

Поверхность. Рентгеновские, синхротронные и нейтронные исследования, 2020, № 11, стр. 84-87
Моделирование диффузии вакансии в кристалле методом гипердинамики
Е. В. Дуда a, b, Г. В. Корнич b, *
a Запорожский государственный медицинский университет
69000 Запорожье, Украина
b Запорожский национальный технический университет
69063 Запорожье, Украина
* E-mail: gkornich@zntu.edu.ua
Поступила в редакцию 12.12.2019
После доработки 22.01.2020
Принята к публикации 24.01.2020
Аннотация
Выполнено моделирование диффузии вакансии в деформированном кристалле алюминия методами гипердинамики и классической молекулярной динамики. Используемый способ построения смещенного потенциала для метода гипердинамики был ранее развит на примере двумерных систем с использованием парных потенциалов. Настоящие результаты свидетельствуют о возможности применения рассматриваемого смещенного потенциала в моделировании реалистичных трехмерных систем с использованием многочастичных потенциалов. Исследована зависимость ускорения моделирования методом гипердинамики по сравнению с молекулярной динамикой от температуры и величины смещения потенциала.
ВВЕДЕНИЕ
Гипердинамика [1] – один из методов ускоренного молекулярно-динамического моделирования. К этим методам относятся также температурно-ускоренная динамика [2] и метод параллельных реплик [3]. Перечисленные подходы были разработаны как способы динамического моделирования процессов, важную роль в которых играют так называемые редкие события. К таким событиям относятся переходы системы между локальными минимумами потенциальной энергии. Моделирование этих процессов методами классической молекулярной динамики (МД) представляет большую вычислительную сложность, так как продолжительность переходов мала по сравнению со временем, в течение которого система находится в окрестности потенциального минимума между переходами. Таким образом, большая часть ресурсов при подобном моделировании расходуется на моделирование поведения системы, находящейся около энергетического минимума, а не на моделирование переходов. Для решения данной проблемы были разработаны методы ускоренной молекулярной динамики. Они используются при моделировании различных систем начиная с кристаллов [4, 5] и заканчивая биомолекулами [6, 7].
Основная идея гипердинамики состоит в понижении барьеров между минимумами потенциальной энергии системы, т.е. в уменьшении энергии активации переходов. Это достигается путем введения, дополнительного слагаемого Ub в потенциал системы:
где U0 – исходный потенциал межатомного взаимодействия в системе, U – смещенный потенциал, используемый при гипердинамическом моделировании. Существуют различные подходы к построению Ub, которые постоянно развиваются. Часть этих подходов представлена в [8–10]. Рассмотрим метод гипердинамики со слагаемым Ub, для построения которого не требуется большого количества вычислительных ресурсов. Такой способ построения потенциала U рассматривали ранее в [11, 12], его тестировали на двумерных атомных системах с применением парного потенциала. В настоящей работе проводится моделирование трехмерной атомной системы с использованием многочастичного потенциала для демонстрации применимости данного способа построения смещенного потенциала при моделировании реалистичных систем. Критерием корректности получаемых результатов является их соответствие данным, полученным методом МД.МЕТОДИКА
При моделировании используется потенциал следующего вида:
где суммирование происходит по атомам системы, слагаемое Ub,i определяется выражением:(3)
${{U}_{{b,i}}} = \left\{ \begin{gathered} {{V}_{0}} \hfill \\ \varphi ({{\rho }_{i}}) \hfill \\ 0 \hfill \\ \end{gathered} \right.\,\,\,\,\begin{array}{*{20}{c}} {{{\rho }_{i}} < {{R}_{1}}} \\ {{{R}_{1}} \leqslant {{\rho }_{i}} < {{R}_{2}}} \\ {{{\rho }_{i}} \geqslant {{R}_{2}}} \end{array},$Особенности данного потенциала, а также ограничения, накладываемые на параметры R1 и R2, рассмотрены в [11]. Для всех вычислений, результаты которых описаны ниже, параметр R1 принимал одно из следующих значений: 0.26, 0.27, 0.28, 0.3 Å, а параметр R2 – 1.0, 1.1, 1.15 Å. Функция φ(ρi) – полиномиальный сплайн пятого порядка, состоящий из двух фрагментов.
Система представляла собой кристалл алюминия, включающий вакансию. Образец содержал 8191 атом и был искусственно сжат в направлениях [110] и [1$\overline 1 $0], так что средние расстояния между атомами в данных направлениях сократились на 1 и 0.5% соответственно. Таким образом, для вакансии в любой момент времени существовали конкурирующие переходы трех типов. Методика модельных сжатий кристалла позволяет проверить, будет ли гипердинамический подход давать такие же частоты для различных переходов, что и классический метод МД. В качестве исходного потенциала U0 был применен многочастичный потенциал Акланда [13]. Для установления и поддержания постоянной температуры кристалла был использован метод “термической ванны” Берендсена [14], который уже применялся нами ранее [15, 16]. Интегрирование уравнений движения выполнялось согласно скоростному методу Верле. Моделирование проводили при температурах 650, 600 и 550 К. При каждой температуре было получено по 1000 переходов для каждого набора параметров R1, R2, V0 методом гипердинамики и столько же методом МД. Для сравнения результатов, полученных различными методами, использовали метод однородности Смирнова [17]. Данный метод применяли для подтверждения гипотезы о подчинении полученных методами МД и гипердинамики выборок времен, проходящих между двумя последовательными переходами системы, одному закону распределения.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Анализ результатов моделирования показал, что выборки времен переходов, которые дает метод гипердинамики, подчиняются тому же закону распределения, что и выборки, получаемые методом МД. Эта гипотеза была подтверждена на уровне значимости 0.1 при V0, не превышающем 0.15 эВ, и на уровне значимости 0.05 при более высоких значениях V0.
На рис. 1 представлена зависимость ускорения τ по времени вычисления, получаемого при использовании метода гипердинамики при сравнении с классическим методом МД, от значения параметра V0. Здесь τ определено следующим образом:
(4)
$\tau = {{{{t}_{{{\text{MD}}}}}} \mathord{\left/ {\vphantom {{{{t}_{{{\text{MD}}}}}} {{{t}_{{{\text{HD}}}}},}}} \right. \kern-0em} {{{t}_{{{\text{HD}}}}},}}$Рис. 1.
Ускорение вычислений τ в зависимости от величины снижения энергетических барьеров V0 при моделируемой температуре: 550 (1); 600 (2); 650 K (3).
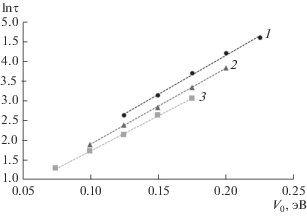
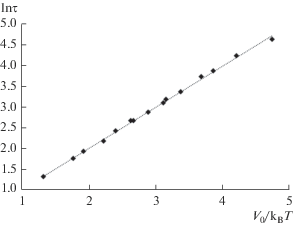
На рис. 1 видно, что при одинаковых значениях V0 вычислительное ускорение увеличивается с ростом температуры. На графике с осью абсцисс V0/kBT (рис. 2), видно, что выполняется следующее соотношение:
ЗАКЛЮЧЕНИЕ
Описанный в [11] способ построения смещенного потенциала был протестирован на трехмерной атомной системе с использованием многочастичного потенциала. Показано, что в этом случае метод гипердинамики позволяет получать результаты, сопоставимые с результатами, которые дает метод классической МД. Получено, что ускорение растет экспоненциально с увеличением параметров V0 и 1/T. Таким образом, при низких температурах, когда метод МД не позволяет моделировать процессы переходов системы между состояниями, метод гипердинамики дает возможность такого моделирования.
Список литературы
Voter A.F. // J. Chem. Phys. 1997. V. 106. № 11. P. 4665.
Sorensen M.R., Voter A.F. // J. Chem. Phys. 2000. V. 112. № 21. P. 9599.
Voter A.F. // Phys. Rev. B. 1998. V. 57. № 22. P. 985.
Montalenti F., Voter A.F. // Phys. Status Sol. B. 2001. V. 226. № 1. P. 21.
Chakraborty S., Zhang J., Ghost S. // Comput. Mater. Sci. 2016. V. 121. P. 23.
Miao Y., Feixas F., Eun C., McCammon J.A. // J. Comput. Chem. 2015. V. 36. № 20. P. 1536.
Andersen O.J., Risor M.W., Poulsen E.C. et al. // Biochem. 2017. V. 56. № 4. P. 634.
Kim S. Y., Perez D., Voter A.F. // J. Chem. Phys. 2013. V. 139. P. 144 110.
Fichthorn K.A., Mubin S. // Comput. Mater. Sci. 2015. V. 100. P. 104.
Hirau H. // J. Chem. Phys. 2014. V. 141. № 23. P. 234 109.
Дуда Е.В., Корнич Г.В. // Поверхность. Рентген., синхротр. и нейтрон. исслед. 2017. № 7. С. 89.
Дуда Е.В., Корнич Г.В. // Физика твердого тела. 2017. № 10. С. 1879.
Vitek V., Ackland G.J., Cserti J. // Mat. Res. Soc. Symp. Proc. 1991. V. 186. P. 237.
Berendsen H.J., Postma J.P.M., Gunsteren W.F.V. et al. // J. Chem. Phys. 1984. V. 81. № 8. P. 3684.
Kornich G.V., Betz G., Bazhin A.I. // Nucl. Instrum. Methods. Phys. Res. B. 1999. V. 153. P. 383.
Kornich G.V., Betz G., Bazhin A.I. // Nucl. Instrum. Methods. Phys. Res. B. 1999. V. 152. P. 437.
Большев Л.Н., Смирнов Н.В. // Таблицы математической статистики. М.: ВЦ АН СССР, 1968. С. 88.
Воеводин В.В., Жуматий С.А., Соболев С.И. и др. // Открытые системы. 2012. № 7. С. 36.
Дополнительные материалы отсутствуют.
Инструменты
Поверхность. Рентгеновские, синхротронные и нейтронные исследования