

Журнал неорганической химии, 2023, T. 68, № 6, стр. 816-823
Синтез конъюгатов катионных мезо-арилпорфиринов с замещенным производным клозо-декаборатного аниона [B10H9NH=C(CH3)CH2C≡CH]–
Ю. С. Бортневская a, Н. С. Захаров a, А. П. Жданов b, М. С. Григорьев c, К. А. Жданова d, *, Н. А. Брагина a, К. Ю. Жижин b, **
a МИРЭА – Российский технологический университет
119571 Москва, пр-т Вернадского, 86, Россия
b Институт общей и неорганической химии им. Н.С. Курнакова РАН
119991 Москва, Ленинский пр-т, 31, Россия
c Институт физической химии и электрохимии им. А.Н. Фрумкина
117342 Москва, Ленинский пр-т, 31, Россия
d Институт “Международный Томографический Центр” СО РАН
630090 Новосибирск, ул. Институтская, 3а, Россия
* E-mail: zhdanova_k@mirea.ru
** E-mail: zhizhin@igic.ras.ru
Поступила в редакцию 13.02.2023
После доработки 07.03.2023
Принята к публикации 07.03.2023
- EDN: UGMJJE
- DOI: 10.31857/S0044457X23600238
Аннотация
Представлен подход к получению водорастворимых конъюгатов катионных мезо-арилпорфиринов типа А3В и производных клозо-декаборатного аниона [B10H9NH=C(CH3)NHCH2C≡CH]– на основе клик-реакции. Строение полученных конъюгатов установлено современными методами анализа. Исследованы их основные спектрально-люминесцентные характеристики.
ВВЕДЕНИЕ
В настоящее время онкологические заболевания являются одной из главных причин смертности во всем мире [1]. Традиционные методы лечения рака, такие как химиотерапия, хирургия и лучевая терапия, имеют ряд тяжелых побочных эффектов, что обусловливает актуальность поиска новых методов лечения раковых заболеваний или повышение эффективности уже существующих [2, 3]. Одними из таких методов являются фотодинамическая (ФДТ) и 10B-нейтронозахватная (БНЗТ) терапия. Оба метода отличаются неинвазивностью и практическим отсутствием токсического воздействия на организм человека, а также отсутствием множественной лекарственной устойчивости [4–9]. Для успешного проведения процедуры ФДТ необходимо совместное действие трех компонентов: фотосенсибилизатора (ФС), лазерного излучения и кислорода в тканях [9]. Для БНЗТ требуется наличие борсодержащего агента и нейтронного излучения [10]. Общей задачей данных подходов является поиск терапевтических агентов и средств их доставки, отвечающих условиям биобезопасности, биосовместимости и таргетности [11–20]. Создание агентов для возможного бинарного применения в ФДТ и БНЗТ может решить сразу несколько задач: придание водорастворимости обычно гидрофобным ФС за счет наличия кластерного аниона бора; появление флуоресцентных свойств у борсодержащих препаратов, позволяющих осуществлять визуализацию тканей in vivo; таргетность за счет эффекта повышенной проницаемости и удержания. Учитывая вышесказанное, создание подобных конъюгатов является актуальной задачей бионеорганической химии.
Ранее нами были разработаны подходы к получению конъюгатов нитрилиевых производных клозо-декаборатного аниона [B10H10]2– и аминосодержащих мезо-арилпорфиринов, изучены их фотофизические свойства [21, 22]. Выход полученных конъюгатов составлял величину ~70%. В настоящее время широкое распространение получили линкеры на основе 1,2,3-триазола, образующегося в ходе биполярного циклоприсоединения ароматических азидов к алкинам или клик-реакции, что может значительно увеличить общий выход в реакциях [23, 24]. В связи с этим цель настоящей работы – синтез водорастворимых конъюгатов мезо-арилпорфиринов и клозо-декаборатного аниона с помощью клик-реакции, установление оптимальных параметров проведения реакции и выделения продуктов, изучение фотофизических свойств полученных соединений.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Реагенты. В работе использовали коммерческие препараты аналитической чистоты фирмы Sigma-Aldrich, силикагель для колоночной хроматографии (Macherey-Nagel), пластины для ТСХ, покрытые силикагелем 60 F254 (Macherey-Nagel), а также органические растворители отечественного производства (ООО “Химмед Синтез”): хлористый метилен (х. ч.), гексан (х. ч.), этилацетат (х. ч.), метанол (х. ч.) и этанол (х. ч.), очищенные по стандартным методикам.
Cпектры ЯМР 1H, 11B, 13C растворов исследуемых веществ в CDCl3 ((CD3)2SO) регистрировали на импульсном Фурье-спектрометре Bruker MSL-300 (Германия) на частотах 300.3, 96.32 и 75.49 МГц соответственно с внутренней стабилизацией по дейтерию. В качестве внешнего стандарта использовали тетраметилсилан или эфират трехфтористого бора.
MALDI масс-спектры записывали на масс-спектрометре Bruker autoflex speed time-of-flight (TOF) (Bruker Daltonics Inc., Germany), оснащенном твердотельным УФ-лазером с длиной волны 355 нм (частота 1 кГц, 1000 импульсов для каждого образца) и рефлектроном и работающем в режиме регистрации положительно заряженных ионов. Для регистрации масс-спектров MALDI использовали стальную мишень MTP 384 ground steel (Bruker Daltonics Inc., Германия).
Электронные спектры поглощения записывали на спектрофотометре TermoSpectronic Helios Alpha в кварцевых кюветах с длиной оптического пути 1 см, стационарные спектры флуоресценции – на спектрофлуориметре Панорама Флюорат-02 (Люмекс) с λex = 420 нм.
Рентгеноструктурный анализ. Рентгенодифракционный эксперимент проводили в ЦКП ИФХЕ РАН на автоматическом четырехкружном дифрактометре с двумерным детектором Bruker KAPPA APEX II (излучение MoKα) с использованием фрагмента кристалла размерами 0.12 × 0.08 × 0.04 мм при температуре 100 K.
Параметры элементарной ячейки уточнены по всему массиву данных. Структура расшифрована прямым методом (SHELXTL) [25] и уточнена полноматричным методом наименьших квадратов по F2 по всем данным в анизотропном приближении для неводородных атомов, кроме разупорядоченных, с помощью программного комплекса OLEX2 [26]. Атомы H боргидридного фрагмента локализованы из разностного Фурье-синтеза электронной плотности и уточнены изотропно без каких-либо ограничений. Атомы H групп NH, CH, CH2 и CH3 размещены в геометрически вычисленных позициях и уточнены с изотропными температурными параметрами, равными 1.2Uэкв атома N или C для NH, CH, CH2 и 1.5Uэкв атома C для CH3.
Кристаллографические данные депонированы в Кембриджском банке структурных данных (CCDС № 2240403).
Синтез 4-(4-бром-н-бутокси)бензальдегида (1). В 30 мл ацетона растворяли 3 г (24.6 ммоль) п-гидроксибензальдегида, 6.89 г (31.9 ммоль) 1,4-дибромбутана и 4.41 г (3.9 ммоль) карбоната калия. Реакционную массу перемешивали при кипячении в течение 12 ч, после чего охлаждали до комнатной температуры и фильтровали через бумажный фильтр. Экстрагировали в системе хлористый метилен–вода с подкислением 1 М раствором соляной кислоты до нейтральной среды. Реакционную массу концентрировали под вакуумом. Целевой продукт очищали колоночной хроматографией на силикагеле G 60, элюировали смесью хлористый метилен–гексан (5 : 1) и сушили в вакууме над P2O5. Выход составил 4.52 г (76%).
Rf = 0.4 (дихлорметан : гексан = 7 : 1). ИК-спектр (ν, см–1): 2928, 2849 (СН2)4, 1675 (С=О), 1467 (Аr), 1160, 1138 (С–О), 642, 550 (С–Br). Спектр ЯМР 1Н (CDCl3, δ, м.д.): 1.73 (2Н, м, СН2СН2Br), 1.87 (2Н, м, ОСН2СН2), 2.55 (2Н, т, СН2СН2Br), 3.43 (2Н, т, ОСН2), 7.51 (2Н, д, 2,6-(ArH)), 8.22 (2Н, д, 3,5-(ArH)), 9.9 (1Н, c, СHO).
Синтез 5-(4-ацетамидофенил)-10,15,20-трис(4-(4-бром-н-бутокси)фенил)порфирина (2). В 200 мл хлористого метилена растворяли 200 мг (3.00 ммоль) пиррола, 0.580 г (2.20 ммоль) 4-(4-бром-н-бутокси)бензальдегида и 0.122 г (0.75 ммоль) 4-ацетаминобензальдегида. Реакционную массу насыщали инертным газом при перемешивании в течение 15 мин, добавляли 40 мкл (0.28 ммоль) эфирата трехфтористого бора и 200 мкл абсолютного этанола и перемешивали 40 мин в токе инертного газа при комнатной температуре, после чего добавляли 0.681 г (3.00 ммоль) DDQ и перемешивали еще в течение 60 мин. Затем реакционную смесь подвергали экстракции в системе хлористый метилен–вода и концентрировали под вакуумом. Целевой продукт очищали колоночной хроматографией на силикагеле G 60, элюировали смесью хлористый метилен–этилацетат (30 : 1) и сушили в вакууме над P2O5. Выход 0.174 г (15%).
Rf = 0.5 (хлористый метилен : этилацетат = 30 : 1). Масс-спектр, m/z: найдено: 1120.434 [M-1]+ (для C58H54Br3N5O4 рассчитано: 1121.79). Электронный спектр (хлористый метилен, λmax, нм (lgε)): 423.4 (6.30), 519.4 (4.79), 558.8 (4.67), 594.4 (4.20), 652.6 (4.32). Спектр ЯМР 1Н (CDCl3, δ, м.д.): –2.75 (2H, c, NH-пиррол), 2.07–2.17 (6Н, м, СН2СН2Br), 2.19–2.28 (6Н, м, ОСН2СН2), 2.30 (3H, с, NHCH3), 3.62 (6Н, т, СН2Br), 4.23 (6Н, т, ОСН2), 7.17 (6Н, д, 3,5-(ArH)), 7.80 (2H, д, β-ArNHAc), 8.03–8.16 (8Н, м, 2,6-(ArH) + α-ArNHAc), 8.86 (8Н, д, CH-пиррол).
Синтез 5-(4-аминофенил)-10,15,20-трис(4-(4-бром-н-бутокси)фенил)порфирина (3). В 8 мл трифторуксусной кислоты растворяли 0.050 г (0.10 ммоль) 5-(4-ацетамидофенил)-10,15,20-трис(4-(4-бром-н-бутоксифенил))порфирина и в течение 30 мин прикапывали 12 мл концентрированной соляной кислоты. Реакционную смесь перемешивали при кипячении на масляной бане в течение 40 ч при температуре 80°С, охлаждали до комнатной температуры, экстрагировали в системе хлористый метилен–вода и промывали водным раствором аммиака до изменения окраски органического слоя с зеленой на бордовую. Реакционную массу концентрировали под вакуумом. Целевой продукт очищали колоночной хроматографией на силикагеле G 60, элюировали смесью хлористый метилен–этилацетат (70 : 1) и сушили в вакууме над P2O5. Выход 0.045 г (87%).
Rf = 0.6 (хлористый метилен : этилацетат = 70 : 1) Масс-спектр, m/z: найдено: 1077.95 [M]+ (для C87H115N5O3 рассчитано: 1077.9). Электронный спектр (хлористый метилен, λmax, нм (lg ε)): 423.8 (6.07), 519.9 (4.76), 559.2 (4.63), 594.7 (4.18), 652.4 (4.29). Спектр ЯМР 1Н (CDCl3, δ, м.д.): ‒2.74 (2H, c, NH-пиррол), 2.12–2.18 (12Н, м, ОСН2СН2СН2СН2Br), 3.76 (6Н, т, СН2Br), 4.27 (6Н, т, ОСН2), 7.06 (2Н, д, β-ArNH2), 7.25 (6H, д, 3,5-ArH), 7.99 (2Н, д, α-ArNH2), 8.11 (6Н, д, 2,6-ArH), 8.85–8.94 (8Н, дд, CH-пиррол).
Синтез 5-(4-азидофенил)-10,15,20-трис(4-(4-бром-н-бутокси)фенил)порфирина (4). В круглодонной колбе растворяли 0.112 г (0.103 ммоль) порфирина 3 в 10 мл ледяной TFA. К полученному раствору в течение 15 мин прикапывали раствор 0.014 г (0.21 ммоль) нитрита натрия в 5 мл воды. Полученную смесь перемешивали на ледяной бане при 0°С 30 мин, после чего к реакционной массе прикапывали в течение 15 мин раствор 0.027 г (0.42 ммоль) азида натрия в 5 мл воды. Вторую стадию реакции проводили при комнатной температуре. После окончания реакции смесь подвергали экстрагированию в системе хлористый метилен–вода и обрабатывали водным раствором аммиака до изменения окраски органического слоя с зеленой на бордовую. Органический слой упаривали на роторном испарителе. Для выделения целевого соединения реакционную массу очищали с помощью колоночной хроматографии на силикагеле G 60, элюировали в системе органических растворителей хлористый метилен–гексан (3 : 1) (целевое соединение элюируется с первой фракцией), сушили в вакууме над P2O5. Выход 0.091 г (80%).
Rf = 0.85 (дихлорметан). ИК-спектр (ν, см–1): 3020, 2950 (C–Н); 2150–2200 (–N3); 1450–1580 (NH, пиррол); 1300 (С–О); 1150–1290 (–N3); 1020 (Ar–O–); 860 (1,4-замещ. Ar); 820 (δ-CH–Ar); 750 (δ-(CH2)4). Электронный спектр (хлористый метилен, λmax, нм (lgε)): 423.6 (6.15), 519.6 (4.78), 558.0 (4.65), 594.4 (4.19), 652.5 (4.31). Спектр ЯМР 1Н (CDCl3, δ, м.д.): –2.74 (2H, c, NH-пиррол), 2.04–2.20 (12Н, м, ОСН2СН2СН2СН2Br), 3.75 (6Н, т, СН2Br), 4.23 (6Н, т, ОСН2), 7.21 (6H, д, 3,5-ArH), 7.32 (2Н, д, β-ArN3), 8.10 (6Н, д, 2,6-ArH), 8.18 (2Н, д, α-ArN3), 8.91–8.98 (8Н, д, CH-пиррол). Спектр ЯМР 13C (CDCl3 δ, м.д.): 158.91, 151.66, 151.00, 148.08, 142.75, 141.53, 134.85, 133.29, 132.13, 131.89, 131.46, 130.73, 126.68, 120.65, 120.00, 116.97, 114.08, 69.64, 33.04, 29.20, 28.93.
Синтез комплекса цинка 5-(4-азидофенил)-10,15,20-трис(4-(4-бром-н-бутокси)фенил)порфирина (5). К раствору 0.030 г (0.023 ммоль) порфирина 4 в 10 мл хлористого метилена приливали раствор 0.062 г (0.249 ммоль) дигидрата ацетата цинка в 5 мл метанола. Полученную смесь перемешивали в течение 3 ч, экстрагировали в системе дихлорметан–вода. Органический слой собирали и упаривали на роторном испарителе. Твердое вещество пурпурного цвета очищали колоночной хроматографией на силикагеле G 60, в качестве элюента использовали смесь растворителей хлористый метилен–гексан (8 : 1). Целевое соединение элюируется с первой фракцией. Продукт перекристаллизовывали из метанола, сушили в вакууме над P2O5. Выход 0.030 г (94%).
Rf = 0.82 (хлористый метилен). Электронный спектр (хлористый метилен, λmax, нм (lg ε)): 424.0 (6.17), 558.3 (4.69), 594.8 (4.22). Спектр ЯМР 1Н (CDCl3, δ, м.д.): 2.08–2.18 (12Н, м, ОСН2СН2СН2СН2Br), 3.69 (6Н, т, СН2Br), 4.43 (6Н, т, ОСН2), 7.26 (6H, д, 3,5-ArH), 7.35 (2Н, д, β-ArN3), 8.15 (6Н, д, 2,6-ArH), 8.19 (2Н, д, α-ArN3), 8.94–8.99 (8Н, д, CH-пиррол).
Синтез производного клозо-декаборатного аниона (Bu4N)[2-B10H9NH=C(CH3)NHCH2C≡CH] (6). К раствору 0.408 г (1.015 ммоль) (Bu4N)[2-B10H9NCCH3] в 20 мл хлористого метилена приливали 0.097 мл (1.514 ммоль) пропаргиламина. Полученный реакционный раствор перемешивали при комнатной температуре в атмосфере сухого аргона в течение 1 ч. После прекращения реакции раствор концентрировали на роторном испарителе, полученный твердый остаток перекристаллизовывали из смеси этанола и изопропилового спирта. Продукт сушили над P2O5. Выход 0.376 г (82%).
Спектр ЯМР 11B (CDCl3, δ, м.д.): 0.8 (д, 1B, B(10), JB–H = 146 Гц), –6.6 (д, 1B, B(1), JB–H = 139 Гц), –17.3 (c, 1B, B(2)), –26.0 (д, 4B, B(3,5,6,9), JB–H = 116 Гц), –29.2 (д, 3B, B(3,7,8), JB–H = 122 Гц); Спектр ЯМР 1Н (CDCl3, δ, м.д.): 2.08–2.18 (12Н, м, ОСН2СН2СН2СН2Br), 3.69 (6Н, т, СН2Br), 4.43 (6Н, т, ОСН2), 7.26 (6H, д, 3,5-ArH), 7.35 (2Н, д, β-ArN3), 8.15 (6Н, д, 2,6-ArH), 8.19 (2Н, д, α-ArN3), 8.94–8.99 (8Н, д, CH-пиррол). Спектр ЯМР 1Н (CDCl3, δ, м.д.): 1.50…–1.00 (м, 9Н, В10Н9), 8.20 (с, 1H, NH–C=NH), 6.25 (с, 1H, NH–C=NH), 3.86 (д, 2H, CH2C≡CH, J = 2.4 Гц), 2.85 (т, 1H, CH2C≡CH, J = 2.0 Гц), 3.03 (м, 8H, Bu4N), 2.30 (с, 3H, NH=C–CH3), 1.46 (м, 8H, Bu4N), 1.28 (м, 8H, Bu4N), 0.82 (м, 12H, Bu4N); Спектр ЯМР 13С (CDCl3, δ, м.д.): 165.4 (С=NH), 81.1 (CH2C≡CH), 70.4 (CH2C≡CH), 33.9 (CH2C≡CH), 58.4 (Bu4N), 24.6 (Bu4N), 23.2 (CH3–C=NH), 20.7 (Bu4N), 14.1 (Bu4N). HRMS (ESI) m/z = 213.3151 а.е.м. (найдено для [B10H9NHC(CH3)HNCH2C≡СH], для {[A]-} вычислено: 213.3124).
Синтез конъюгата 7. К раствору 0.050 г (0.043 ммоль) соединения 5 в 15 мл ТHF добавляли 0.020 г (0.043 ммоль) соединения 6. К полученной смеси приливали раствор 0.013 г (0.051 ммоль) пентагидрата сульфата меди(II) и 0.013 г (0.064 ммоль) раствора аскорбата натрия в 3 мл воды. Реакционную массу кипятили в течение 15 ч, затем экстрагировали в системе хлористый метилен–вода. Органический слой концентрировали при пониженном давлении. Твердое вещество пурпурного цвета очищали колоночной хроматографией на силикагеле G 60. В качестве элюента использовали смесь хлористого метилена и метанола в объемном соотношении 30 : 1. Целевое соединение элюируется со второй фракцией. Сушили в вакууме над P2O5. Выход 0.042 г (85%).
Rf = 0.4 (дихлорметан : метанол = 30 : 1). ESI-MS масс-спектр, m/z: найдено: 691.1524 [M+]2+(для C61H65B10Br3N9O3Zn рассчитано: 1382.3040). ЭСП (CH2Cl2, λmax, нм (lgε)): 426 (5.62), 554 (4.18), 595 (3.87). Спектр ЯМР 1Н (CDCl3, δ, м.д.): 0.76–1.35 (9H, уш. м, B10H9), 1.88–1.99 (6H, м, CH2Br), 3.35 (6Н, с), 2.91 (6Н, м, CH2CH2Br), 3.61 (6Н, м, OCH2CH2), 4.09 (6Н, м, OCH2), 5.78 (1Н, м, NH=CH), 7.12 (8Н, м, 3,5-ArH), 8.03 (8Н, уш с, 2,6-ArH), 8.39 (1Н, м, NH), 8.69 (2Н, c, βPyr), 8.89 (6Н, уш. с, βPyr). Спектр ЯМР 13C (CDCl3, δ, м.д.): 165.26, 158.33, 150.63, 150.44, 149.49, 144.06, 142.09, 135.77, 134.74, 132.29, 131.22, 121.30, 120.73, 118.43, 117.72, 112.28, 67.12, 58.63, 44.93, 29.44, 26.73, 23.61, 19.37, 13.39.
Синтез конъюгата 8. Навеску 0.030 г (0.021 ммоль) соединения 7 растворяли в 15 мл безводного пиридина. Реакционную массу кипятили с обратным холодильником в течение 10 ч. Осадок, образовавшийся в ходе реакции, отфильтровывали и промывали диэтиловым эфиром. Затем растворяли в метаноле и переосаждали диэтиловым эфиром. Целевое соединение сушили при пониженном давлении над P2O5. Выход 0.022 г (73%).
Rf = 0.1 (метанол). ESI-MS масс-спектр, m/z: найдено: 668.2897 [M-Zn + Na]2+ (для C76H76B10N12O3Na рассчитано: 1335.7064). ЭСП (ДМФА, λmax, нм (lgε)): 429 (5.70), 562 (4.24), 610 (3.93). Спектр ЯМР 1Н (CD3OD, δ, м.д.): 1.18 (3Н, м, СН3СН=), 1.87–1.99 (9Н, м, B10H9), 2.22 (6Н, м, СН2СН2Py), 3.79 (6Н, м, ОСН2СН2), 4.25 (6Н, м, ОСН2), 4.76 (6Н, м, СН2СН2Py), 5.27 (2Н, с, –CH2NH–), 7.29 (8Н, м, 3,5-ArH), 8.01 (8Н, м, 2,6-ArH), 8.21 (6Н, м, 3,5-Py), 8.32 (4Н, м, 4-Py), 8.38 (2Н, д, J = 8 Гц), 8.62 (2H, м, β-Pyr), 8.76 (6Н, м, β-Pyr), 9.06 (1Н, c, CH=NH), 9.17 (6Н, м, β-Pyr). Спектр ЯМР 13C (DMSO-d6, δ, м.д.): 158.58, 150.03, 149.35, 146.16, 144.96, 143.46, 142.62, 135.33, 131.43, 128.60, 122.50, 120.67, 117.88, 113.50, 67.14, 60.78, 46.11, 29.48, 28.45, 26.44, 25.54, 16.15.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
В настоящей работе предложен подход к получению новых конъюгатов катионных мезо-арилпорфиринов с производными аниона [B10H10]2– на основе клик-реакции. Для этого на первом этапе работы получены ω-бромзамещенные порфирины типа А3В, содержащие реакционноспособную аминогруппу (схема 1 ). Бензальдегид 1 получен по стандартному методу О-алкилирования 4-гидроксибензальдегида 1,4-дибромбутаном в ацетоне в присутствии избытка основания карбоната калия (5 экв) с выходом 76%.
Ранее для получения аминопорфиринов типа А3В нами предложен удобный подход по модифицированному методу Адлера [21], когда синтез проводили при кипячении в смеси трех растворителей: нитробензола, уксусной и пропионовой кислот. Однако оказалось, что использование данного подхода для синтеза порфирина 2 приводит к образованию трудноразделимой смеси продуктов. По всей видимости, в этих условиях (кипячение выше 150°С) происходит отщепление молекулы бромоводорода от терминальной метиленовой группы с образованием смеси побочных продуктов. В связи с этим для синтеза мезо-арилпорфирина 2 была использована монопиррольная конденсация по Линдсею [27]. Данный метод является более мягким: реакция протекает при комнатной температуре в хлороформе с использованием катализатора Льюиса – эфирата трехфтористого бора (BF3 · Et2O). После хроматографической очистки выход соединения 2 составил 13%.
Для получения аминосодержащего порфирина 3 удаляли ацетильную защитную группу в соединении 2 при кипячении и перемешивании в трифторуксусной кислоте (TFA) с медленным введением по каплям концентрированной соляной кислоты. Очистку целевого соединения проводили колоночной хроматографией на силикагеле. Выход соединения 3 составил 87%, его строение и индивидуальность подтверждены методами ТСХ, ЯМР 1Н, ИК- и УФ-спектроскопии, а также масс-спектрометрии MALDI-TOF. В спектре ЯМР 1Н соединения 3 присутствуют сигналы от протонов в β-положениях пиррольных групп при 8.85–8.91 м.д., а также сигналы в форме дублетов от протонов мета-фенильных групп при 7.85 и 7.25 м.д. с интегральной интенсивностью 1 : 3, что свидетельствует о наличии несимметричной системы протонов в порфирине типа А3В. По сравнению с порфирином 2 было зафиксировано исчезновение сигналов от протонов ацетильной группы в области сильного поля, что подтверждало исчерпывающее удаление ацетильной защиты. В ИК-спектре соединения 3 в диапазонах 3380–3310 и 1650–1590 см–1 присутствуют полосы валентных и деформационных колебаний первичной аминогруппы.
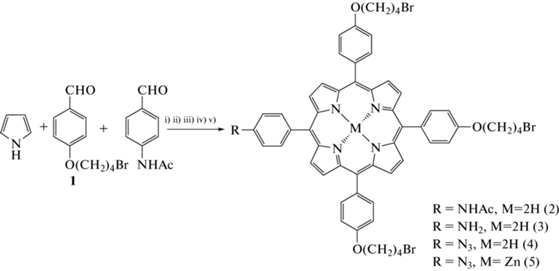
Схема 1 . Реагенты и условия проведения реакций: i) ацетон, K2CO3, t°C; ii) CHCl3, BF3 · Et2O, DDQ, rt; iii) TFA, HCl, 80°C; iv) TFA, NaNO2, 0°C, затем NaN3 rt; v) ZnOAc2, CH2Cl2–CH3OH.
Для получения целевого конъюгата с помощью клик-реакции требовалось получить азидосодержащий порфирин 4 по методу Хьюсгена–Мельдаля–Шарплесса [23]. На начальном этапе аминопорфирин обрабатывали смесью TFA и водного раствора нитрита натрия при 0°С во избежание получения побочных продуктов и сильного экзотермического эффекта. Образовавшееся диазосоединение обрабатывали водным раствором азида натрия при комнатной температуре (схема 1 ) для получения целевого продукта 4. Выход соединения 4 составил 80%. Наличие азидной группы в полученном соединении подтверждено методом ИК-спектроскопии, в диапазонах 2150–2200 и 1150–1290 см–1 наблюдаются полосы симметричных и асимметричных валентных колебаний азидной группы. В спектре ЯМР 13С азида 4 наблюдается пик при 70 м.д., соответствующий сигналу фенильной группы, связанной с N3. Для проведения клик-реакции необходимо защитить полость порфиринового макроцикла, так как в клик-реакции участвуют соли меди, способные к комплексообразованию с исходным порфирином. Поэтому на основе порфирина 4 стандартным методом был получен его комплекс цинка 5.
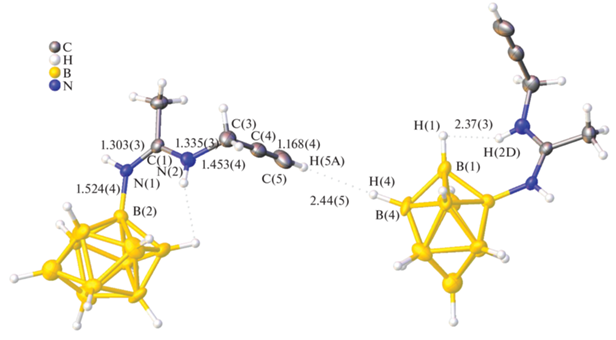
Для синтеза соединения 6 использовали подход, основанный на процессе нуклеофильного присоединения первичных аминов к активированным тройным связям нитрилиевых производных клозо-декаборатного аниона [13]. В данном случае в качестве нуклеофила использовали пропаргиламин. Реакция протекает в мягких условиях и позволяет селективно получать производные с терминальной тройной связью. Соединение было охарактеризовано методами мультиядерной ЯМР-спектроскопии и ESI-масс-спектрометрии. Кроме того, структура полученного соединения была установлена методом рентгеноструктурного анализа (рис. 1). Так, структура амидина 6 построена из тетрабутиламмониевых катионов и замещенных клозо-декаборатных анионов. Экзополиэдрический заместитель в анионе расположен в экваториальном поясе. Длина связи B(1)N(1) составляет 1.524 Å, что соответствует ординарной связи бор–азот [28]. Амидиновый заместитель плоский, длины связей N(1)C(1) и C(1)N(2) составляют 1.303 и 1.335 Å соответственно, что указывает на наличие сопряжения в заместителе. Пропаргильная группа имеет линейную геометрию и параметры связей, характерные для данного типа заместителей. Стоит отметить, что структура стабилизирована за счет диводородных связей. Так, Z-конфигурация амидинового фрагмента обусловлена образованием внутримолекулярной связи между протоном аминогруппы и апикальным атомом водорода кластера N(2)H(2D)…H(1)B(1) (2.37 Å). Протон терминальной тройной связи участвует в образовании межмолекулярного протон-гидридного взаимодействия с атомом водорода соседнего кластера C(5)H(5A)…H(4')B(4') (2.44 Å). Данный тип связи обусловливает образование полимерных цепей из замещенных анионов.
Ключевой этап в синтезе конъюгата порфирина с замещенным производным клозо-декаборатного аниона состоял в проведении реакции 1,3-биполярного циклоприсоединения производных кластерных анионов бора с терминальной тройной связью к азидопорфирину. В качестве ацетиленовой компоненты использовали соединение 6 (схема 2 ). Катализатором реакции служила водная смесь медного купороса и аскорбата натрия, генерирующая соль одновалентной меди in situ. Были подобраны оптимальные условия проведения данной реакции. Так, использовали два существующих подхода: кипячение в системе вода–ТГФ или перемешивание при небольшом нагреве (40°С) в системе ДМФА–трет-бутиловый спирт. Реакционную массу подвергали экстракции. Выход соединения 6 составил 88%, что свидетельствует о высокой эффективности синтеза.
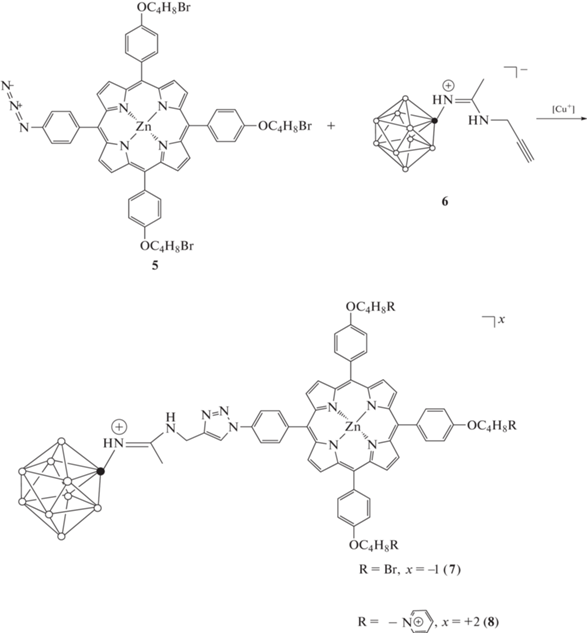
Схема 2 .
Конечная стадия получения положительно заряженного производного борсодержащего порфиринового конъюгата (соединение 8) заключается в проведении реакции кватернизации пиридином бромзамещенного порфирина 7. Катионный порфирин с терминальными пиридиниевыми группами, отделенными от макроцикла алифатическими спейсерами, синтезировали в соответствии со стандартной методикой кватернизации при кипячении в пиридине. Целевой конъюгат в ходе реакции выпадает в осадок. Продукты синтеза очищали и выделяли путем перекристаллизации в системе диэтиловый эфир–метанол. Выход соединения 8 составил 95%. Структура и индивидуальность полученных соединений подтверждены методами ТСХ, 1Н ЯМР- и УФ-спектроскопии, а также ESI масс-спектрометрии.
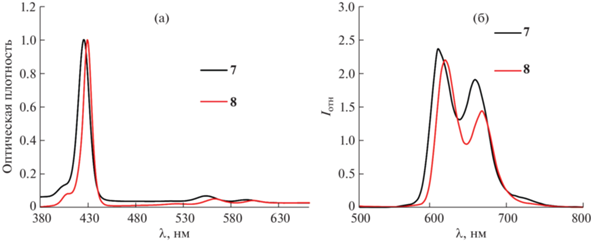
В электронных спектрах поглощения полученных борсодержащих конъюгатов наблюдается батохромный сдвиг полос порядка 8–10 нм по сравнению со спектрами исходных порфиринов (рис. 2а). Так, значение полосы Соре для соединения 5 составляет 420 нм, тогда как для конъюгата 8 – 429 нм. Данное явление можно объяснить сопряжением между кластерным анионом бора и порфирином. В спектрах флуоресценции для конъюгата 8 также наблюдается батохромный сдвиг полос (10–15 нм) и небольшое уменьшение интенсивности флуоресценции (рис. 2б).
ЗАКЛЮЧЕНИЕ
Таким образом, разработана стратегия, позволяющая получать водорастворимые конъюгаты порфиринов и производных клозо-декаборатного аниона, установлены оптимальные условия проведения реакции, выделения продуктов, все полученные соединения исследованы современными физико-химическими методами.
Клик-реакции являются перспективными методиками для дальнейшего получения конъюгатов с азидозамещенными порфиринами, позволяющими получать целевые соединения с высоким выходом.
Список литературы
Vaccarella S., Georges D., Bray F. et al. // The Lancet Regional Health – Europe. 2022. P. 100551. https://doi.org/10.1016/j.lanepe.2022.100551
Prasad R., Jain N.K., Conde J. et al. // Mater. Today Adv. 2020. 100087. https://doi.org/10.1016/j.mtadv.2020.100087
Trotti A., Pajak T.F., Gwede C.K. et al. // The Lancet Oncology. 2007. V. 8. № 7. P. 613. https://doi.org/10.1016/S1470-2045(07)70144-4
Chen C., Wu C., Yu J. et al. // Coord. Chem. Rev. 2022. V. 461. P. 214495. https://doi.org/10.1016/j.ccr.2022.214495
Zhdanova K.A., Ivantsova A.V., Vyalba F.Yu. et al. // Pharmaceutics. 2023. V. 15. P. 269. https://doi.org/10.3390/pharmaceutics15010269
Barth R., Mi P., Yang W. et al. // Cancer Commun. 2018. V. 38. № 35. P. 1. https://doi.org/10.1186/s40880-018-0299-7
Suzuki M. // Int. J. Clin. Oncol. 2020. V. 25. P. 43. https://doi.org/10.1007/s10147-019-01480-4
Dąbrowski J.M., Arnaut L.G. // Photochem. Photobiol. Sci. 2015. V. 14. № 10. P. 1765. https://doi.org/10.1039/c5pp00132c
Mironov A.F., Zhdanova K.A., Bragina N.A. // Russ. Chem. Rev. 2018. V. 87. P. 859. https://doi.org/10.1070/RCR4811
Worm D.J., Hoppenz P., Els-Heindl S. et al. // J. Med. Chem. 2020. V. 63. № 5. P. 2358. https://doi.org/10.1021/acs.jmedchem.9b01136
Zhou Z., Zhang L., Zhang Z. et al. // Asian J. Pharm. 2021. V. 16. P. 668. https://doi.org/10.1016/j.ajps.2020.12.003
Matveev E.Yu., Novikov S.S., Levitskaya V.Y. et al. // Fine Chemical Technologies. 2022. V. 17. № 5. P. 427. https://doi.org/10.32362/2410-6593-2022-17-5-427-438
Нелюбин А.В., Соколов М.С., Селиванов Н.А. и др. // Журн. неорган. химии. 2022. Т. 67. С. 1562.
Нелюбин А.В., Селиванов Н.А., Быков А.Ю. и др. // Журн. неорган. химии. 2022. Т. 67. С. 1588.
Kanygin V., Zaboronok A., Taskaeva I. et al. // J. Fluor. 2021. V. 31. P. 73. https://doi.org/10.1007/s10895-020-02637-5
Matsumoto Y., Hattori K., Arima H. et al. // Appl. Radiat. Isot. 2020. V. 163. P. 109201. https://doi.org/10.1016/j.apradiso.2020.109201
Li Y., Chyan M.-K., Hamlin D.K. et al. // Nucl. Med. Biol. 2021. V. 92. P. 217. https://doi.org/10.1016/j.nucmedbio.2020.04.005
Lee W., Sarkar S., Ahn H. et al. // Biochem. Biophys. Res. Commun. 2020. V. 522. № 3. P. 669. https://doi.org/10.1016/j.bbrc.2019.11.144
Korolkov I.V., Ludzik K., Kozlovskiy A.L. et al. // Mater. Today Commun. 2020. V. 24. P. 101247. https://doi.org/10.1016/j.mtcomm.2020.101247
Авдеева В.В., Гараев Т.М., Малинина Е.А. и др. // Журн. неорган. химии. 2022. Т. 67. С. 35.
Zhdanova K.A., Zhdanov A.P., Ezhov A.V. et al. // Russ. Chem. Bull. 2014. V. 63. № 1. P. 194. https://doi.org/10.1007/s11172-014-0413-1
Ezhov A.V., Vyal’ba F.Yu., Kluykin I.N. et al. // Macroheterocycles. 2017. V. 10. № 4–5. P. 505. https://doi.org/10.6060/mhc171254z
Rostovtsev V.V., Green L.G., Fokin V.V. et al. // Angew. Chem. Int. Ed. 2002. V. 41. № 14. P. 2596. https://doi.org/10.1002/1521-3773(20020715)41:14<2596:: AID-ANIE2596>3.0.CO;2-4
Polevaya Y.P., Tyurin V.S., Beletskaya I.P. // J. Porphyrins Phthalocyanines. 2014. V. 18. № 1. P. 20. https://doi.org/10.1142/S1088424613500636
Sheldrick G.M. // Acta Crystallogr. 2015. V. 71. P. 3. https://doi.org/doi.org/10.1107/S2053273314026370
Dolomanov O.V., Bourhis L.J., Gildea R.J. et al. // J. Appl. Crystallogr. 2009. V. 42. № 2. P. 339. https://doi.org/10.1107/S0021889808042726
Lindsey J.S., Hsu H.C., Schreiman I.C.// Tetrahedron Lett. 1986. V. 27. P. 4969. https://doi.org/10.1016/s0040-4039(00)85109-6
Allen F.H., Kennard O., Watson D.G. et al. // J. Chem. Soc., Perkin Trans. II. 1987. P. S1. https://doi.org/10.1039/p298700000s1
Дополнительные материалы отсутствуют.
Инструменты
Журнал неорганической химии