

Поверхность. Рентгеновские, синхротронные и нейтронные исследования, 2019, № 8, стр. 53-55
Потенциальная энергия взаимодействия атома с атомной плоскостью
В. П. Кощеев 1, *, Ю. Н. Штанов 2
1 НИУ МАИ, филиал “Стрела”
140180 Московская область, Жуковский, Россия
2 Тюменский индустриальный университет, филиал ТИУ в г. Сургуте
628400 Сургут, Россия
* E-mail: koshcheev1@yandex.ru
Поступила в редакцию 26.12.2018
После доработки 20.01.2019
Принята к публикации 23.01.2019
Аннотация
Фурье-компонента потенциальной энергии взаимодействия атома и атомной плоскости представлена в виде многочлена четвертой степени от атомного форм-фактора. Численный расчет выполнен в приближении экранированного кулоновского потенциала. Показано, что учет принципа Паули приводит к потенциальному барьеру и дополнительной области притяжения атома и атомной плоскости. Показано, что данная модель удовлетворительно описывает потенциальную энергию взаимодействия атома углерода с листом графена.
ВВЕДЕНИЕ
В работе [1] была исследована проблема применения метода функционала плотности (например, [2]) к задаче вычисления потенциальной энергии взаимодействия атомов и ионов. В статье [3] был предложен альтернативный вариант решения этой задачи, в котором учет принципа Паули приводит к потенциальному барьеру и дополнительной области притяжения двух атомов. Дальнейшее развитие подхода [3] представлено в настоящей работе.
ВЫВОД ВЫРАЖЕНИЯ ДЛЯ ПОТЕНЦИАЛЬНОЙ ЭНЕРГИИ ВЗАИМОДЕЙСТВИЯ
Рассмотрим потенциальную энергию взаимодействия атома с зарядом ${{Z}_{1}}e$ с атомом, заряд которого ${{Z}_{2}}e$:
(1)
$\begin{gathered} U = \frac{{{{Z}_{1}}{{Z}_{2}}{{e}^{2}}}}{{\left| {{{{\mathbf{r}}}_{1}} - {{{\mathbf{r}}}_{2}}} \right|}} + \sum\limits_{{{j}_{1}} = 1}^{{{Z}_{1}}} {\sum\limits_{{{j}_{2}} = 1}^{{{Z}_{2}}} {\frac{{{{e}^{2}}}}{{\left| {{{{\mathbf{r}}}_{{1{{j}_{1}}}}} - {{{\mathbf{r}}}_{{2{{j}_{2}}}}}} \right|}}} } - \\ - \,\,\sum\limits_{{{j}_{2}} = 1}^{{{Z}_{2}}} {\frac{{{{Z}_{1}}{{e}^{2}}}}{{\left| {{{{\mathbf{r}}}_{1}} - {{{\mathbf{r}}}_{{2{{j}_{2}}}}}} \right|}}} - \sum\limits_{{{j}_{1}} = 1}^{{{Z}_{1}}} {\frac{{{{Z}_{2}}{{e}^{2}}}}{{\left| {{{{\mathbf{r}}}_{2}} - {{{\mathbf{r}}}_{{1{{j}_{1}}}}}} \right|}}} , \\ \end{gathered} $Флуктуации потенциальной энергии взаимодействия (1) вызываются квантовыми флуктуациями, которые испытывают атомные электроны. Усреднение по квантовым флуктуациям местоположения атомных электронов будем осуществлять с помощью метода [4], который Бете использовал для вычисления атомного форм-фактора. Произведем усреднение (1) по квадратам модулей волновых функций атомов. Соответствующие средние будем обозначать ${{\left\langle {...} \right\rangle }_{{e1}}},$ ${{\left\langle {...} \right\rangle }_{{e2}}}{\text{:}}$
(2)
$\begin{gathered} {{\left\langle U \right\rangle }_{{e1,e2}}} = \int {\frac{{{{d}^{3}}{\mathbf{k}}}}{{{{{\left( {2\pi } \right)}}^{3}}}}} \frac{{4\pi {{e}^{2}}}}{{{{k}^{2}}}}\left( {{{Z}_{1}} - {{F}_{1}}\left( k \right)} \right) \times \\ \times \,\,\left( {{{Z}_{2}} - {{F}_{2}}\left( k \right)} \right)exp\left( {i{\mathbf{k}}\left( {{{{\mathbf{r}}}_{1}} - {{{\mathbf{r}}}_{2}}} \right)} \right), \\ \end{gathered} $Аналогично тому, как это делается в кинетической теории [5], добавим к выражению для Фурье-компоненты потенциальной энергии взаимодействия двух атомов множитель $\left( {1 - {{F\left( k \right)} \mathord{\left/ {\vphantom {{F\left( k \right)} Z}} \right. \kern-0em} Z}} \right),$ с помощью которого будем учитывать принцип Паули. Величина ${{F(k)} \mathord{\left/ {\vphantom {{F(k)} Z}} \right. \kern-0em} Z}$ является Фурье-компонентой плотности распределения атомных электронов, которая нормирована на единицу. Ожидается, что сомножитель $\left( {1 - {{F\left( k \right)} \mathord{\left/ {\vphantom {{F\left( k \right)} Z}} \right. \kern-0em} Z}} \right)$ будет действовать аналогично функции распределения Ферми−Дирака, с помощью которой учитывают принцип Паули в системах частиц с полуцелым спином [4]. В работе [3] принцип Паули был учтен для обоих атомов с помощью двух дополнительных сомножителей:
(3)
$\begin{gathered} U\left( k \right) = \frac{{4\pi {{Z}_{1}}{{Z}_{2}}{{e}^{2}}}}{{{{k}^{2}}}}{{\left[ {1 - {{{{F}_{1}}\left( k \right)} \mathord{\left/ {\vphantom {{{{F}_{1}}\left( k \right)} {{{Z}_{1}}}}} \right. \kern-0em} {{{Z}_{1}}}}} \right]}^{2}} \times \\ \times \,\,{{\left[ {1 - {{{{F}_{2}}\left( k \right)} \mathord{\left/ {\vphantom {{{{F}_{2}}\left( k \right)} {{{Z}_{2}}}}} \right. \kern-0em} {{{Z}_{2}}}}} \right]}^{2}}. \\ \end{gathered} $Численный расчет выполним для случая Z1 = $ = {{Z}_{2}} = Z.$
Атомный форм-фактор выберем в приближении экранированного кулоновского потенциала:
(4)
$F\left( k \right) = {{Z{{\mu }^{2}}} \mathord{\left/ {\vphantom {{Z{{\mu }^{2}}} {({{\mu }^{2}} + {{k}^{2}})}}} \right. \kern-0em} {({{\mu }^{2}} + {{k}^{2}})}},$Потенциальная энергия взаимодействия атома с атомной плоскостью кристалла имеет вид:
(5)
$U\left( x \right) = n\int\limits_{ - \infty }^\infty {U\left( {{{k}_{x}}} \right)\exp \left( {i{{k}_{x}}x} \right)\frac{{d{{k}_{x}}}}{{2\pi }}} ,$С помощью теории вычетов [6] по формулам (3) и (4) вычислим интеграл (5) получим:
(6)
$\begin{gathered} U\left( x \right) = \frac{{2i\pi n{{{\left( {Ze} \right)}}^{2}}}}{3}\mathop {\lim }\limits_{{{k}_{x}} \to i\mu } \frac{{{{d}^{3}}}}{{dk_{x}^{3}}} \times \\ \times \,\,\left( {\frac{{k_{x}^{6}}}{{{{{\left( {{{k}_{x}} + i{\mu }} \right)}}^{4}}}}\exp \left( {i{{k}_{x}}x} \right)} \right), \\ \end{gathered} $Результат вычисления по формуле (6) запишется как
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
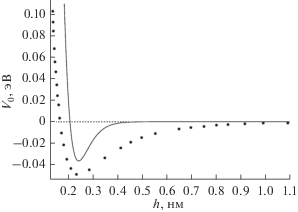
На рис. 1a представлен график потенциальной энергии взаимодействия атома с атомной плоскостью. Видно, что учет принципа Паули приводит как к потенциальному барьеру (рис. 1б), так и к дополнительной области притяжения (рис. 1в) атома и атомной плоскости. На рис. 2 показано сравнение результатов расчета по формуле (7) с данными, которые были получены в работе [7]. Для атомов углерода в графене длина экранирования составляет: ${{\mu }^{{ - 1}}} = a$ = $0.258$ Å, $({{{{{(Ze)}}^{2}}} \mathord{\left/ {\vphantom {{{{{(Ze)}}^{2}}} a}} \right. \kern-0em} a})$ ≈ ≈ 2.011 кэВ, $n$ = ${4 \mathord{\left/ {\vphantom {4 {(3\sqrt 3 a_{0}^{2})}}} \right. \kern-0em} {(3\sqrt 3 a_{0}^{2})}},$ ${{a}_{0}} = 1.42$ Å. Длина экранирования является единственным варьируемым параметром в формуле (7). Было принято, что $a = {{a}_{{T - F}}}$ = $0.885{{a}_{0}}{{Z}^{{{{ - 1} \mathord{\left/ {\vphantom {{ - 1} 3}} \right. \kern-0em} 3}}}}$ = $0.258$ Å, ${{a}_{0}} = 0.529$ Å. Используя (рис. 1б), можно оценить высоту потенциального барьера, которая препятствует сближению атома углерода с листом графена $U\left( {x = 4.255a} \right)$ = 1.403 эВ. Видно, что данная модель удовлетворительно описывает потенциальную энергию взаимодействия атома углерода с листом графена.
Рис. 1.
Потенциальная энергия взаимодействия атома с атомной плоскостью в различных масштабах: a − ${x \mathord{\left/ {\vphantom {x a}} \right. \kern-0em} a} \in \left[ {0;{\text{ }}10} \right],$ б − ${x \mathord{\left/ {\vphantom {x a}} \right. \kern-0em} a} \in \left[ {3;{\text{ }}10} \right],$ в − ${x \mathord{\left/ {\vphantom {x a}} \right. \kern-0em} a} \in \left[ {7;{\text{ }}19} \right].$
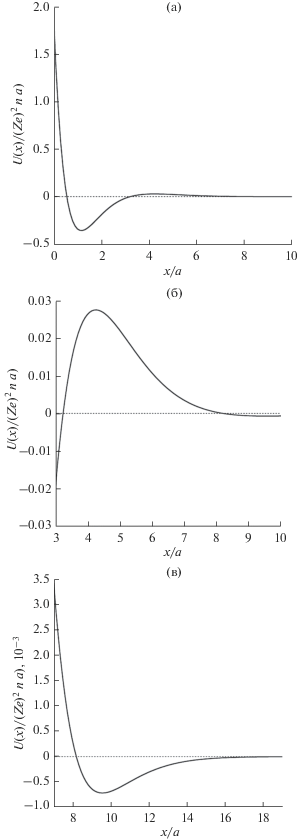
Рис. 2.
Зависимость потенциала взаимодействия углерода с подложкой от его удаленности от плоской подложки, образованной листом графена. Темные точки − результат работы [7], черная линия – результат расчета $U(x)$ по формуле (7).
ЗАКЛЮЧЕНИЕ
В работе представлены первые результаты нового метода расчета потенциальной энергии взаимодействия атома с атомной плоскостью в непрерывном приближении. Продемонстрировано удовлетворительное согласие с результатами работы [7]. С другой стороны, в работе [3] было показано, что новый метод расчета потенциальной энергии взаимодействия двух изолированных атомов кремния приводит к результату, который значительно меньше парной энергии взаимодействия двух атомов кремния по данным работы [8]. Следует ожидать [3], что более реалистические модели электронной плотности (например, [9–11]) позволят получить количественное согласие нового подхода к вычислению потенциальной энергии взаимодействия двух атомов с результатами расчета с помощью теории функционала плотности, но не нарушат удовлетворительное согласие с результатами работы [7], которое было достигнуто в настоящей работе.
Список литературы
Medvedev M.G. et al. // Science. 2017. V. 355. № 6320. P. 49. https://doi.org/10.1126/science.aah5975
Сарры А.М., Сарры М.Ф. // ФТТ. 2012. Т. 54. № 6. С. 1237.
Кощеев В.П., Штанов Ю.Н. // Письма в ЖТФ. 2018. Т. 44. № 13. С. 28.
Бете Г. Квантовая механика. М.: Мир, 1965. 333 с.
Лифшиц Е.М., Питаевский Л.П. Физическая кинетика. М.: Наука, 1979. 528 с.
Свешников А.Г., Тихонов А.Н. Теория функций комплексной переменной. М.: Наука, 1974. 319 с.
Савин А.В., Мазо М.А. // ФТТ. 2017. Т.59. № 6. С. 1234.
Заводинский В.Г., Горкуша О.А. // ФТТ. 2014. Т. 56. В. 11. С. 2253.
Molière G. // Zeitschrift Naturforsch. Tl. A. 1947. V. 2. P. 133.
Doyle P.A. Turner P.S. // Acta Crystallogr., Sect. A. 1968. V. 24. P. 390.
Kirkland E.J. Advanced computing in electron microscopy. Springer Science & Business Media, 2010. 293 p.
Дополнительные материалы отсутствуют.
Инструменты
Поверхность. Рентгеновские, синхротронные и нейтронные исследования