

Журнал неорганической химии, 2023, T. 68, № 6, стр. 768-773
Исследование процесса гидролиза нитрилиевых производных клозо-додекаборатного аниона (Et4N)[B12H11N≡C–R], где R = Me, Et, nPr, iPr
А. В. Нелюбин a, И. Н. Клюкин a, Н. А. Селиванов a, А. Ю. Быков a, А. С. Кубасов a, А. П. Жданов a, *, К. Ю. Жижин a, Н. Т. Кузнецов a
a Институт общей и неорганической химии им. Н.С. Курнакова РАН
119991 Москва,
Ленинский пр-т, 31, Россия
* E-mail: zhdanov@igic.ras.ru
Поступила в редакцию 02.12.2022
После доработки 23.12.2022
Принята к публикации 27.12.2022
- EDN: UFEDHV
- DOI: 10.31857/S0044457X22602310
Аннотация
В результате взаимодействия нитрилиевых производных клозо-додекаборатного аниона (Et4N)[B12H11N≡C–R] (R = Me, Et, nPr, iPr) с водой получен ряд иминолов состава (Et4N)[B12H11NH=C(OH)–R]. Установлено, что продукты гидролиза находятся в кислотно-основном равновесии иминол–амид, которое можно контролировать путем изменения кислотности среды. Продукты реакций идентифицированы и охарактеризованы методами 11В, 1Н, 13С ЯМР-спектроскопии, ИК-спектроскопии, ESI-масс-спектрометрии. Строение анионов [B12H11(Z-NH=C(OH)nC3H7)]− и [B12H11(E-NH–C(O)nC3H7)]2− установлено методом РСА.
ВВЕДЕНИЕ
Клозо-додекаборатный анион является одним из наиболее изученных кластерных анионов бора. Благодаря ряду уникальных свойств, присущих данному классу соединений, они представляют интерес как исходные платформы для получения различных лигандов в координационной химии [1–6], катализаторов [2, 7], материалов для электрохимических устройств [8, 9], высокоэнергетических веществ [10–14], потенциальных медицинских препаратов [15–17]. Разнообразие применения кластерных анионов связано с возможностью получения производных, содержащих один или несколько экзополиэдрических заместителей. Одной из важнейших областей химии данных соединений является химия производных, содержащих связь бор–азот. Борилированные амиды на основе клозо-додекаборатного аниона и находящиеся с ними в равновесии борилированные иминолы впервые были получены как побочные продукты при попытке синтеза нитрилиевых производных [18]. В дальнейшем они нашли свой применение в качестве лигандов [19] и исходных соединений для синтеза полизамещенных производных, обладающих противомикробной активностью [20, 21]. Основные недостатки методов получения данных соединений – малый выход целевых продуктов и необходимость трудоемкой очистки целевых соединений.
В настоящей работе нами предложен и оптимизирован метод получения борилированных иминолов на основе реакции гидролиза нитрилиевых производных клозо-додекаборатного аниона.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Элементный анализ. Содержание углерода, водорода и азота в образцах определяли на элементном CHNS-анализаторе Eurovector EuroEA 3000, содержание бора – на атомно-эмиссионном спектрометре с индуктивно связанной плазмой iCAP 6300 Duo (Thermo Scientific).
ИК-спектры соединений записывали на ИК-фурье-спектрофотометре Инфралюм ФТ-08 (НПФ АП “Люмекс”) в области 4000–600 см–1 с разрешением 1 см–1. Образцы готовили в виде раствора в CH2Cl2.
Cпектры ЯМР 1H, 11B, 13C растворов исследуемых веществ в CD2Cl2 или в CD3CN записывали на импульсном фурье-спектрометре Bruker AVANCE 300 (ФРГ) на частотах 300.3, 96.32 и 75.49 МГц соответственно с внутренней стабилизацией по дейтерию. В качестве внешних стандартов использовали тетраметилсилан или эфират трехфтористого бора.
ESI-масс-спектры растворов исследуемых веществ в CH3CN записывали на спектрометре LСМS-IT-TOF (Shimadzu, Japan). Спектры HRMS были получены в режиме прямого введения. Масс-спектры получали в диапазоне m/z от 120 до 700 Да. Напряжение детектора 1.55 кВ, распыляющий газ 1.50 л/мин, температура CDL 200.0°C, напряжение ЭСИ 4.50 кВ. Настройку прибора (калибровку массы и проверку чувствительности) проводили перед анализом.
Рентгеноструктурный анализ. Набор дифракционных отражений для кристалла получен в Центре коллективного пользования ИОНХ РАН на автоматическом дифрактометре Bruker Smart Apex2 (λMoKα, графитовый монохроматор, ω–φ-сканирование). Данные были проиндексированы и интегрированы с помощью программы SAINT. Применяли поправку на поглощение, основанную на измерениях эквивалентных отражений (SADABS) [22]. Структуры расшифрованы прямым методом с последующим расчетом разностных синтезов Фурье. Все неводородные атомы уточнены в анизотропном приближении, все атомы водорода – по модели “наездника” с тепловыми параметрами Uизо = 1.2Uэкв (Uизо) соответствующего неводородного атома (1.5Uизо для СН3-групп).
Все расчеты проводили с использованием программы SHELXTL [23]. Структура расшифрована и уточнена с помощью программного комплекса OLEX2 [24].
Тетрафенилфосфониевые соли для РСА были получены добавлением эквимолярного количества PPh4Cl к соответствующим производным в минимальном количестве ацетонитрила.
Кристаллографические данные депонированы в Кембриджском банке структурных данных (CCDС № 2231708, 2231709).
Синтез. Нитрилиевые производные клозо-додекаборатного аниона вида (Et4N)[B12H11N≡C–R], где R = Me (Et4N)(1a), Et (Et4N)(1b), nPr (Et4N)(1c), iPr (Et4N)(1d), были получены по оптимизированной литературной методике [25, 26].
(Et4N)[B12H11(NHC(OH)CH3)] – (Et4N)(2a). Растворяли 0.312 г (1.0 ммоль) (Et4N)(1a) в 5 мл ацетонитрила и 5 мл дистиллированной воды. Реакционную смесь кипятили с обратным холодильником в течение 2 ч. После охлаждения до комнатной температуры к раствору прибавляли 1 мл 1 н раствора HCl. Полученный раствор концентрировали на роторном испарителе до объема 2 мл и отфильтровывали целевое вещество. Полученный продукт сушили в вакууме. Выход (Et4N)[B12H11(NHC(OH)CH3)] ((Et4N)(2a)) 0.30 г (91%).
11B{H} ЯМР-спектр (CD3CN, δ, м.д.): –8.2 (с, 1B, B–N), –15.3, –16.0 (с, 11B, B–H(B2–12)). 1H ЯМР-спектр (CD3CN, δ, м.д.): 2.5–0.0 (м, 11H, B–H), 3.17 (8H, Et4N), 1.22 (12H, Et4N), 10.7 (с, 1H, OH), 8.52 (т, 1H, NH=C), 2.18 (с, 3H, C–CH3). 13C{H} ЯМР-спектр (CD3CN, δ, м.д.): 53.1 (Et4N), 7.7 (Et4N), 176.3 (NH=C), 20.1 (NH=C–CH3). ИК-спектр (CH2Cl2, см−1): 3323, 3291, 3245 ν(N–H), 2492 ν(B–H), 1653 ν(C=N). MS(ESI) m/z: 200.2421 (найдено для [B12H11(NHC(OH)CH3)]–, вычислено для {[A]–} 200.2423). Данные элементного анализа приведены в табл. 1.
Таблица 1.
Данные элементного анализа синтезированных соединений
| Соединение | С, % | H, % | N, % | B, % | ||||
|---|---|---|---|---|---|---|---|---|
| вычислено | найдено | вычислено | найдено | вычислено | найдено | вычислено | найдено | |
| (Et4N)(2a) | 36.38 | 36.40 | 10.99 | 10.85 | 8.49 | 8.56 | 39.3 | 39.0 |
| (Et4N)(2b) | 38.39 | 38.46 | 11.13 | 11.04 | 8.14 | 8.17 | 37.7 | 37.5 |
| (Et4N)(2с) | 40.24 | 40.13 | 11.26 | 11.36 | 7.82 | 7.70 | 36.2 | 36.0 |
| (Et4N)(2d) | 40.24 | 40.10 | 11.26 | 11.30 | 7.82 | 7.65 | 36.2 | 35.8 |
| (Et4N)2(3c) | 49.28 | 49.01 | 12.20 | 12.29 | 8.62 | 8.55 | 26.6 | 26.1 |
(Et4N)[B12H11(NHC(OH)C2H5)] ((Et4N)(2b)) получали по аналогичной методике. Из 0.329 г (Et4N)(1b) получено 0.313 г (Et4N)[B12H11(NHC(OH)C2H5)] ((Et4N)(2b)). Выход 91%.
11B{H} ЯМР-спектр (CD3CN, δ, м.д.): –8.1 (с, 1B, B–N), –15.1, –15.8 (с, 11B, B–H(B2–12)). 1H ЯМР-спектр (CD3CN, δ, м.д.): 2.5–0.0 (м, 11H, B–H), 3.17 (8H, Et4N), 1.22 (12H, Et4N), 10.8 (с, 1H, OH), 8.4 (т, 1H, NH=C), 2.47 (к, 2H, CH2CH3, J = 7.57 Гц), 1.16 (т, 3H, CH2CH3, J = 7.54 Гц). 13C{H} ЯМР-спектр (CD3CN, δ, м.д.): 53.1 (Et4N), 7.7 (Et4N), 179.1 (NH=C), 27.7 (CH2CH3), 9.84 (CH2CH3). ИК-спектр (CH2Cl2, см−1): 3328, 3271 ν(N–H), 2495 ν(B–H), 1653 ν(C=N). MS(ESI) m/z: 214.2573 (найдено для [B12H11(NHC(OH)C2H5)], вычислено для {[A]-} 214.2578).
(Et4N)[B12H11(NHC(OH)nC3H7)] ((Et4N)(2с)) получали по аналогичной методике. Из 0.340 г (Et4N)(1с) получено 0.344 г (Et4N)[B12H11(NHC(OH)nC3H7)] ((Et4N)(2с)). Выход 96%.
11B{H} ЯМР-спектр (CD3CN, δ, м.д.): –8,1 (с, 1B, B–N), –15.0, –15.8 (с, 11B, B–H(B2–12)). 1H ЯМР-спектр (CD3CN, δ, м.д.): 2.5–0.0 (м, 11H, B–H), 3.17 (8H, Et4N), 1.22 (12H, Et4N), 10.8 (с, 1H, OH), 8.46 (т, 1H, NH=C), 2.42 (т, 2H, CH2CH2CH3, J = 7.33 Гц), 1.65 (м, 2H, CH2CH2CH3), 0.92 (т, 3H, CH2CH2CH3, J = 7.40). 13C{H} ЯМР-спектр (CD3CN, δ, м.д.): 53.1 (Et4N), 7.7 (Et4N), 178.9 (NH=C), 35.8 (CH2CH2CH3), 19.8 (CH2CH2CH3), 13.2 (CH2CH2CH3). ИК-спектр (CH2Cl2, см−1): 3302, 3279 ν(N–H), 2496 ν(B–H), 1653 ν(C=N). MS(ESI) m/z: 228.2740 (найдено для [B12H11(NHC(OH)nC3H7)], вычислено для {[A]-} 228.2734).
(Et4N)[B12H11(NHC(OH)iC3H7)] ((Et4N)(2d)) получали по аналогичной методике. Из 0.341 г (Et4N)(1d) получено 0.330 г (Et4N)[B12H11(NHC(OH)iC3H7)] ((Et4N)(2d)). Выход 92%.
11B{H} ЯМР-спектр (CD3CN, δ, м.д.): –8.1 (с, 1B, B–N), –14.9, –15.8 (с, 11B, B–H(B2–12)). 1H ЯМР-спектр (CD3CN, δ, м.д.): 2.5–0.0 (м, 11H, B–H), 3.17 (8H, Et4N), 1.22 (12H, Et4N), 10.8 (с, 1H, OH), 8.33 (т, 1H, NH=C), 2.72 (гепт., 1H, CH(CH3)2, J = 6.89 Гц), 1.18 (д, 6H, CH(CH3)2, J = 6.97 Гц). 13C{H} ЯМР-спектр (CD3CN, δ, м.д.): 53.1 (Et4N), 7.7 (Et4N), 182.4 (NH=C), 34.3 (CH(CH3)2), 18.9 (CH(CH3)2). ИК-спектр (CH2Cl2, см−1): 3311, 3276 ν(N–H), 2498 ν(B–H), 1647 ν(C=N). MS(ESI) m/z: 228.2737 (найдено для [B12H11(NHC(OH)nC3H7)], вычислено для {[A]-} 228.2734).
(Et4N)2[B12H11(NHC(O)nC3H7)] – (Et4N)2(3с). Навеску 0.186 г (0.5 ммоль) (Et4N)(2a) растворяли в 5 мл CH2Cl2. К полученному раствору приливали 5 мл 0.1 н раствора NaOH и добавляли 0.09 г (0.55 ммоль) (Et4N)Cl. Реакционную массу перемешивали в течение 2 ч, органическую часть отделяли, упаривали на роторном испарителе досуха. Полученный продукт перекристаллизовывали из смеси этанол/2-пропанол и сушили в вакууме.
Получено 0.245 г (Et4N)2[B12H11(NHC(O)nC3H7)] ((Et4N)2(3с)). Выход 90%.
11B{H} ЯМР-спектр (CD3CN, δ, м.д.): –4.0 (с, 1B, B–N), –14.8, –16.2, –19.4 (с, 11B, B–H(B2–12)). 1H ЯМР-спектр (CD3CN, δ, м.д.): 2.5–0.0 (м, 11H, B–H), 3.17 (8H, Et4N), 1.22 (12H, Et4N), 7.7 (с, 1H, NH–C), 2.44 (т, 2H, CH2CH2CH3, J = 7.33 Гц), 1.46 (м, 2H, CH2CH2CH3), 0.86 (т, 3H, CH2CH2CH3, J = 7.40). 13C{H} ЯМР-спектр (CD3CN, δ, м.д.): 53.1 (Et4N), 7.7 (Et4N), 177.7 (NH–C=O), 35.4 (CH2CH2CH3), 20.0 (CH2CH2CH3), 13.3 (CH2CH2CH3).
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Реакция гидролиза нитрилиевых производных клозо-додекаборатного аниона протекает по схеме:
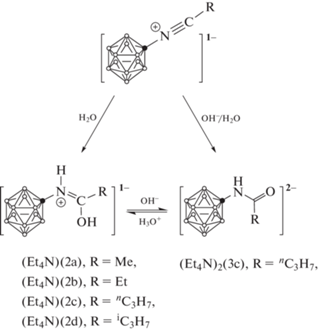
Схема 1 .
В отличие от реакции нитрилиевых производных с аминами, процесс гидролиза нитрилиевых производных протекает с заметной скоростью только при повышенной температуре [25, 27, 28].
Образующийся на первой стадии реакции иминол находится в равновесии с соответствующим амидным производным клозо-додекаборатного аниона. При гидролизе нитрилиевых производных в водно-ацетонитрильной среде образуется смесь двух продуктов с преобладанием иминольной формы (10 : 1). Дальнейшая обработка полученной смеси продуктов кислотой или основанием позволяет получать соответственно иминольную или амидную форму производного.
Полноту протекания реакции контролировали с помощью 11B{1H} ЯМР-спектроскопии. В спектрах полученных иминолов сигнал от замещенного атома бора наблюдается в области –8.1…–8.2 м.д. Сигналы от незамещенных атомов бора лежат в области –15.0…–17.0 м.д. Равновесие между иминольной и амидной формой полученных производных было изучено при титровании раствора борилированного амида трифторуксусной кислотой (шаг – 0.1 экв. трифторуксусной кислоты). Полученные спектры соотносятся с литературными данными [29, 30]: сигналы от замещенного атома бора в амидах лежат в области –5.1 м.д. и при добавлении избытка кислоты смещаются в область слабого поля (9.0 м.д.) в продукте иминольной структуры (рис. 1).
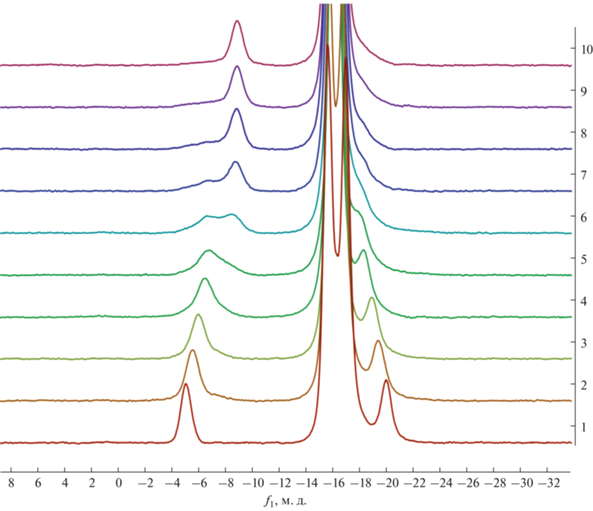
Полученные иминолы исследованы методами 1H и 13C{H} ЯМР-спектроскопии, ИК-спектроскопии и ESI-HR масс-спектрометрии. В спектрах 1Н ЯМР наблюдается один уширенный сигнал от гидридных атомов водорода клозо-додекаборатного аниона в области 2.50…0.00 м.д. Иминольный фрагмент представлен группой из двух сигналов: сигналом протонов гидроксильной группы в области 11.0…10.0 м.д. и протоном, связанным с атомом азота, в области 9.0…8.0 м.д. Сигнал протонов, связанных с α-атомом углерода нитрилиевого заместителя, лежит в более сильном поле по сравнению с продуктами присоединения спиртов [31]. Положение данного сигнала аналогично сигналам в продуктах присоединения первичных аминов, что указывает на образование продуктов в Z-конфигурации.
Производные иминольной (NEt4)[B12H11(Z-NH=C(OH)nC3H7)] и амидной структуры (PPh4)2[B12H11(E-NH–C(O)nC3H7)] были изучены методами РСА (рис. 2).
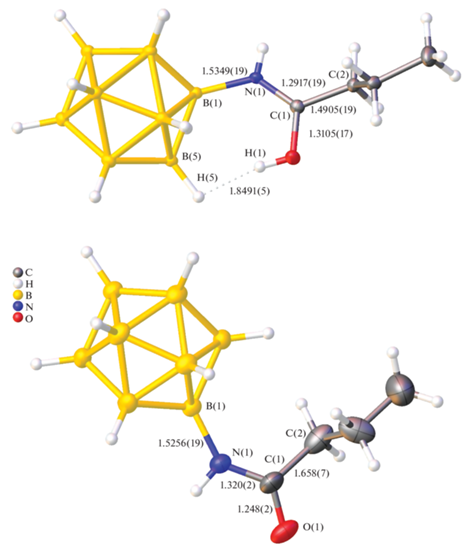
Установлено, что в структуре продукта, находящегося в форме иминола, длина связи N(1)–C(1) составляет 1.292 Å, а C(1)–O(1) – 1.310 Å, данные значения соответствуют промежуточным порядкам связей C–N и C–O, что указывает на наличие сопряжения в молекуле исследуемого иминола. В отличие от продуктов присоединения спиртов к нитрилиевым производным клозо-додекаборатного аниона, реакция присоединения воды протекает с образованием продукта в Z-конфигурации при двойной связи N(1)–C(1). Стереоселективность процесса обусловлена возможностью образования внутримолекулярной диводородной связи между атомом водорода гидроксильной группы и одним из гидридных атомов кластера. Длина диводородной связи в полученной структуре составляет 1.849 Å, это значение меньше средней длины диводородных связей в молекулах борилированных амидинов.
В продукте амидной структуры длина связи N(1)–C(1) составляет 1.320 Å, а C(1)–O(1) – 1.248 Å, что хорошо согласуется с литературными данными, в том числе и для органических амидов. Разница в длинах связей двух кристаллических структур объясняется более выраженной делокализацией электронов в молекуле иминола.
ЗАКЛЮЧЕНИЕ
Предложен и оптимизирован метод получения борилированных иминолов и амидов на основе реакции гидролиза нитрилиевых производных клозо-додекаборатного аниона. Реакция характеризуется простотой синтетических операций и выходом целевых продуктов, близким к количественным. Показано, что иминолы и амиды могут претерпевать взаимное превращение в зависимости от pH среды.
Список литературы
Geis V., Guttsche K., Knapp C. et al. // Dalton Trans. 2009. № 15. P. 2687. https://doi.org/10.1039/b821030f
Bolli C., Derendorf J., Jenne C. et al. // Chem. A Eur. J. 2014. V. 20. № 42. P. 13783. https://doi.org/10.1002/chem.201403625
Bolli C., Derendorf J., Jenne C. et al. // Eur. J. Inorg. Chem. 2017. V. 2017. № 38–39. P. 4552. https://doi.org/10.1002/ejic.201700620
Zhang Y., Liu J., Duttwyler S. // Eur. J. Inorg. Chem. 2015. V. 2015. № 31. P. 5158. https://doi.org/10.1002/ejic.201501009
Kirchmann M., Wesemann L. // Dalton Trans. 2008. № 4. P. 444. https://doi.org/10.1039/B715305H
Matveev E.Y., Avdeeva V.V., Zhizhin K.Y. et al. // Inorganics. 2022. V. 10. № 12. P. 238. https://doi.org/10.3390/inorganics10120238
Messina M.S., Axtell J.C., Wang Y. et al. // J. Am. Chem. Soc. 2016. V. 138. № 22. P. 6952. https://doi.org/10.1021/jacs.6b03568
Gigante A., Duchêne L., Moury R. et al. // ChemSusChem. 2019. V. 12. № 21. P. 4832. https://doi.org/10.1002/cssc.201902152
Duchêne L., Lunghammer S., Burankova T. et al. // Chem. Mater. 2019. V. 31. № 9. P. 3449. https://doi.org/10.1021/acs.chemmater.9b00610
Derdziuk J., Malinowski P.J., Jaroń T. // Int. J. Hydrogen Energy. 2019. V. 44. № 49. P. 27030. https://doi.org/10.1016/j.ijhydene.2019.08.158
Rao M.H., Muralidharan K. // Polyhedron. 2016. V. 115. P. 105. https://doi.org/10.1016/j.poly.2016.03.062
Hagemann H. // Molecules. 2021. V. 26. № 24. P. 7425. https://doi.org/10.3390/molecules26247425
Sharon P., Afri M., Mitlin S. et al. // Polyhedron. 2019. V. 157. P. 71. https://doi.org/10.1016/j.poly.2018.09.055
Zhilitskaya L.V., Shainyan B.A., Yarosh N.O. // Molecules. 2021. V. 26. № 8. P. 2190. https://doi.org/10.3390/molecules26082190
Barth R.F., Zhang Z., Liu T. // Cancer. Commun. 2018. V. 38. № 1. P. 36. https://doi.org/10.1186/s40880-018-0280-5
Hattori Y., Kusaka S., Mukumoto M. et al. // J. Med. Chem. 2012. V. 55. № 15. P. 6980. https://doi.org/10.1021/jm300749q
Hatanaka H. // J. Neurol. 1975. V. 209. № 2. P. 81. https://doi.org/10.1007/BF00314601
Wiersema R.J., Middaugh R.L. // Inorg. Chem. 1969. V. 8. № 10. P. 2074. https://doi.org/10.1021/ic50080a009
Guangxian X., Jimei X., Technology S. New Frontiers in Rare Earth Science and Applications. 1985. https://doi.org/10.1016/c2013-0-11730-8
Varkhedkar R., Yang F., Dontha R. et al. // ACS Cent. Sci. 2022. V. 8. № 3. P. 322. https://doi.org/10.1021/acscentsci.1c01132
Sun Y., Zhang J., Zhang Y. et al. // Chem. A Eur. J. 2018. V. 24. № 41. P. 10364. https://doi.org/10.1002/chem.201801602
Krause L., Herbst-Irmer R., Sheldrick G.M. et al. // J. Appl. Crystallogr. 2015. V. 48. № 1. P. 3. https://doi.org/10.1107/S1600576714022985
Sheldrick G.M. // Acta Crystallogr., Sect. C: Struct. Chem. 2015. V. 71. № 1. P. 3. https://doi.org/10.1107/S2053229614024218
Dolomanov O.V., Bourhis L.J., Gildea R.J. et al. // J. Appl. Crystallogr. 2009. V. 42. № 2. P. 339. https://doi.org/10.1107/S0021889808042726
Nelyubin A.V., Selivanov N.A., Bykov A.Y. et al. // Int. J. Mol. Sci. 2021. V. 22. № 24. P. 13391. https://doi.org/10.3390/ijms222413391
Nelyubin A.V., Klyukin I.N., Novikov A.S. et al. // Inorganics. 2022. V. 10. № 11. P. 196. https://doi.org/10.3390/inorganics10110196
Нелюбин А.В., Соколов М.С., Селиванов Н.А. и др. // Журн. неорган. химии. 2022. Т. 67. С. 1562.
Нелюбин А.В., Селиванов Н.А., Клюкин И.Н. и др. // Журн. неорган. химии. 2021. Т. 66. С. 1297.
Zhang Y., Sun Y., Wang T. et al. // Molecules. 2018. V. 23. № 12. P. 1. https://doi.org/10.3390/molecules23123137
Bogdanova E.V., Stogniy M.Y., Suponitsky K.Y. et al. // Molecules. 2021. V. 26. № 21. P. 6544. https://doi.org/10.3390/molecules26216544
Laskova J., Ananiev I., Kosenko I. et al. // Dalton Trans. 2022. V. 51. № 8. P. 3051. https://doi.org/10.1039/D1DT04174F
Дополнительные материалы отсутствуют.
Инструменты
Журнал неорганической химии