5, 2JC–P = = 7.5 Гц); 132.12 д (о-C6H5, 2JC–P = 7.9 Гц); 210.75 д (C=O, 3JC–P = 13.9 Гц).
Спектр ЯМР 31P{1H} (δP, м.д.): 39.05 с.
Спектры ЯМР 1H, 13С{1H} и 31P{1H} фосфорилкарбонильных лигандов I, III регистрировали на приборе Bruker AV-500 (рабочая частота 500.13 (1H), 125.77 (13C{1H}) и 202.46 MГц (31P{1H})) в растворе CDСl3 (С = 0.1 моль/л). Внутренний эталон для спектров ЯМР 1H – сигналы остаточных протонов дейтерированного растворителя, а для спектров ЯМР 13С{1H} – сигналы ядер атомов углерода дейтерированного растворителя; внешний эталон для спектров ЯМР 31Р{1H} – 85%-ная H3PO4. Отнесение сигналов в спектрах ЯМР 1H и 13С{1H} было проведено с использованием корреляций COSY, HMQC и HMBC.
Исследование экстракции металлов. В работе использовали хлороформ (х. ч.), арсеназо III (ч. д. а.), HNO3 (ос. ч.), ГСО 8363-2003 закись-окись урана, аттестовано на содержание урана 84.784 ± 0.016%, La(NO3)3 · 6H2O (х. ч.), Nd(NO3)3 · 6H2O (х. ч.), Ho(NO3)3 · 6H2O (х. ч.), Yb(NO3)3 · 6H2O (х. ч.). Растворы готовили объемно-весовым методом: водные растворы – в бидистиллированной воде, растворы нитратов исследуемых элементов – растворением навески соответствующего нитрата в 0.01 моль/л растворе HNO3. Концентрацию растворов нитратов металлов (0.1 ммоль/л) уточняли спектрофотометрически по методике [23] с использованием спектрофотометра Cary 5000 Scan (Varian). Концентрацию растворов HNO3 определяли потенциометрическим титрованием 0.1 моль/л NaOH с использованием рН-метра/кондуктометра S470 SevenExcellence™ (MettlerToledo) с точностью ±0.01 ед. рН.
Исследование экстракции катионов металлов выполняли по следующей методике. В пробирку с притертой пробкой вносили 1.5 мл раствора азотной кислоты, концентрация которой варьировалась от 0.052 до 5.0 моль/л; 0.5 мл 0.1 М раствора нитрата металла, 2 мл 0.01 М раствора лиганда в хлороформе. Фазы перемешивали в течение 20 мин в ротаторе. Время установления равновесия экстракции проверяли, увеличивая время контакта фаз до 120 мин, коэффициенты распределения при этом не изменялись. Расслаивание фаз осуществляли центрифугированием. После разделения фаз концентрацию катионов металлов в водной фазе определяли спектрофотометрическим методом [23]. Для каждой концентрации проводили не менее пяти независимых опытов. Суммарная погрешность результатов составляла ~20%, учитывая не исключенную и случайную составляющие. Соответственно доверительный интервал определяемых концентраций металлов в эксперименте составлял 0.002 ммоль/л. Все эксперименты проводили при температуре 20 ± 1°С. Коэффициенты распределения при экстракции (D = [M]орг/[M]в) определяли по разности при постоянных концентрациях экстрагента (0.01 моль/л в хлороформе) и исходных концентрациях металла в эксперименте (0.025 ммоль/л в водной фазе).
Все расчеты проводили с использованием программы Природа [24, 25] (функционал PBE0 [26, 27]). Для всех систем было выполнено разложение электронной плотности во вспомогательном базисе. Геометрию всех соединений оптимизировали без ограничений по симметрии. Анализ колебательных спектров использовали для идентификации стационарных точек.
Изучение комплексов с f-элементами проводили с использованием базиса cc-pVDZ [24]. Геометрию лиганда и его комплексов с f-элементами оптимизировали без ограничений по спину системы.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
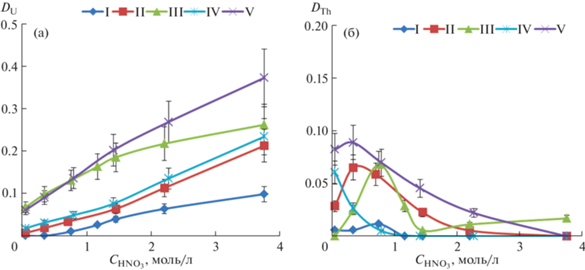
При анализе зависимостей коэффициентов распределения (D) от структуры фосфорилкетонов I–V исследовали влияние длины линкера, а также геометрически мягкую и жесткую конформации мостика, разделяющего фосфорильный и карбонильный фрагменты. Лиганд II, содержащий алкен-1,2-диильный фрагмент, обладает жесткой транс-конфигурацией [14]. В то же время введение циклического жесткого фрагмента в линкер фосфорилкетона I, который заставляет сближаться фосфорильную и карбонильную группы, создает тем самым потенциально бидентатный ансамбль при координации к иону металла. Однако наличие в структуре лигандов I и II конформационно жестких фрагментов как циклического, так и алкенового характера не сказывается на эффективности экстракции актинидов U(VI) и Th(IV) (рис. 1а и 1б). Коэффициенты распределения урана(VI) при экстракции раствором лиганда I на 70% ниже по сравнению с DU при использовании в качестве экстрагентов IV и V с гибкими алкандиильными линкерами. Возможно, наблюдаемый эффект связан со стерическими затруднениями, препятствующими потенциально наиболее эффективной координации при комплексообразовании. В то же время лиганд III, имеющий гибкую подвижную конфигурацию и циклический фрагмент, включающий карбонильную группировку, сравним по эффективности экстракции урана(VI) с IV и V (рис. 1а).
Рис. 1.
Зависимость коэффициентов распределения при экстракции урана(VI) (а) и тория(IV) (б) в раствор хлороформа, содержащий 0.01 моль/л фосфорилкетонов (I–V), от концентрации азотной кислоты в водной фазе.
На примере комплекса родственного фосфорилкетона (2-метил-4-оксопент-2-ил)дифенилфосфиноксида Ph2P(O)CMe2CH2C(O)Me и нитрата уранила [28, 29] установлено монодентатное координирование лиганда к иону металла посредством кислорода фосфорильной группы. Вследствие этого по аналогии с комплексом, описанным в работе [29], вполне предсказуема монодентатная координация фосфорилкетонов I–V при участии лишь кислорода фосфорильной группы. При сравнении экстракционной способности лигандов с жесткой (I и II) и гибкой (III–V) конформацией, координирующихся к иону металла как в одном, так и в другом случае монодентатно, коэффициенты распределения урана(VI) в органическую фазу при использовании III–V выше (рис. 1а). Возможно, этот эффект связан также с лучшей совместимостью с растворителем экстрагентов III–V с алкандиильными мостиками [30]. Необходимо отметить, что по эффективности экстракции урана(VI) лиганд IV сравним c II, несмотря на метиленовый линкер, разделяющий Р=О- и С=О-группировки и в целом подвижную конфигурацию молекулы лиганда IV.
Для уточнения типа координации фосфорилкетонов I–V было проведено квантово-химическое моделирование строения комплексов с нитратом уранила. Все исследованные фосфорилкетоны образуют однотипные комплексы с нитратом уранила за счет координации иона металла с атомом кислорода фосфиноксидной группы. На рис. 2 представлена оптимизированная структура комплекса фосфорилкетона II, комплексы других фосфорилкетонов изоструктурны (табл. 1) и подобны родственному комплексу (2-метил-4-оксопент-2-ил)дифенилфосфиноксида (L) с нитратом уранила [29]. Как показано на примере комплексов лиганда II, координация с атомом кислорода кетогруппы энергетически менее выгодна, чем с атомом кислорода фосфиноксидного фрагмента, разница в энергиях соответствующих изомерных комплексов составляет 30.35 ккал/моль. Расчет геометрии известного из литературы комплекса фосфорилкетона L с нитратом уранила (табл. 1) показал, что моделирование хорошо воспроизводит строение комплекса. Межатомные расстояния, по данным моделирования, несколько завышены по сравнению со структурными параметрами в кристалле, это связано с моделированием в приближении изолированной молекулы, что приблизительно соответствует условиям газовой фазы и не учитывает межмолекулярного взаимодействия при формировании кристаллической упаковки. Во всех исследованных комплексах координационный полиэдр иона урана является гексагональной бипиримидой с атомами кислорода группы уранила в ее вершинах и координированными лигандами в базальной плоскости. Два нитрат-аниона координированы по бидентатно-хелатному типу. Фосфорилкетоны I–V имеют большее расстояние U–OP по сравнению с модельным комплексом лиганда L, что может свидетельствовать об ухудшении координационной способности атома кислорода по мере роста стерической нагруженности лиганда. Для фосфорилкетонов с мостиком из двух атомов углерода между фосфиноксидной и карбонильной группами наименьшее расстояние U–OP наблюдается в случае циклогексан-1,2-диильного мостика (I), а наибольшее – в случае конформационно жесткого транс-этиленового мостика (II). Интересно отметить, что включение карбонильной группы в конформационно более жесткое циклогексановое кольцо слабо сказывается на координации фосфорильной группы. Так, расстояния U–OP для комплекса фосфорилкетона III очень близки к величинам, рассчитанным для модельного комплекса с известным из литературных данных лигандом L. Это может свидетельствовать о слабом влиянии конформационной подвижности фосфорилкетонов на их способность координировать ион уранила. Следует отметить, что реагенты II–IV, проявляющие умеренную экстракционную способность по отношению к уранил-катиону, образуют комплексы с наибольшими длинами связей U–OP, в то время как их аналоги с циклогексан-1,2-диильным (I) и этан-1,2-диильным (V) мостиками образуют комплексы с существенно более короткими расстояниями. Таким образом, можно предположить, что комплексы уранила в экстракционной системе соответствуют составу металл : лиганд = 1 : 2 и их строение хорошо коррелирует с данными РСА для родственного (2-метил-4-оксопент-2-ил)дифенилфосфиноксида, в то время как в случае более конформационно жесткого фосфорилкетона (II), вероятно, из-за стерических препятствий происходит образование комплекса другой стехиометрии, что сказывается на резком понижении его экстракционной способности.
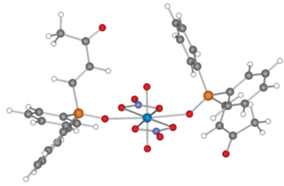
Таблица 1.
Межатомные расстояния и углы в ближайшей координационной сфере иона урана для комплексов фосфорилкетонов I–V с нитратом уранила. Данные РСА комплекса (2-метил-4-оксопент-2-ил)дифенилфосфиноксида (L) с нитратом уранила [29] приведены для сравнения
| Связь, угол | UO2(I)2 | UO2(II)2 | UO2(III)2 | UO2(IV)2 | UO2(V)2 | UO2(L)2 | |
|---|---|---|---|---|---|---|---|
| РСА | DFT | ||||||
| U=O | 1.804 1.810 | 1.798 1.807 | 1.806 1.811 | 1.804 | 1.807 1.808 | 1.774(2) | 1.809 1.810 |
| U–OP | 2.395 2.396 | 2.425 2.428 | 2.389 2.415 | 2.414 | 2.398 2.402 | 2.330(2) | 2.389 2.406 |
| U–${{{\text{O}}}_{{{\text{N}}{{{\text{O}}}_{3}}}}}$ | 2.507 2.499 2.540 2.530 |
2.505 2.506 2.516 2.518 |
2.512 2.510 2.509 2.523 |
2.519 2.519 |
2.506 2.508 2.530 2.531 |
2.542(2) 2.536(2) | 2.518 2.515 2.506 2.507 |
| P=O | 1.536 1.537 | 1.526 1.533 | 1.538 1.541 | 1.536 | 1.536 | 1.510(2) | 1.537 |
| O=U=O | 178.60 | 179.47 | 178.88 | 179.97 | 178.44 | 180.0 | 178.99 |
| OPUOP | 179.24 | 175.95 | 175.52 | 179.88 | 179.68 | 180.0 | 176.11 |
Соединения I–III обладают низкой экстракционной способностью по отношению к торию(IV) (рис. 1б), что вполне согласуется с родственными фосфорилкетонами, исследованными ранее [17, 19, 20].
В отличие от урана(VI), к которому фосфорилкетоны координируются только монодентатно, лантаниды взаимодействуют с фосфорилированными алканонами не только посредством атома кислорода фосфорильной группы, но связывая и карбонильную группировку [29]. Так, в ИК-спектре твердого комплекса Nd(L)2(NO3)3 наблюдается смещение ΔνСО = 20 см–1, что, по-видимому, связано с координацией С=О-группы. Подобный сдвиг при координации С=О-группы для родственных потенциально бидентатных фосфорилкарбонильных лигандов Ph2P(O)CH2C(O)Ph [31] и (i-PrO)2P(O)CH2C(O)NEt2 [32–35] в комплексах с нитратами лантанидов составляет 25–30 см–1. Схожие спектральные характеристики ΔνСО отмечены также при образовании водородной связи карбонильной группы с координированной молекулой воды, например в комплексе нитрата эрбия с (i-PrO)2P(O)CH2C(O)NEt2 [35]. При этом для комплекса Ce[Ph2P(O)CH2C(O)Ph]3(NO3)3, где, по данным РСА, одна молекула лиганда координирована с металлом бидентатно, вторая – монодентатно, а третья находится в промежуточном состоянии, в ИК-спектре наблюдаются две полосы С=О-группы: при 1675 и 1655 см–1 по сравнению с 1680 см–1 для свободного лиганда [31]. Вследствие этого можно выдвинуть обоснованное предположение, что лантаниды экстрагируются в органическую фазу в виде моно- и полифункциональных комплексов.
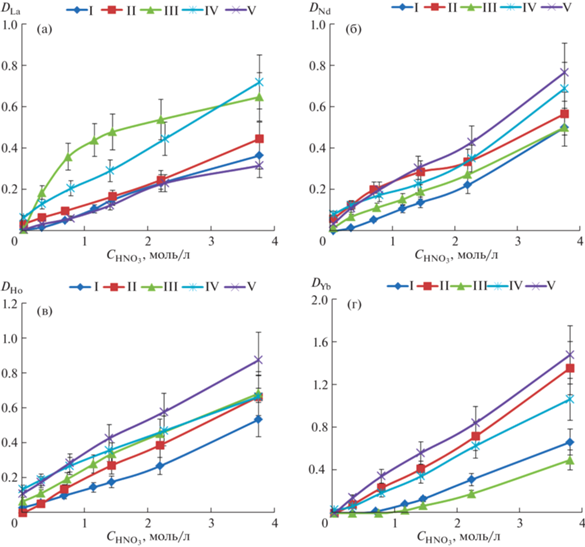
Экстракционная способность исследованных фосфорилкетонов по отношению к лантанидам выше, чем к актинидам (рис. 3). Коэффициент распределения лантана (DLa) при экстракции растворами лигандов I и II в хлороформе, имеющих конформационно жесткие фрагменты, сравним с DLa при использовании соединения V, однако ниже, чем для фосфорилкетонов III и IV. Так, при экстракции лантана лигандами I, II и V степень извлечения изменяется в диапазоне от 25 до 30% в одну стадию. При этом степень извлечения при экстракции лантана растворами соединений III и IV в СHCl3 достигает ~40% (рис. 3а). Намного меньше сказывается влияние строения исследуемых фосфорилкетонов при экстракции неодима(III) (рис. 3б) и гольмия(III) (рис. 3в). Степень извлечения находится в пределах от 30 до 40%. При экстракции иттербия степень извлечения существенно возрастает, причем наблюдается заметное влияние строения лиганда на экстракционную способность. Так, соединения I и III, содержащие циклические группировки в структуре, экстрагируют иттербий на ~40% (рис. 3г). Неожиданным результатом стал рост экстракционной способности лигандов II и V по отношению к иттербию. Извлечение этого лантанида в одну стадию в случае лигандов II и V составляет >60%. При этом необходимо отметить, что в случае лигандов II и V в структурах молекул экстрагентов содержатся стерически не нагруженные этилен-1,2-диильный и этан-1,2-диильный радикалы линкера, которые не создают препятствий при монодентатной координации нескольких молекул экстрагента вокруг меньшего по ионному радиусу иона иттербия, что может объяснять их большую экстракционную эффективность по сравнению со стерически нагруженными аналогами, для которых такая координация становится проблематичной.
ЗАКЛЮЧЕНИЕ
Показано, что введение в структуру молекулы фосфорилкетона конформационно жестких циклоалкандиильных и алкендиильных линкеров, разделяющих фрагмент с фосфорильной группой и карбонилсодержащую группировку, снижает экстракционную способность соединений как для актинидов, так и для лантанидов. В то же время введение в структуру линкера гибких алкильных радикалов повышает комплексообразующую и экстракционную способность.
Список литературы
Assessment of partitioning processes for transmutation of actinides IAEA. Vienna, 2010 IAEA-TECDOC-1648. 96 p.
Зильберман Б.Я., Пузиков Е.А., Рябков Д.В. и др. // Атомная энергия. 2009. Т. 107. № 5. С. 273. https://doi.org/10.1007/s10512-010-9233-z
Ion Exchange and Solvent Extraction: A Series of Advances / Ed. Moyer. B.A. N.Y.: CRCPress, 2010. V. 19. 673 p.
Розен А.М. // Рос. хим. журн. Журн. Рос. хим. об-ва им. Д.И. Менделеева. 1996. Т. 40. № 6. С. 42.
Мастрюкова Т.А., Артюшин О.И., Одинец И.Л., Тананаев И.Г. // Рос. хим. журн. Журн. Рос. хим. об-ва им. Д.И. Менделеева. 2005. № 2. С. 86.
Розен А.М., Крупнов Б.В. // Успехи химии. 1996. Т. 65. № 11. С. 1052.
Matveev P., Mohapatra P.K., Kalmykov S.N. et al. // Solvent Extr. Ion Exch. 2020. P. 679. https://doi.org/10.1080/07366299.2020.1856998
Медведь Т.Я., Чмутова М.К., Нестерова Н.П. и др. // Изв. АН СССР. Сер. хим. 1981. № 9. С. 2112.
Chmutova M.K., Kochetkova N.E., Koiro O.E. et al. // J. Radioanal. Chem. 1983. V. 80. № 1–2. P. 63. https://doi.org/10.1007/BF02517648
Myasoedov B.F., Chmutova M.K., Kochetkova N.E. et al. // Solv. Extr. Ion Exch. 1986. № 4. P. 61. https://doi.org/10.1080/07366298608917853
Litvina M.N., Chmutova M.K., Kulyako Yu.M., Myasoedov B.F. // Radiochemistry. 2001. V. 43. № 1. P. 66. https://doi.org/10.1023/A:1012878106998
Шарова Е.В., Артюшин О.И., Одинец И.Л. // Успехи химии. 2014. Т. 83. № 2. С. 95.
Татаринов Д.А., Миронов В.Ф., Костин А.А. и др. // Журн. общ. химии. 2010. Т. 80. № 7. С. 1211.
Бодрин Г.В., Горюнов Е.И., Горюнова И.Б. и др. // Докл. АН. 2012. Т. 447. № 4. С. 401.
Горюнов Е.И., Бодрин Г.В., Горюнова И.Б. и др. // Изв. АН. Сер. хим. 2013. № 3. С. 779.
Сафиулина А.М., Матвеева А.Г., Лизунов А.В. и др. // Докл. АН. 2015. Т. 460. № 6. С. 673.
Сафиулина А.М., Борисова Н.Е., Лизунов А.В. и др. // Журн. неорган. химии. 2022. Т. 67. № 4. С. 513.
Елистратова Ю.Г., Мустафина А.Р., Татаринов Д.А. и др. // Изв. АН. Сер. хим. 2011. № 5. С. 779.https://doi.org/10.1007/s11172-011-0126-7
Матвеева А.Г., Горюнов Е.И., Ту А.М. и др. // Изв. АН. Сер. хим. 2014. № 11. С. 2493.
Матвеева А.Г., Ту А.М., Сафиулина А.М. и др. // Изв. АН. Сер. хим. 2013. № 6. С. 1309.
Mazzotta S., Gramigna L., Bernardi L. et al. // Org. Proc. Res. Dev. 2010. V. 14. № 3. P. 687. https://doi.org/10.1021/op1000308
Giguere R.J., Herberich B. // Synth. Commun. 1991. V. 21. № 21. P. 2197. https://doi.org/10.1080/00397919108055453
Саввин С.Б. Органические реагенты группы арсеназо III. М.: Атомиздат, 1971. 352 с.
Laikov D.N. // Chem. Phys. Lett. 2005. V. 416. № 1–3. P. 116. https://doi.org/10.1016/j.cplett.2005.09.046
Laikov D.N. // Chem. Phys. Lett. 1997. V. 281. № 1–2. P. 151. https://doi.org/10.1016/S0009-2614(97)01206-2
Perdew J.P., Burke K., Ernzerhof M. // Phys. Rev. Lett. 1996. V. 77. № 18. P. 3865. https://doi.org/10.1103/PhysRevLett.77.3865
Perdew J.P., Burke K., Ernzerhof M. // Phys. Rev. Lett. 1997. V. 78. № 7. P. 1396. https://doi.org/10.1103/PhysRevLett.78.1396
Сафиулина А.М., Матвеева А.Г., Дворянчикова Т.К. и др. // Изв. АН. Сер. хим. 2012. № 2. С. 390.
Матвеева А.Г., Григорьев М.С., Дворянчикова Т.К. и др. // Изв. АН. Сер. хим. 2012. № 2. С. 397.
Рейхардт К. Растворители и эффекты среды в органической химии. М.: Мир, 1991. 763 с.
Babecki R., Platt A.W.G., Russell D.R. // Inorg. Chim. Acta. 1990. V. 171. № 1. P. 25. https://doi.org/10.1016/S0020-1693(00)84658-7
Casellato U., Vigato P.A., Vidali M. // Coord. Chem. Rev. 1981. V. 36. № 3. P. 183. https://doi.org/10.1016/S0010-8545(00)80500-7
McCabe D.J., Duesler E.N., Paine R.T. // Inorg. Chim. Acta. 1988. V. 147. № 2. P. 265. https://doi.org/10.1016/S0020-1693(00)83383-6
Bowen S.M., Duesler E.N., Paine R.T. // Inorg. Chem. 1982. V. 21. № 1. P. 261. https://doi.org/10.1021/ic00131a048
Bowen S.M., Duesler E.N., Paine R.T. // Inorg. Chim. Acta. 1982. V. 61. № 1. P. 155. https://doi.org/10.1016/S0020-1693(00)89134-3
Дополнительные материалы отсутствуют.
Инструменты
Журнал неорганической химии