

Поверхность. Рентгеновские, синхротронные и нейтронные исследования, 2019, № 8, стр. 86-96
Структурные, морфологические и сорбционные характеристики нанокристаллического дефектного гидроксиапатита кальция, основы для создания стоматологических биомиметических композитов
Д. Л. Голощапов 1, *, А. С. Леньшин 1, К. А. Никитков 1, В. Н. Бартенев 1, Д. В. Савченко 2, 3, Е. А. Тутов 4, П. В. Середин 1, 5, **
1 Воронежский государственный университет
394018 Воронеж, Россия
2 Institute of Physics of the Czech Academy of Sciences (CAS)
18221 Prague, Czech Republic
3 National Technical University of Ukraine “Igor Sikorsky Kyiv Polytechnic Institute”
03056 Kyiv, Ukraine
4 Воронежский государственный технический университет
394006 Воронеж, Россия
5 Уральский федеральный университет
620002 Екатеринбург, Россия
* E-mail: goloshchapov@phys.vsu.ru
** E-mail: paul@phys.vsu.ru
Поступила в редакцию 28.01.2019
После доработки 25.02.2019
Принята к публикации 28.02.2019
Аннотация
С применением комплекса структурно-спектроскопических методов анализа изучены взаимосвязи между структурой, морфологией и сорбционными характеристиками образцов нанокристаллического карбонатзамещенного гидроксиапатита кальция, синтезированного с использованием биогенного источника кальция (яичной скорлупы птиц) методом химического осаждения из раствора. Методами рентгеновской дифрактометрии и рентгеноспектрального микроанализа установлено, что примеси попадают в синтезированный материал из яичной скорлупы птиц, что ведет к изменению параметров элементарной ячейки кристалла гидроксиапатита без образования дополнительных фосфатных фаз. Методами оптической спектроскопии и электронного парамагнитного резонанса установлено, что в результате наследования набора примесей карбонат-ионов образуется структура гидроксиапатита В-типа. Увеличение содержания фосфатных групп PO4 в процессе синтеза материалов в атмосфере приводит к уменьшению содержания структурно связанных групп СО3. Понижение величины рН раствора на этапе синтеза, связанное с повышением содержания анионов ${\text{Р О }}_{4}^{{3-}},$ влияет на морфологию образцов за счет увеличения размера дефектов – нанопор на поверхности нанокристаллов. Подобное изменение морфологии материалов приводит к изменениям в сорбционных характеристиках образцов, определяемых методом тепловой десорбции азота. Площадь удельной поверхности порошков составляет ~55.5 ± 0.9 м2/г, что превышает в разы известные аналоги. Несмотря на развитую поверхность, образцы карбонатзамещенного гидроксиапатита кальция остаются стабильными в атмосфере насыщенных паров воды, а основные потери при поляризации связаны с потерями Максвелла–Вагнера. Анализ характеристик полученных из биогенного источника кальция образцов карбонатзамещенного гидроксиапатита показывает их потенциальную значимость для создания биомиметических материалов, имитирующих структуру, морфологию и анизотропию нативных твердых тканей человеческого зуба.
ВВЕДЕНИЕ
Использование наноструктурированных композитов при решении задач терапевтической стоматологии, связанных с реставрацией и регенерацией нативной твердой ткани, обусловлено иерархической природой и функциональными характеристиками зубной эмали и дентина [1–3]. Органоминеральные биокомпозиты неродственного твердой ткани зуба состава активно используются при создании стоматологических цементов и керамик [3, 4]. Следует отметить, что их морфология, прочностные и износостойкие характеристики отличаются от свойств нативной ткани зуба. Терапия с использованием этих материалов часто приводит к появлению трещин, деминерализации, вторичному кариесу, эрозии и механической деформации зубов [1, 5]. Несмотря на интенсивные исследования в области биоматериаловедения, данная проблема остается неразрешенной и по-прежнему актуальной.
Современные исследования минеральной и органической составляющей зубов демонстрируют перспективность использования биокомпозитов с максимально близкими к зубной ткани свойствами [1, 6–8]. Поэтому в состав разрабатываемых, а также уже созданных органоминеральных биомиметических композитов для стоматологии непременно входит аналог минеральной составляющей твердой ткани зуба – гидроксиапатит кальция [3, 8, 9]. Он не только обладает уникальной структурой, но и широким диапазоном требуемых характеристик в различных формах благодаря самым разнообразным способам и методикам получения, что позволяет ему выступать перспективным модификатором стоматологических керамик и цементных масс [8, 10, 11]. Хорошо известно, что физико-химические свойства гидроксиапатита кальция и его морфологическая организация могут достаточно широко варьироваться за счет способа получения и типа вносимых в структуру примесей [3, 12–14], а также используемых для его получения источников [15–17].
Поэтому важной задачей в области современного стоматологического материаловедения и фундаментальной медицины является поиск методик синтеза гидроксиапатита кальция, который может стать основой высокотропных к интактной ткани биомиметических композитов, воспроизводящих структурные, физико-химические, морфологические, оптические и биомеханические свойства, имитирующие структуру (в том числе на наноуровне), иерархию и особенности анизотропии твердых тканей зуба человека, а также способных образовывать устойчивую функциональную связь с нативной тканью [7, 8, 18].
Одним из возможных способов получения гидроксиапатита кальция является использование в качестве источника кальция биогенных материалов [15, 16, 19]. Целесообразность данного подхода заключается в соответствии получаемых материалов по структурным и морфологическим характеристикам и элементному составу апатиту костной ткани человеческого организма. Однако физико-химические свойства подобных образцов гидроксиапатита кальция требуют всестороннего исследования, так как исходные биогенные материалы, обладающие собственной морфологией, структурой, разнообразным элементным и фазовым составом, оказывают огромное влияние на финальные свойства синтезируемых образцов [20, 21]. Литературный обзор показывает, что существует огромное количество работ, посвященных исследованиям морфологии, размерного фактора, структурной организации, влияния типа внедренных примесей на свойства гидроксиапатита [13, 22–24]. Однако мало исследованной областью остаются сорбционные свойства синтезированных гидроксиапатитов кальция [25], которые играют огромную роль и имеют важное значение при создании стоматологических цементов нового класса, поскольку данная характеристика определяет его усадку и адгезию к нативной ткани при реставрации зуба.
Как было показано ранее [26–31], гидроксиапатит кальция с использованием яичной скорлупы может быть получен методом химического осаждения в наноформе. Однако такие образцы могут содержать примеси различных ионов. В частности, в ряде работ [28, 31] указывается на возможное включение карбонат-аниона в структуру гидроксиапатита кальция при получении образцов на воздухе. Следует отметить, что при использовании карбоната кальция и ортофосфорной кислоты возможен синтез карбонатзамещенного гидроксиапатита с содержанием карбонат-аниона ~8 мас. % [32]. Морфологически, с использованием яичной скорлупы могут быть получены различные материалы со средними размерами нанокристаллов 40–100 нм [30, 31], в том числе пластинчатой формы, толщиной 8–15 нм и диаметром 30–5 нм [26]. Следует отметить, что с применением методов “мокрой” химии возможно создание материалов с более развитой удельной поверхностью, чем при высокотемпературной обработке натуральных костей животных [33], что является преимуществом данной методики при обработке биогенных источников.
Поэтому целью настоящей работы было установление зависимости сорбционных, морфологических и структурных характеристик нанокристаллического гидроксиапатита кальция от параметров синтеза и последующей обработки, а также возможностей использования финального материала для создания стоматологических биомиметических композитов.
МАТЕРИАЛЫ И МЕТОДЫ ИССЛЕДОВАНИЯ
Технология синтеза. Нанокристаллический гидроксиапатит кальция был получен с использованием яичной скорлупы птиц по методике, описанной в [15]. Изменение концентрации включенных карбонат-групп контролировали по изменению содержания заранее рассчитанного количества раствора ортофосфорной кислоты 0.3 M и значения pH конечного раствора от 9 до 7 с шагом 0.5 с применением рН-метра/ионометра ИПЛ 111-1. Поскольку при создании стоматологических цементов, а также биомиметических материалов образцы гидроксиапатита кальция зачастую подвергают прессованию, порошкообразные материалы прессовали при давлении 250 бар в стальной пресс-форме без использования пластификатора.
Рентгеноструктурный анализ. Дифрактометрические исследования с высоким разрешением были проведены на канале ANKA-PDIFF синхротрона ANKA центра KIT KNMF (Karlsruhe, Германия). Источником излучения служил поворотный магнит, генерирующий магнитное поле с индукцией 1.5 Tл (Ес = 6 кэВ). В эксперименте использовали монохроматическое излучение, соответствующее Kα1-излучению кобальта с длиной волны λ = 1.7902 Å. Поток на образец сфокусированного излучения с энергией 10 кэВ составлял ~2 × 1016 Вт/м2 при токе в канале 100 мА. Размер поперечного сечения пучка, падающего на образец, ~0.5 × 0.5 мм. Рентгенофазовый анализ (РФА) проводили с использованием базы данных JCPDS–ICDD 2018 г.
Рентгеноспектральный микроанализ. Элементный анализ полученных образцов исследовали методом рентгеноспектрального анализа (РСМА) с помощью приставки Inca-250 к растровому электронному микроскопу JEOL JSM-6380LV.
Инфракрасная спектроскопия и комбинационное рассеяние света. ИК-спектры образцов были получены с использованием приставки Diamond ATR с алмазной призмой к ИК-фурье-спектрометру VERTEX 70 BRUKER для изучения нарушенного полного внутреннего отражения. Спектры пропускания были зарегистрированы с разрешением 4 см–1 в диапазоне 400–4000 см–1. Спектры комбинационного рассеяния света (КРС) были зарегистрированы с помощью спектрометра Raman Microscope RamMics M532 EnSpectr в области 350–3650 см–1 с использованием лазера с длиной волны 532 нм.
Электронный парамагнитный резонанс. С целью определения радикалов в структуре полученных образцов нанокристаллического гидроксиапатита кальция были записаны спектры стационарного и эхо-детектированного электронного парамагнитного резонанса с помощью спектрометра Bruker E580. Метод эхо-детектирования применяли с целью разделения перекрывающихся компонент электронных парамагнитных спектров при g = 2.0000.
Просвечивающая электронная микроскопия. Метод просвечивающей электронной микроскопии (ПЭМ) был использован для непосредственной оценки размеров нанокристаллов синтезированных образцов. Исследование проводили в электронном микроскопе Libra 120 Carl Zeiss. При подготовке к исследованию образцы подвергали ультразвуковому воздействию (~1 ГГц).
Тепловая десорбция азота. Для оценки пористости порошкообразных образцов использовали метод тепловой десорбции азота. Результаты измерений объема газа (азота), сорбируемого на исследуемых образцах при четырех различных значениях парциального давления, рассчитывали с использованием программного обеспечения UniSorbi для прибора СОРБИ-MS по теории Брунауера–Эммета–Теллера (БЭТ).
Диэлектрическая спектроскопия. Для проведения экспериментов образцы гидроксиапатита кальция спрессовывали под давлением 250 бар с оловянной фольгой для формирования плоского конденсатора. Образованный конденсатор помещали в сосуд с насыщенными парами воды, давление которых постоянно контролировали. Диэлектрические характеристики изучали на приборе LCR-meter GW Insteck LCR-819.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Рентгенофазовый анализ. Влияние концентрации фосфатных групп в конечном растворе на фазовый состав получаемых образцов было изучено методом РФА. Результаты показали, что для всех порошкообразных образцов, полученных при значениях рН раствора от 9 до 7 с шагом 0.5 и отожженных при температуре 400°С, характерно образование единственной фазы – гидроксиапатита кальция (рис. 1 кривые 1 и 2). Анализ данных рентгеновской дифракции и данных из базы стандартов JCPDS–ICDD (карточка № 01-074-0565) показывает, что в образцах возникают искажения кристаллической решетки, вызванные включением в элементарную ячейку примесных атомов, входящих в состав прекурсора гидроксиапатита – биогенного карбоната кальция яичной скорлупы птиц.
Рис. 1.
Дифрактограммы образцов гидроксиапатита, полученных при рН 7 (1) и 9 (2) и отожженных при температуре 400°С.
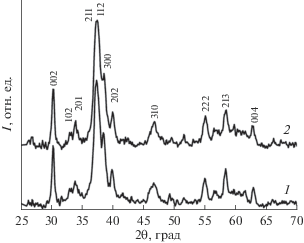
Анализ структурных изменений, вызванных прессованием исходного порошкообразного гидроксиапатита для проведения сорбционных характеристик, был выполнен по данным рентгеновской дифрактометрии. В качестве примера на рис. 2 представлена типичная дифрактограмма порошка гидроксиапатита кальция, полученного из раствора с рН 8.5 (кривая 1) и спрессованной таблетки (кривая 2). Результаты показывают, что после прессования порошка на дифрактограмме образца хорошо видно перераспределение интенсивностей основных дифракционных рефлексов. Эксперимент демонстрирует уменьшение относительной интенсивности дифракционных рефлексов 002 и 004 и возрастание интенсивности рефлекса 300. Данный факт связан с образованием текстуры в образцах гидроксиапатита кальция после прессования под давлением 250 бар.
Рис. 2.
Дифрактограммы образца гидроксиапатита, полученного из раствора с рН 8.5 и отожженного при температуре 400°С, в форме: порошка (1); спрессованной таблетки (2).

Рентгеноспектральный микроанализ. В табл. 1 представлены результаты РСМА скорлупы яиц, усредненной по 10 образцам. Как видно из полученных данных, в составе исходного реагента (яичной скорлупы) присутствуют атомы магния, серы, калия и хлора, которые способны встраиваться в структуру синтезируемого материала и образовывать дефекты. Исследования полученных по описанной технологии образцов гидроксиапатита кальция методом РСМА продемонстрировали, что в составе синтезированных материалов обнаруживаются только атомы магния в следовой концентрации, тогда как атомы серы, калия и хлора указанным методом не регистрируются. Данный факт свидетельствует о том, что в процессе синтеза часть содержащихся в исходном растворе примесных атомов удаляется из конечного материала или присутствует в нем еще в более низкой концентрации.
Таблица 1.
Элементный состав образцов яичной скорлупы птиц, использованной для получения карбонатзамещенного гидроксиапатита кальция
| Концентрация элементов, ат. % | |||||||
|---|---|---|---|---|---|---|---|
| Ca | P | O | C | Mg | S | K | Cl |
| 10.6 ± 2.3 | 1.4 ± 1.1 | 55.7 ± 4.60 | 30.9 ± 4.70 | 1.0 ± 0.40 | 0.1 ± 0.04 | 0.1 ± 0.00 | 0.1 ± 0.00 |
ИК-спектроскопия. Оптические исследования синтезированных и подготовленных образцов гидроксиапатита кальция позволили уточнить молекулярный состав материалов, а также их тонкие структурные свойства. На рис. 3 представлены ИК-спектры образцов, полученных при значениях рН 7 (кривая 1) и 9 (кривая 2) и отожженных при температуре 400°С. Как видно из рис. 3, в ИК-спектрах образцов наблюдается один и тот же набор колебательных полос. Частота основных колебаний не изменяется, а также не происходит и перераспределение их интенсивности. Этот результат находится в хорошем соответствии с данными РФА о существовании во всех образцах (отожженных при 400°С) лишь одной фазы – гидроксиапатита кальция.
Рис. 3.
ИК-спектры образцов гидроксиапатита, полученных при рН 7 (1) и 9 (2) и отожженных при температуре 400°С.

Следует отметить, что для всех полученных образцов характерной особенностью является присутствие в спектрах колебательных полос группы СО3 при 1415 и 1450 см–1. Хорошо известно, что появление этих колебательных мод в спектрах гидроксиапатита кальция связано с замещением в кристаллической решетке гидроксиапатита группы РО4 группой СО3 и образованием карбонатзамещенного гидроксиапатита B-типа. Об этом свидетельствует и тот факт, что с понижением величины рН раствора, использованного при получении материалов, уменьшается и интенсивность колебательной полосы включенного в структуру гидроксиапатита кальция карбонат-аниона.
Спектроскопия КРС. В ряде работ по исследованию карбонатзамещенного гидроксиапатита кальция [34, 35] было показано, что в случае появления искажения в кристаллической решетке гидроксиапатита кальция и включении аниона СО3 в его структуру в спектрах КРС наблюдался сдвиг полос РО4. Образцы, полученные в настоящей работе при различных значениях рН 9, 8.5, 8, 7.5, 7, в которых по данным ИК-спектроскопии содержание группы СО3 может варьироваться в широких пределах, были исследованы методом спектроскопии КРС.
Как видно из рис. 4, в спектрах КРС образцов, полученных при различных значениях рН раствора, кроме основных мод, соответствующих фосфорно-кислородной группе PO4 и локализованных около 961, 587 и 431 см–1, наблюдается дополнительная мода в области ~1070 см–1. Ее появление в спектрах можно соотнести с карбонат-анионом ${\text{CO}}_{3}^{{2 - }}$. Детальное рассмотрение формы и интенсивности колебаний, наблюдаемых в области 1000–1150 см–1 и относимых к группе РО4 (1030, 1048 см–1) и группе СО3 (1073 см–1) показывает, что с увеличением интенсивности моды РО4 в спектре (образец, полученный при рН 7, т.е. с большим содержанием фосфатных групп) снижается интенсивность полосы группы СО3. Этот факт хорошо коррелирует с данными ИК-спектроскопии о природе карбонатзамещенного гидроксиапатита кальция В-типа. Наблюдаемые в ИК- и спектрах КРС колебательные моды представлены в табл. 2.
Рис. 4.
Спектры КРС образцов, полученных при рН 7 (1) и 9 (2) и отожженных при температуре 400°С, λвозб = 532 нм.

Таблица 2.
Наблюдаемые в спектрах ИК и КРС моды
| Мода КРС | Сдвиг КРС, см–1 | Молекулярная группа | ИК-полоса | ω, см–1 |
|---|---|---|---|---|
| υ2 PO4 | 431 | Изгиб O–P–O, υ2 | – | – |
| υ2 PO4 | 447 | Изгиб O–P–O, υ2 | – | – |
| υ4 PO4 | 579 | Изгиб O–P–O, υ4 | PO4 ν 4 | 573 |
| υ4 PO4 | 590 | Изгиб O–P–O, υ4 | PO4 ν 4 | 602 |
| υ4 PO4 | 607 | Изгиб O–P–O, υ4 | – | – |
| υ4 PO4 | 614 | Изгиб O–P–O, υ4 | – | – |
| – | – | OH | OH | 627 |
| P–(OH) | 880–885 | HPO4 | CO3 (ν3) | 872 |
| υ1 PO4 | 962 | Растяжение P–O | PO4 ν1 | 963 |
| HPO4 | 1005 | Симметричное растяжение | – | – |
| υ3 PO4 | 1028 | PO4 | – | – |
| υ3 PO4 | 1040 | Плечо P–O, антисимметричное растяжение | – | – |
| υ3 PO4 | 1047 | Самая интенсивная мода υ3 PO4 | – | – |
| υ3 PO4 | 1052 | Плечо P–O, антисимметричное растяжение | – | – |
| υ1 СO3, B-тип | 1070–1072 | B-тип СO3 | – | – |
| υ3 PO4 | 1076–1077 | Антисимметричное растяжение P–O | – | – |
| υ1 СO3, A-тип | 1106 | А-тип | – | – |
| – | – | PO4 | PO4 ν3 | 1090 |
| – | – | CO3 | CO3 (ν3) | 1415 |
| – | – | CO3 | CO3 (ν3) | 1451 |
| OH | 3570 | OH | OH | 3570 |
Электронный парамагнитный резонанс. Для уточнения данных о радикалах, присутствующих в образцах, был использован метод ЭПР. Следует отметить, что сигналы ЭПР в полученных образцах карбонатзамещенного гидроксиапатита кальция до облучения не наблюдались. Для регистрации сигнала ЭПР высушенные при 400°С (1 ч) образцы были подвергнуты облучению электронами с дозой 10 кГр. Результаты были получены методами стационарного и эхо-детектированного ЭПР через семь дней после облучения.
Для определения природы радикалов было проведено моделирование спектров ЭПР всех образцов. Это позволило установить, что в общей сложности в спектрах ЭПР наблюдаются пять сигналов: от радикалов O–, CO2–, CO3–, ${\text{CO}}_{3}^{{3--}}$ [36] и от органического радикала CH3 [37]. Параметры парамагнитных радикалов, наблюдаемых в спектрах ЭПР исследуемых образцов, интерпретировали в соответствии с [36–39], они приведены в табл. 3.
Таблица 3.
Параметры парамагнитных радикалов, наблюдаемых в спектрах ЭПР в образцах гидроксиапатита кальция при Т = 300 К
| Парамагнитный радикал | Парамагнитный центр [4] | Положение [1–4] | g1 | g2 | g3 | A1 = A2= A3, мТл | Ссылка |
|---|---|---|---|---|---|---|---|
| O– | О2– | В прослойках
вблизи поверхности нанокристаллов |
2.0422(5) | 2.0333(5) | 2.0023(5) | – | [36, 38] |
| ${\text{CO}}_{2}^{--}$ | ${{{\text{С О }}_{2}^{0}} \mathord{\left/ {\vphantom {{{\text{С О }}_{2}^{0}} {{\text{С О }}_{3}^{{2--}}}}} \right. \kern-0em} {{\text{С О }}_{3}^{{2--}}}}$ | 2.0032(3) | 2.0020(3) | 1.9977(3) | – | [36] | |
| ${\text{CO}}_{3}^{--}$ | ${\text{CO}}_{3}^{{2--}}$ | 2.016(1) | 2.0100(3) | 2.004(1) | – | [39] | |
| ĊH3 | – | 2.0029(3) | 2.0029(3) | 2.0029(3) | 2.23 | [37] | |
| ${\text{CO}}_{3}^{{3--}}$ | ${\text{CO}}_{3}^{{2--}}$ | В позициях групп ${\text{PO}}_{4}^{{3--}}$ | 2.0048(3) | 2.0032(3) | 2.0017(3) | – | [36] |
Было установлено, что при изменении содержания фосфорно-кислородных групп в образцах спиновая концентрация парамагнитных карбонатных радикалов CO2–, CO3–, ${\text{CO}}_{3}^{{3--}}$ (так же как и интенсивность их парамагнитных центров по данным ИК- и спектроскопии КРС) возрастает, в то время как спиновая концентрация радикалов O– и ĊH3 уменьшается.
Просвечивающая электронная микроскопия. Внутрирешеточные замещения в полученных образцах и образование карбонатзамещенного гидроксиапатита кальция должны обязательно отразиться на наноструктуре материала. Для того чтобы уточнить кристаллическую структуру, а также установить фазовый состав отдельного нанокристаллита в синтезированных образцах, был использован метод ПЭМ высокого разрешения. Были изучены образцы, полученные с различными значениями рН 9, 8.5, 8, 7.5, 7 и отожженные при 400°С, которые дополнительно подвергались ультразвуковому диспергированию в водной среде. Эксперимент показал, что образовавшиеся нанокристаллы всех исследованных образцов имеют стержневидную форму, их диаметр ~20 нм и длина ~50 нм. На рис. 5 и 6 представлены изображения нанокристаллов, полученных из растворов с pH 7 и 9 и отожженных при 400°С. Детальное исследование образцов карбонатзамещенного гидроксиапатита кальция методом ПЭМ с нанометровым разрешением показало пористую структуру этих материалов (рис. 5, 6).


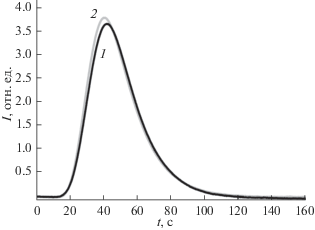
Тепловая десорбция азота. Хорошо известно, что развитая поверхность пористых карбонатзамещенных гидроксиапатитов кальция является важной характеристикой такого рода образцов, используемых в стоматологическом материаловедении, в том числе при создании биомиметических материалов. Поэтому были определены удельные площади поверхности синтезированных порошков с использованием метода тепловой десорбции азота. Оценки удельных площадей проведены в приближении одинакового размера частицы. На рис. 7 представлены кривые тепловой десорбции азота из материалов, синтезированных при значениях рН 7 и 9 и отожженных при 400°С (кривые 1 и 2 соответственно). Расчет удельной поверхности для всех порошков методом БЭТ показал, что образцы, полученные при различных значениях рН 9, 8.5, 8, 7.5, 7, имеют одинаковую площадь удельной поверхности ~55.4 ± 0.9 м2/г.
Рис. 7.
Зависимости тепловой десорбции азота из образцов карбонатзамещенного гидроксиапатита кальция, полученных при значениях рН 7 (1) и 9 (2), а также отожженных при 400°С, p/p0 = 0.09.
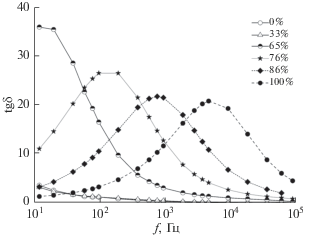
Диэлектрическая спектроскопия. Развитая поверхность образцов карбонатзамещенного гидроксиапатита кальция, а также тот факт, что гидроксиапатит является широкозонным диэлектриком (Eg = ~7.7 эВ), однозначно должны отразиться на его электрохимических свойствах. Наиболее удобным методом исследования выступает импеданс-спектроскопия, с использованием которой уже было установлено, что в отожженных материалах гидроксиапатита вид зависимости тангенса угла диэлектрических потерь tg δ определяется наличием структурных дефектов, возникающих в ходе его получения [40]. Однако при использовании материалов на основе гидроксиапатита кальция в реальных условиях часто имеют дело с влажными средами. Поэтому для изучения зависимости tg δ от частоты электрического поля полученные образцы исследовали в условиях контролируемой сорбции паров воды.
Полученные результаты измерения тангенса угла диэлектрических потерь показали (рис. 8), что в интервале частот 10 Гц–100 кГц и диапазоне относительной влажности p/ps 0–100% проводимость образцов карбонатзамещенного гидроксиапатита определяется сорбированной водой. Особенность тангенса угла диэлектрических потерь, вызванных поляризацией Максвелла–Вагнера, заключается в появлении в экспериментальных спектрах широкой полосы с явно выраженным максимумом. Положение этого максимума смещается в сторону высоких частот при возрастании относительной влажности p/ps в диапазоне 65–100%. Подобный эффект отмечается и для схожих по конструкции систем на основе гидроксиапатита кальция [41].
Рис. 8.
Зависимость тангенса угла диэлектрических потерь tg δ от частоты f в образцах карбонатзамещенного гидроксиапатита кальция при различной влажности p/ps.
Обсуждение результатов. Сопоставление полученных методом РФА результатов для всех образцов позволяет сделать заключение, что изменение концентрации фосфатных групп в исходном растворе не приводит к изменению фазового состава конечного материала, однако проявляется в искажении кристаллической решетки гидроксиапатита. Для всех образцов гидроксиапатита кальция были определены параметры их элементарной ячейки. Полученные результаты, а также данные из базы данных JCPDS–ICDD представлены в табл. 4.
Таблица 4.
Параметры элементарной ячейки полученных образцов гидроксиапатита в сравнении с образцами из базы данных JCPDS–ICDD
| Гидроксиапатит | a, b, Å | c, Å |
|---|---|---|
| ICDD № 09-432 | 9.418 | 6.884 |
| ICDD № 01-074-0565 | 9.424 | 6.879 |
| рН 9 | 9.406 | 6.871 |
| рН 7 | 9.404 | 6.864 |
Из сопоставления параметров элементарной ячейки видно, что синтезированные в работе материалы имеют меньшие значения параметров, чем аналогичные образцы, представленные в базе JCPDS–ICDD. Хорошо известно, что условия синтеза гидроксиапатита кальция влияют на процессы роста кристаллов, а это в свою очередь может приводить к изменению параметров кристаллической структуры [42]. Поэтому меньшие значения параметров элементарной ячейки a, b и c полученных в работе образцов можно связать с особенностью использованной методики синтеза гидроксиапатита кальция из яичной скорлупы птиц, а также с наличием примесей в ее составе (табл. 1). В различных работах показано, что скорлупа птиц содержит магний и фосфор в концентрации от ~1% [43]. Исходя из представленных экспериментальных данных видно, что в составе гидроксиапатита кальция также присутствуют примеси атомов S, K, Cl на уровне 0.1–0.2 ат. %. Эти примеси и группы в составе скорлупы способствуют созданию биомиметического композита, наиболее схожего по элементному составу с гидроксиапатитом костной и зубной ткани [44]. Изменение содержания фосфатных групп в процессе получения образцов и изменение величины рН на ±0.5 влияет на отношение Са/Р, что также обнаружено в ходе количественного анализа, проведенного методом РСМА, который подтвердил данные РФА и ИК-спектроскопии.
Данные ИК-спектроскопии (рис. 3 и табл. 2) согласуются с данными рентгеновской дифракции о том, что причиной искажений кристаллической решетки гидроксиапатита является внедрение примесных атомов и групп в его структуру. По данным ИК-спектроскопии установлено влияние включения карбонат-аниона в гидроксиапатит в случае пониженного содержания фосфатных групп в образце (рис. 3, кривые 1, 2). Полученные результаты соответствуют литературным данным [9, 32, 45, 46].
Аналогичные корреляции обнаруживаются при рассмотрении спектров КРС полученных образцов (рис. 4, кривые 1 и 2). На врезке выделен участок спектра, соответствующий модам РО4 и СО3 и наглядно показывающий, что при введении большего количества фосфатных групп в раствор интенсивность моды колебаний карбонат-аниона уменьшается, что совпадает с данными ИК-спектроскопии. Следует отметить, что как в ИК-спектрах зубной ткани, так и в спектрах КРС [47, 48] обнаруживаются полосы, соответствующие карбонат-аниону [7, 9], поэтому его наличие в образцах подтверждает возможность использования данных материалов в качестве основы для создания стоматологических биомиметических композитов.
Методом ЭПР, который является чувствительным к разнообразного рода структурным дефектам и часто применяется для изучения гидроксиапатита кальция и его замещенных форм [36, 38, 39, 49], подтверждено образование карбонатзамещенного гидроксиапатита B-типа для всех полученных образцов (табл. 3). Более того, согласно данным ЭПР с уменьшением отношения Ca/P в образцах с рН 9, 8.5, 8, 7.5, 7 интенсивность сигналов от радикалов CO2– и ${\text{CO}}_{3}^{{3--}}$ возрастает. Этот факт дополняет данные ИК- и спектроскопии КРС о зависимости содержания карбонат-аниона в кристаллической решетке гидроксиапатита.
Развитая поверхность полученных образцов карбонатзамещенного гидроксиапатита, обнаруженная методом ПЭМ, может быть использована для увеличения сорбционной емкости материалов и образования биоактивных композитов. Пористая структура характерна для образцов гидроксиапатита кальция, получаемых методами химического осаждения [50]. Нанопоры в карбонатзамещенном гидроксиапатите кальция являются 3D дефектом. Из рис. 5, 6 видно, что при уменьшении значения рН раствора размеры нанопор увеличиваются.
Высокая удельная поверхность полученных образцов может быть связана не только с особенностями состава и структуры используемого источника кальция при его получении (яичной скорлупы), но и с малыми размерами и особенностями морфологии нанокристаллов. Поскольку карбонатзамещенный гидроксиапатит кальция часто применяют в качестве наполнителя цементных паст пломбировочных материалов, высокая удельная поверхность может способствовать лучшему связыванию материала в составе цемента.
Что же касается характера изменения тангенса угла диэлектрических потерь в образцах, то следует отметить, что в известных аналогичных работах [51–55] по изучению беспримесного гидроксиапатита кальция изменения связывают с переориентацией групп ОН. Объяснение полученным в настоящей работе данным можно дать исходя из следующих предположений. Полагаем, что заряды в образцах карбонатзамещенного гидроксиапатита кальция под действием внешнего электрического переменного поля будут образовываться на границах раздела проводящих и изоляционных слоев. В данном случае границу между нанокристаллами гидроксиапатита и сорбированной водой можно рассматривать как совокупность диполей, направление моментов которых меняется вместе с изменением приложенного поля. Из эксперимента следует, что диэлектрические потери в образцах возрастают, когда период колебаний напряженности электрического поля становится сопоставим со временем релаксации поверхностной поляризации.
ЗАКЛЮЧЕНИЕ
На основании экспериментальных данных, полученных с применением комплекса структурно-спектроскопических методов анализа, удалось показать, что с использованием биогенного источника кальция (яичной скорлупы птиц) возможно получение наноразмерного карбонатзамещенного гидроксиапатита кальция с высокими сорбционными характеристиками. Методами просвечивающей электронной микроскопии установлено, что изменение величины рН раствора в процессе синтеза материалов ведет к изменению поверхности нанокристаллов и влияет на величину нанопор на их поверхности. Обнаруженные методом ЭПР в нанокристаллах структурные дефекты связаны с примесями, существующими в исходном реагенте – яичной скорлупе. Присутствие этих примесей в структуре конечного материала приводит к изменению параметров элементарной кристаллической ячейки. Наличие карбонат-ионов в структуре карбонатзамещенного гидроксиапатита кальция В-типа подтверждается методами ИК-спектроскопии и спектроскопии КРС и также заметно влияет на морфологию поверхности.
Расчет удельной поверхности порошков методом БЭТ по данным тепловой десорбции азота показал, что площадь удельной поверхности полученных образцов карбонатзамещенного гидроксиапатита кальция при значениях рН от 9 до 7 (шаг 0.5) составляет ~55.4 ± 0.9 м2/г, что превышает известные аналоги примерно в три раза и является важной характеристикой синтезированных материалов. Обнаруженная сорбционная способность порошков связана с методом получения и определяется развитой морфологией материала.
Исследования синтезированных и подвергнутых прессованию нанокристаллов карбонатзамещенного гидроксиапатита кальция, проведенные методом рентгеновской дифракции и импеданс-спектроскопии, позволяют предположить, что предобработанные, таким образом, порошки гидроксиапатита не изменяют своего фазового состава, однако весьма чувствительны к содержанию в них OH-групп.
Благодаря своим характеристикам синтезированные по представленной технологии из биогенного источника кальция образцы карбонатзамещенного гидроксиапатита кальция потенциально значимы для создания биомиметических материалов, близких по своей структуре к нативным твердым тканям человеческого зуба.
Список литературы
Xavier J.R., Desai P., Varanasi V.G. et al. // Nanotechnology in Endodontics / Ed. Kishen A. Springer International Publishing, 2015. P. 5.
Eliaz N., Metoki N. // Materials. 2017. V. 10. № 4. P. 334.
Dorozhkin S.V. Hydroxyapatite and Other Calcium Orthophosphates: Bioceramics, Coatings and Dental. N.Y.: Applications Nova Science Publishers, 2017. 462 p.
Mikhail S.S., Azer S.S., Schricker S.R. // Handbook of Nanomaterials Properties / Ed. Bhushan B., Luo D. et al. Berlin–Heidelberg: Springer, 2014. P. 1377.
Hamouda I.M., Shehata S.H. // J. Biomed. Res. 2011. V. 25. № 6. P. 418.
Hannig M., Hannig C. // Adv. Dental Res. 2012. V. 24. № 2. P. 53.
Seredin P.V., Goloshchapov D.L., Prutskij T., Ippolitov Y.A. // Res. Phys. 2017. V. 7. P. 1086.
Lübke A., Enax J., Wey K. et al. // Bioinspiration Biomimetics. 2016. V. 11. № 3. P. 035001.
Seredin P.V., Goloshchapov D.L., Kashkarov V.M. et al. // J. Surf. Invest.: X-ray, Synchrotron Neutron Tech. 2018. V. 12. № 3. P. 442.
Galamboš M., Suchánek P., Rosskopfová O. // J. Radioanal. Nucl. Chem. 2012. V. 293. № 2. P. 613.
Barandehfard F., Kianpour Rad M., Hosseinnia A. et al. // Ceramics Int. 2016. V. 42. № 15. P. 17866.
Kolmas J., Groszyk E., Kwiatkowska-Rozycka D. // BioMed Res. Int. 2014. V. 2014. P. 1.
Mouriño V., Cattalini J.P., Boccaccini A.R. // J. R. Soc. Interface. 2012. V. 9. № 68. P. 401.
Domashevskaya E.P., Al-Zubad A.A.i, Goloshchapov D.L. et al. // J. Surf. Invest. X-ray, Synchrotron Neutron Tech. 2014. V. 8. № 6. P. 1128.
Goloshchapov D.L., Kashkarov V.M., Rumyantseva N.A. et al. // Ceramics Int. 2013. V. 39. № 4. P. 4539.
Akram M., Ahmed R., Shakir I. et al. // J. Mater. Sc. 2014. V. 49. № 4. P. 1461.
Laonapakul T. // KKU Engin. J. 2015. V. 42. № 3. P. 269.
Seredin P.V., Goloshchapov D.L., Kashkarov V.M. et al. // Res. Phys. 2016. V. 6. P. 447.
Liu Q., Huang S., Matinlinna J.P. et al. // BioMed Res. Int. 2013. V. 2013. P. 1.
Matsumoto T., Tamine K., Kagawa R. et al. // J. Ceram. Soc. Jpn. 2006. V. 114. № 1333. P. 760.
Manoj M., Subbiah R., Mangalaraj D. et al. // Nanobiomedicine. 2015. V. 2. P. 1.
Šupová M. // Ceram. Int. 2015. V. 41. P. 9204.
Lakhkar N.J., Lee I.-H., Kim H.-W. et al. // Adv. Drug Delivery Rev. 2013. V. 65. № 4. P. 405.
Bose S., Fielding G., Tarafder S., Bandyopadhyay A. // Trends Biotechnol. 2013. V. 31. № 10. P. 594.
Turon P., del Valle L., Alemán C., Puiggalí J. // Appl. Sci. 2017. V. 7. № 1. P. 60.
Siva Rama Krishna D., Siddharthan A., Seshadri S.K., Sampath Kumar T.S. // J. Mater. Sci. Mater. Medicine. 2007. V. 18. № 9. P. 1735.
Prabakaran K., Balamurugan A., Rajeswari S. // Bull. Mater. Sci. 2005. V. 28. № 2. P. 115.
Sasikumar S., Vijayaraghavan R. // Trends Biomater. Artificial Organs. 2006. V. 19. № 2. P. 70.
Sopyan A.P.D.I., Raihana M.F., Hamdi M., Ramesh S. // Proceed. 4th Kuala Lumpur Int. Conf. on Biomedical Engineering. Kuala Lumpur, 2008. V. 21. P. 333.
Rivera-Muñoz E., Curiel R., Rodríguez R. // Mater. Res. Innov. 2003. V. 7. № 2. P. 85.
Khandelwal H., Prakash S. // J. Miner. Mater. Char. Engin. 2004. V. 4. № 2. P. 119.
Ishikawa K., Matsuya S., Lin X. et al. // J. Ceram. Soc. Jpn. 2010. V. 118. № 1377. P. 341.
Sobczak-Kupiec A., Malina D., Kijkowska R., Wzorek Z. // Digest J. Nanomater. Biostruct. 2012. V. 7. № 1. P. 385.
Penel G., Delfosse C., Rey C. et al. // Dental Med. Problems. 2003. V. 40. P. 37.
Khan A.F., Awais M., Khan A.S. et al. // Appl. Spectr. Rev. 2013. V. 48. № 4. P. 329.
Callens F., Vanhaelewyn G., Matthys P., Boesman E. // Appl. Magn. Resonance. 1998. V. 14. № 2. P. 235.
Gilinskaya L.G. // J. Struct. Chem. 2010. V. 51. P. 471.
Moens P., Callens F., Matthys P. et al. // J. Chem. Soc. Faraday Transac. 1991. V. 87. № 19. P. 3137.
De Oliveira L.M., Rossi A.M., Lopes R.T., Rodrigues L.N. // Rad. Protection Dosimetry. 2002. V. 101. № 1–4. P. 539.
Itoh S., Nakamura S., Kobayashi T. et al. // Calcified Tissue Int. 2006. V. 78. № 3. P. 133.
Goloshchapov D.L., Lenshin A.S., Tutov E.A. et al. // Smart Nanocomposites. 2013. V. 4. № 1. P. 101.
Dorozhkin S.V. // Mater. 2009. V. 2. № 4. P. 1975.
King`ori A.M. // Int. J. Poultry Sci. 2011. V. 10. № 11. P. 908.
Buddhachat K., Klinhom S., Siengdee P. et al. // PLoS One. 2016. V. 11. № 5. P. e0155458.
Yusufoglu Y., Akinc M. // J. Am. Ceram. Soc. 2008. V. 91. № 1. P. 77.
De Araújo J.C., Moreira E.L., Moraes V.C.A. et al. // Mater. Res. 2011. V. 14. № 3. P. 376.
Goloshchapov D.L., Kashkarov V.M., Ippolitov Y.A. et al. // Res. Phys. 2018. V. 10. P. 346.
Seredin P., Goloshchapov D., Prutskij T., Ippolitov Y. // PLoS One. 2015. V. 10. № 4. P. 1.
Ishii H., Ikeya M. // Appl. Rad. Isotopes. 1993. V. 44. № 1. P. 95.
Mujahid M., Sarfraz S., Amin S. et al. // Mater. Res. 2015. V. 18.№ 3. P. 468.
Teterskij A.V., Morozov V.A., Stefanovich S.Y., Lazoryak B.I. // Russ. J. Inorgan. Chem. 2005. V. 50. № 7. P. 986.
Gittings J.P., Bowen C.R., Dent A.C.E. et al. // Acta Biomaterialia. 2009. V. 5. № 2. P. 743.
Mahabole M.P., Aiyer R.C., Ramakrishna C.V. et al. // Bull. Mater. Sci. 2005. V. 28. № 6. P. 535.
Mensah-Darkwa K., Gupta R.K., Kumar D. // J. Mater. Sci. Technol. 2013. V. 29. № 9. P. 788.
Jogiya B.V., Jethava H.O., Tank K.P. et al. // AIP Conf. Proceed. 2016. V. 1728. № 1. P. 020227.
Дополнительные материалы отсутствуют.
Инструменты
Поверхность. Рентгеновские, синхротронные и нейтронные исследования