

Поверхность. Рентгеновские, синхротронные и нейтронные исследования, 2021, № 12, стр. 47-73
Радиационная карбонизация поливинилиденфторида: Часть I (Обзор)
Л. А. Песин *
Южно-Уральский государственный гуманитарно-педагогический университет
454080 Челябинск, Россия
* E-mail: pesinla@mail.ru
Поступила в редакцию 22.02.2021
После доработки 23.04.2021
Принята к публикации 28.04.2021
Аннотация
Обсуждаются как известные из литературы, так и ранее не опубликованные результаты исследований, посвященных модификации поливинилиденфторида (ПВДФ) под воздействием ионов инертных газов и мягкого рентгеновского излучения лабораторных источников с магниевым и алюминиевым анодом. Наряду с экспериментальными данными представлены модельные расчеты кинетики дегидрофторирования полимера и градиента концентрации остаточного фтора в анализируемом приповерхностном слое. Особенности деградации ПВДФ при бомбардировке электронами и облучении фотонами синхротронного излучения будут рассмотрены в следующем обзорном материале, который в настоящее время находится в стадии подготовки.
ВВЕДЕНИЕ
Поливинилиденфторид (ПВДФ) – это полимер с цепочечной молекулярной структурой. Его структурная формула (–CH2–CF2–)n, характерные температуры плавления, кристаллизации и стеклования составляют, соответственно, 171–180, 141–151 и –40°С. ПВДФ нерастворим в обычных растворителях, только в специальных (диметилацетамид, диметилформамид, диметилсульфоксид) [1, 2].
ПВДФ относится к классу кристаллизующихся полимеров, в котором сосуществуют упорядоченные и аморфные области. Реальные образцы, как правило, содержат множество фаз и произвольно ориентированных относительно друг друга кристаллов. Даже в пределах одной цепи возможны соединения типа “голова к голове”, “хвост к хвосту” и другие. Поэтому точно определить кристаллическую структуру ПВДФ, и, следовательно, координаты атомов, очень сложно. Доля кристаллической фазы обычно составляет 0.5 и ниже. ПВДФ может кристаллизоваться в четырех полиморфных модификациях (α, αр, β и γ), которые различаются как типом решетки, так и конформацией цепи [3].
ПВДФ обладает ценными пироэлектрическими, пьезоэлектрическими и нелинейными оптическими свойствами [4]. β-конформация (плоский зигзаг) является сегнетоэлектрическим полимером, диполи которого могут быть “выстроены” внешним электрическим полем. После поляризации этот материал становится сегнетоэлектриком, и в нем значительно усиливаются пьезоэлектрические эффекты [5]. Кроме того, по сравнению с керамическими пьезоэлектриками, ПВДФ обладает целым рядом преимуществ, таких как: слабое акустическое сопротивление, отсутствие хрупкости, высокая гибкость и относительная дешевизна производства изделий, имеющих разнообразное практическое применение. В [6], например, описаны технология изготовления и поляризации датчика динамического давления на основе ПВДФ.
Широкое применение в различных областях техники находят сополимеры на основе ПВДФ, в частности, с тетра- и трифторэтиленом [7, 8]. В [9] приведены результаты исследований диэлектрической дисперсии для нанокристаллов (толщиной 10–20 нм и диаметром 100–200 нм) сополимера ПВДФ с трифторэтиленом состава 70/30. Обнаружено, что фазовый переход в сегнетоэлектрических нанокристаллах имеет место при 100°С. Кроме того, авторы наблюдали широкий температурный гистерезис, что указывает на фазовый переход первого рода.
Различные сополимеры ПВДФ находят применение в технологиях изготовления мембран. В частности, на основе ПВДФ получают протонные мембраны высокой проводимости, гибридные органо-неорганические катионообменные мембраны, ионные сетчатые мембраны [10], которые перспективны для изготовления биомедицинских устройств, роботов, сенсоров и активаторов.
ПВДФ используется как прекурсор в многочисленных попытках синтеза линейной формы углерода – карбина [11]. В настоящее время чистые кристаллы карбина синтезировать не удается, поскольку одномерные образования малоустойчивы. Продукты синтеза (карбиноиды) содержат в различных количествах различные включения, вакансии, искривления цепи и межцепочечные сшивки [11, 12]. Перспективность ПВДФ для синтеза карбиноидных структур определяется его строением. В структуре равное количество атомов фтора и водорода, которые в результате радиационного или химического воздействия удаляются из полимера в виде фтористого водорода. Это открывает возможность как слабой [13], так и глубокой, хотя и не исчерпывающей [14], карбонизации за счет вариации дозы в результате реакций дегидрофторирования [15–17] без разрушения углеродной цепочечной структуры.
Интересные перспективы имеют и иные способы модификации ПВДФ, не связанные с сополимеризацией. В частности, поскольку уменьшение концентрации фтора в полимере сопровождается увеличением его электрической проводимости [13], возникает возможность создавать слоистые гетероструктуры путем частичного дефторирования поверхности. Например, сэндвич из полупроводниковой карбонизированной пленки заданной толщины и прозрачной непроводящей пьезоэлектрической подложки исходного полимера может быть использован для конструирования опто- и акустоэлектронных наноустройств.
Известны два основных пути карбонизации ПВДФ: радиационный (облучение фотонами и бомбардировка микрочастицами различной массы и энергии) [13, 14, 16–24] и химический [11, 15, 25–32]. Соседние атомы углерода, оставшись без фтора и водорода, могут соединяться друг с другом одинарными и кратными связями, образуя кумуленовые или полииновые структуры [32], а также межцепочечные сшивки. Неполнота карбонизации приводит к сохранению некоторого количества молекулярных комплексов CF2 (CH2) или/и CF (CH) внутри частично “голой” цепи. Эти комплексы могут играть стабилизирующую роль, удерживая карбонизированные фрагменты соседних цепей на расстоянии, достаточном для предотвращения немедленного коллапса линейного углерода и образования структур более высокой размерности.
Наиболее простым и продуктивным методом карбонизации ПВДФ, позволяющим модифицировать достаточно большое количество полимера, является химическая обработка. Преимущества же радиационной карбонизации заключаются в возможности более точного управления дозой и другими характеристиками облучения, а также мониторинга протекания реакции дегидрофторирования в режиме реального времени. В частности, основная часть настоящего обзора будет посвящена описанию экспериментов, в которых мягкое рентгеновское облучение использовалось для модификации поверхности ПВДФ и одновременного контроля ее химического состояния.
Настоящий обзор носит материаловедческий характер. В нем обсуждаются результаты экспериментальных исследований производных ПВДФ, карбонизированных при радиационном воздействии трех типов: бомбардировкой ионами инертных газов и облучением фотонами лабораторных источников MgKα- и AlKα-излучений. Нельзя было оставить без внимания вопрос о влиянии на поверхность полимерной мишени вторичных электронов из фильтрующей металлической фольги, которые возникают естественным образом при использовании лабораторных немонохроматических источников мягкого рентгеновского излучения. Кроме того, обсуждаются свойства производных ПВДФ, карбонизированных в результате комбинаций облучения фотонами с предварительной или последующей ионной бомбардировкой. Особенности деградации ПВДФ при бомбардировке электронами и облучении фотонами синхротронного излучения будут рассмотрены в следующем обзорном материале, который в настоящее время находится в стадии подготовки.
1. ПРИМЕНЕНИЕ РЕНТГЕНОВСКОЙ ФОТОЭЛЕКТРОННОЙ СПЕКТРОСКОПИИ ДЛЯ АНАЛИЗА ТОНКОЙ СТРУКТУРЫ СПЕКТРОВ С1s ПВДФ И ПРОДУКТОВ ЕГО РАДИАЦИОННОЙ КАРБОНИЗАЦИИ
Метод рентгеновской фотоэлектронной спектроскопии (РФЭС), с одной стороны, предоставляет возможность радиационной карбонизации поверхности полимера и одновременного мониторинга изменения ее химического состава и электронной структуры. С другой стороны, если карбонизация проводится химическим способом или бомбардировкой электронами и/или ионами, при измерениях спектров происходит дополнительная модификация поверхности мягким рентгеновским излучением, используемым для возбуждения фотоэмиссии. При интерпретации данных в случаях использования немонохроматического рентгеновского излучения необходимо также учитывать влияние вторичных электронов фильтрующей фольги на некоторые параметры спектров [33].
Компонентный анализ заключается в представлении экспериментального спектра в виде суммы модельных кривых (компонент), каждой из которых соответствует определенное энергетическое состояние остовных электронов. Решение этой задачи позволяет найти доли атомов данного элемента, находящихся в тех или иных энергетических состояниях. Зная величины первичного и вторичного химического сдвига для различных пар химических элементов, можно при помощи компонентного анализа определять доли атомов данного элемента, находящихся в тех или иных функциональных группах и фрагментах молекул.
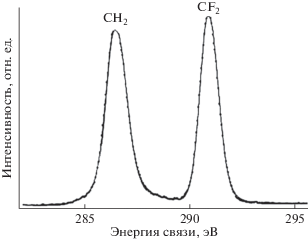
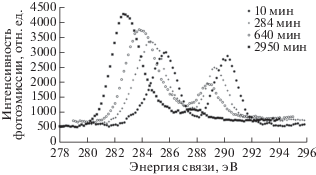
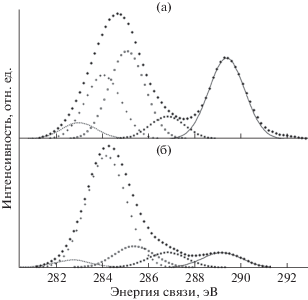
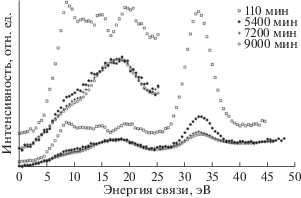
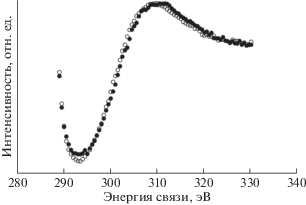
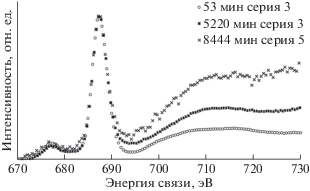
Метод РФЭС и компонентный спектральный анализ тонкой структуры спектров С1s электронов применялись в ряде пионерских работ по изучению радиационного воздействия на поверхность ПВДФ [13, 18, 34]. Целый ряд исследований, например [18, 34], свидетельствует о наличии в спектре исходного ПВДФ двух пиков, площади под которыми почти равны, вызванных фотоэмиссией C1s-электронов из приблизительно одинакового количества групп CH2 и CF2 в полимерных цепях (рис. 1 и 2а). Сравнение результатов этих измерений демонстрирует большое сходство формы спектров. Низкоэнергетический пик дублета несколько ниже и шире высокоэнергетического, а зазор между ними близок к 4.5 эВ. Эта величина определяется разностью первичных и вторичных химических сдвигов пика С1s вследствие влияния электроотрицательных атомов фтора на энергию связи остовных электронов углерода. В обеих обсуждаемых работах для возбуждения спектров, представленных на рис. 1 и 2а, использовали фотоны монохроматизированного Kα-излучения алюминиевого анода (hν = 1486.6 эВ). В [18] изменений спектра не замечено, возможно, вследствие небольшого времени сканирования. В исследовании [34] наблюдалось уменьшение атомного отношения F/C до 0.85 при мощности источника 1.40 Вт и времени экспозиции 500 мин. Следует отметить, что в [18, 34] изучали образцы ПВДФ разных производителей. Начальные соотношения площадей под пиками СН2 и CF2 равны, соответственно, 53.18/46.82 и 51/49.
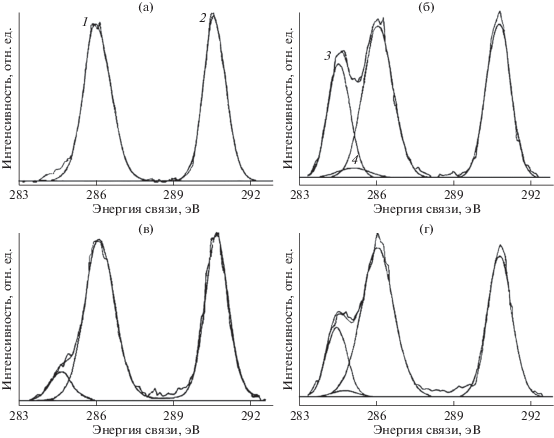
В [18] исследовано также воздействие бомбардировки быстрыми ионами Kr+ и облучения немонохроматическим рентгеновским излучением, сопровождаемым потоком вторичных электронов из фильтрующей алюминиевой фольги. Образец полукристаллического ПВДФ марки Foraflon 1000 фирмы ATOCHEM представлял собой цилиндрический диск диаметром 18 мм и толщиной 100 мкм и состоял из неполярной α-фазы, не обладающей пьезо- и пироэлектрическими свойствами. Предварительно образец очищали ацетоном в ультразвуковой ванне. Измерения РФЭС были проведены на спектрометре V.G. ESCALAB MK II, оснащенном монохроматическим источником рентгеновского излучения AlKα (hν = = 1486.6 эВ) и полусферическим 150°-ным анализатором. Монохроматор был изготовлен из сферически изогнутого кристалла кварца. Энергия пропускания анализатора была постоянной и равна 20 эВ, разрешающая способность в этих условиях составляла 0.5 эВ. Калибровка энергоанализатора была выполнена по линии 4 f7/2 поликристаллического золота (энергия связи 84 эВ). Давление в камере поддерживали на уровне 5 × × 10–11 Торр. Для облучения ионами Kr с энергией 10 МэВ/а.е.м. (доза 109 см–2) был использован ускоритель CANIL (г. Кан, Франция). Источник рентгеновского излучения и электронная пушка (LEG 200) были установлены в камере анализатора ESCALAB. Энергия электронного пучка пушки могла варьироваться от 0 до 10 кэВ, а ток от 0.1 до 100 нА. Для снятия электростатического заряда применяли потоковую электронную пушку, ток нити накала которой составлял 450 мкА, а энергия электронов – 0.5 эВ.
Результаты этого исследования представлены на рис. 2 и в табл. 1, в которой сохранена авторская интерпретация наблюдаемых спектральных особенностей. При воздействии ионов Kr+ интегральная интенсивность пиков остовных электронов, эмитированных из атомов углерода CF2- и CН2-групп, уменьшается, и появляются новые пики при 284.60 и 285.12 эВ (рис. 2б), которые авторы относят, соответственно, к эмиссии из атомов углерода, соединенных двойной связью ‒C=C– и связанных с атомом водорода. После воздействия потока электронов и рентгеновского излучения (AlKα, 10 кВ, 10 мА) в течение 6 ч форма спектра также изменяется, особенно заметным становится уменьшение пика (–CF2–) и появление пика около 284.70 эВ (рис. 2в). Эти изменения усиливаются при увеличении времени (12 ч) и мощности (10 кВ, 40 мА) воздействия, пик при 284.60 эВ становится больше (рис. 2г). Из рис. 2г видно, что деконволюция спектра выявляет новую небольшую компоненту около 284.8 эВ, не обсуждаемую авторами в дальнейшем. Авторскую интерпретацию компоненты при 285.12 эВ вряд ли можно признать однозначной, так как согласно [35] связь с водородом не вызывает химического сдвига. Более вероятно смещение пика эмиссии С1s-электронов атомов углерода, не связанных ковалентной связью с фтором, в сторону меньших значений энергии связи за счет полного или частичного дефторирования соседних с ними атомов, что уменьшает величину вторичных химических сдвигов. В [18] постулируется отсутствие атомов углерода, ковалентно связанных лишь с одним атомом фтора. Однако это противоречит данным рис. 2в, 2г, на которых отчетливо видно небольшое увеличение эмиссии электронов с энергией связи 287–288 эВ.
Таблица 1.
Интенсивности и энергетические положения компонентов C1s-линии в зависимости от радиационного воздействия [18]
| Образец | C1s-линия | Энергетическое положение компонент C1s-линии, эВ | Относительная интенсивность, %* |
|---|---|---|---|
| Необлученный | –CF2– | 290.60 | 46.82 |
| –CH2– | 286.13 | 53.18 | |
| Ионы Kr, 10 МэВ/а.е.м., 109 см–2 |
–CF2– | 290.58 | 33.96 |
| –CH2– | 286.09 | 40.69 | |
| –C=C– | 284.60 | 22.49 | |
| ≡C–H | 285.12 | 2.86 | |
| e– + X, 6 ч, 10 кэВ, 1014 см–2 |
–CF2– | 290.70 | 41.66 |
| –CH2– | 286.20 | 51.70 | |
| –C=C– | 284.70 | 6.64 | |
| e– + X, 12 ч, 10 кэВ, 1.6 × 1014 см–2 |
–CF2– | 290.50 | 34.68 |
| –CH2– | 286.01 | 49.11 | |
| –C=C– | 284.47 | 14.56 |
В [13] исследована модификация поверхности ПВДФ при воздействии немонохроматического рентгеновского излучения в течение 800 мин. Авторы регистрировали РФЭС через каждые 100 мин. Это дало им возможность мониторинга содержания химических элементов и изменений тонкой структуры спектров C1s-электронов в процессе восьми последовательных измерений. В течение эксперимента наблюдались энергетические сдвиги спектров. Поэтому авторы осуществили привязку положений спектральных пиков на шкале энергии связи к пику CF2-групп в C1s-спектре, полученном через 100 мин после начала рентгеновского облучения. Его приняли равным 290.90 эВ по аналогии с [34].
ПВДФ Kynar производства Goodfellow Cambridge Ltd имел α-конформацию цепей и не проявлял пьезоэлектрических и пироэлектрических свойств. Образец в форме диска диаметром 10 мм и высотой 0.8 мм был очищен перед измерениями в ультразвуковой ванне с изопропанолом в течение 30 мин, а затем высушен в вакууме при комнатной температуре. Измерения были выполнены с использованием рентгеновского фотоэлектронного спектрометра Kratos XSAM 800. Спектры возбуждали излучением магниевого анода (12 кВ, 20 мА), в котором доминируют фотоны с энергией 1253.6 эВ. Система энергетического анализа состояла из полусферического электростатического электронного энергоанализатора радиусом 127 мм с трехканальным детектором. Постоянная энергия пропускания составляла 20 эВ, что обеспечивало разрешение порядка 1 эВ. Давление остаточных газов в камере при измерениях РФЭС не превышало 3.5 × 10–11 Торр. Время регистрации одного спектра не превышало 20 мин.
Перед началом измерений образец в течение 14 ч выдерживали в камере при давлении около 5 × 10–11 Торр для очистки поверхности от адсорбированных газов. Для частичного снятия и стабилизации электростатического заряда образца использовали потоковую электронную пушку, энергия электронов составляла 6 эВ. Специальный эксперимент такой же длительности показал, что при выключенном рентгеновском источнике электроны, генерируемые потоковой пушкой, никоим образом не влияют на форму и интенсивность РФЭС исходного материала, и, следовательно, на физико-химическое состояние его поверхности.
Для вычисления относительной атомной концентрации фтора F/C (количества атомов фтора, приходящихся на один атом углерода) использовали отношение интегральных интенсивностей спектров F1s и C1s, измеренных после вычитания фоновых составляющих. Спектральный фон аппроксимировали линейными функциями. Для учета факторов, приводящих к различному количеству регистрируемых остовных электронов, эмитированных из атомов углерода и фтора, авторы использовали эмпирический множитель 5.263. К сожалению, методика определения величины этого множителя в обсуждаемой работе не описана.
Экспериментальные спектры представлены на рис. 3. Из рисунка видно закономерное убывание интенсивности эмиссии F1s-электронов и СF2-компоненты в полосе С1s. Заметно смещение максимумов и спектров в целом в сторону меньших значений энергии связи, что свидетельствует об уменьшении электростатического заряда вследствие увеличения электропроводности поверхности образца при дефторировании.
Рис. 3.
Вариации формы и энергетического положения спектров остовных электронов углерода (а) и фтора (б) ПВДФ в процессе продолжительных измерений РФЭС. Нумерация спектров от 1 до 8 соответствует измерениям спустя 100, 200, 300, 400, 500, 600, 700 и 800 мин от начала облучения. Адаптировано из [13].
Для исследования тонкой структуры С1s-полосы использовали метод компонентного анализа. Модельные спектральные линии были сконструированы в виде суммы кривых Гаусса и Лоренца, доли которых составляли, соответственно, 80 и 20%. Метод позволил выявить две основные компоненты при 290.9 (СF2) и 286.5 эВ (СН2) в соответствии с [18, 34], а также небольшой пик при 285.2 эВ, интенсивность которого составляла около 12% от общей интегральной интенсивности С1s-спектра. Очень маленький пик был также выявлен при 288.3 эВ. Авторы полагают, что происхождение этих пиков связано с загрязнениями углеродом поверхности образца. Второй пик может быть связан с появлением групп СF при регистрации первого спектра на самом начальном этапе дефторирования поверхности. К сожалению, в [13] такие существенные детали процедуры компонентного анализа, как ширины и количество используемых модельных линий, не приведены. Можно, однако, догадываться, что использовали либо трех-, либо четырехкомпонентное приближение.
Величина отношения интегральных интенсивностей спектров F1s и C1s уменьшилась на 12.3% при увеличении продолжительности эксперимента в интервале 100–800 мин. Это уменьшение происходило практически линейно. Изменения в C1s-спектрах также указывают на уменьшение содержания фтора. При воздействии рентгеновского излучения пик CF2 уменьшается, в то время как компонента, соответствующая углероду, не связанному с фтором (–CH2–, >СН–, =CH–) практически не изменяется. Эволюцию формы спектра C1s-электронов авторы объясняют тем, что уменьшение числа компонент, соответствующих группам CF2, от 41 до 30% от всей площади под пиками в спектре С1s в том же временном интервале происходит за счет образования в образце CF-групп. Эффект проявляется в возрастании пика при 288.3 эВ от 6 до 17%. Эти изменения также линейно зависят от продолжительности экспозиции.
Экспериментальные результаты приводят к заключению, что основным следствием воздействия мягкого рентгеновского излучения и вторичных электронов, эмитируемых фильтрующей фольгой, в данном временнóм интервале является распад дифторметиленовых (CF2) групп до фторметиновых (CF) вследствие дегидрофторирования. Предлагаемая авторами молекулярная модель включает две стадии этого процесса. Начальная реакция заключается в расщеплении связей С–H и C–F и формировании промежуточных продуктов в виде радикалов или ионов:
Схема 1 .
По мнению авторов, расщепление связей C–C менее вероятно. Промежуточные радикалы могут реагировать с радикалами в смежных цепочках, образуя сшивки полимерных цепей:
Схема 2 .
В силу высокой прочности связей в молекулах HF атомы H и F, образовавшиеся в начальных реакциях, могут увлекать за собой другие атомы F или H из полимерной цепи, что приводит к элиминированию молекулы HF и формированию в цепи других радикалов. Атомы H и F также могут извлекать атомы F или H из групп CF2 или CH2 соседних звеньев одной и той же цепи (эффект аллильной активации), что также приводит к элиминированию молекулы HF и формированию двойных связей (–СF2–CH=CF–CH2–) в основных цепях полимера. Кроме этого, авторы допускают возможность отрыва от цепи атома водорода другим атомом водорода и, соответственно, генерации нового радикала, но считают маловероятным подобный процесс с участием двух атомов фтора.
Приведенная в [13] интерпретация вызывает некоторые сомнения. Во-первых, как указывают сами авторы, в экспериментальных спектрах не обнаружен пик, характерный для полиеновых структур. Однако при образовании двойной связи в основной цепи вторичный химический сдвиг энергии остовных электронов углерода группы ‒СН= должен уменьшаться, поскольку его вызывают уже не четыре, а лишь три атома фтора. Это должно приводить к возникновению дополнительного пика С1s эмиссии при меньшей, чем 286.5 эВ, энергии связи либо, если разрешение анализатора недостаточно, к соответствующему уширению особенности с центром около 286.5 эВ. Вот почему было бы полезно указать ширины и число модельных компонент, используемых для исследования тонкой структуры экспериментальных спектров.
Во-вторых, в работе описаны два различных механизма образования СF-групп в окружении атомов углерода, не связанных ковалентной связью с фтором. Один из них – сшивка соседних цепей, в которых реализуется ковалентная связь атомов углерода CF- и CH-групп. Тем не менее точно такую же вероятность имеют межцепочечные сшивки, осуществляемые альтернативным образом:
Схема 3 .
За счет вторичного сдвига при такой сшивке должна наблюдаться либо дополнительная особенность с энергией связи, несколько большей, чем пик эмиссии С1s-электронов из CF-групп фторзамещенной полиеновой структуры, либо соответствующее уширение этого пика.
В более поздних работах [14, 17, 21, 22, 30, 33, 36–38] изучали кинетику карбонизации пленки ПВДФ Kynar производства фирмы Атофина, Франция (тип 720, толщина 50 мкм). Пленка содержит около 50% кристаллической фазы, погруженной в аморфную. Свойства аморфной фазы подобны свойствам переохлажденной жидкости. Доминирующий тип конформации цепей и той, и другой фазы – α-конформация. Кристаллическая часть материала состоит из пластин толщиной порядка 10–6 см и длиной 10–5 см.
Для изучения кинетики карбонизации пленки ПВДФ Kynar проведено множество серий экспериментов, в которых варьировались способы воздействия, продолжительность облучения фотонами (экспозиция) и доза ионной бомбардировки. Некоторые серии были проведены практически в идентичных условиях с единственной целью проверки воспроизводимости получаемых данных. Во избежание излишней громоздкости обзора автор решил ограничиться описанием и анализом результатов лишь восьми наиболее информативных независимых серий измерений, информация о которых приведена в табл. 2.
Таблица 2.
Отличительные особенности серий измерений кинетики карбонизации пленки ПВДФ Kynar методом РФЭС
| Номер серии | Тип воздействия | Максимальная экспозиция, мин | Доза, 1015 см–2 | Энергия пропускания, эВ | Ссылка |
|---|---|---|---|---|---|
| 1 | MgKα | 832 | 20 | [36] | |
| 2 | MgKα | 6000 | 20 | [37] | |
| 3 | AlKα + е– | 5400 | 320, 70 | [14, 33] | |
| 4 | AlKα + е–, Ar+ | 7200 | 11–96 | 320 | [17, 21, 22] |
| 5 | AlKα + е– | 9000 | 320 | * | |
| 6 | AlKα | 820 | 320 | [33] | |
| 7 | Ar+, (AlKα + е–)** | 1790 | 0.04–4.5 | 320 | [33] |
| 8 | Ar+, (AlKα + е–)** | 2032 | 0.04–4.5 | 320 | [33] |
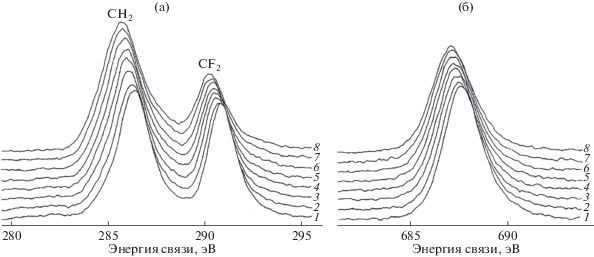
В первых двух из них [36, 37] пленка подвергалась непрерывному воздействию немонохроматического излучения магниевого анода (12 кВ, 20 мА) и потока вторичных электронов от фильтрующей фольги в течение, соответственно, 832 и 6000 мин. Давление остаточных газов в камере не превышало 3.5 × 10–10 Торр. Использовался спектрометр ESCALAB Mk II. Регистрация С1s-электронов, эмитированных в направлении, перпендикулярном поверхности пленки, проведена в режиме постоянной энергии пропускания (20 эВ) в интервале значений энергии связи 278–300 эВ с шагом 0.1 эВ. Время регистрации одного спектра составляло 226 с, включая “мертвое” время между сканами. В первой серии измерений получены 123 спектра, во второй надежное выделение компонент оказалось возможным лишь в 50 спектрах, полученных до 4000 мин. Это позволило осуществить достаточно подробный мониторинг модификации формы С1s-спектров вследствие воздействия MgKα-излучения. На рис. 4 представлены примеры этих спектров на различных стадиях радиационной карбонизации в серии 2.
Рис. 4.
Эволюция С1s-спектров пленки ПВДФ Kynar при увеличении продолжительности радиационного воздействия MgKα-излучения (серия 2).
В сериях 3–6 ту же исходную пленку облучали немонохроматическим AlKα-излучением, фильтрованным алюминиевой фольгой, в течение различных промежутков времени. В серии 4 после облучения фотонами в течение 7200 мин пленку подвергали бомбардировке расфокусированным пучком ионов Ar+ с энергией ~1 кэВ. В сериях 7 и 8 бомбардировка расфокусированным пучком ионов Ar+ с энергией 600 эВ чередовалась с облучением AlKα-фотонами при измерениях спектров эмиссии. Для возбуждения и регистрации спектров использовали аппаратурный комплекс фотоэлектронного спектрометра ЭС ИФМ-4 с магнитным анализатором, спроектированного и изготовленного в Институте физики металлов УрО РАН (Екатеринбург) [39]. В данном спектрометре задерживающее напряжение приложено к держателю образца, в то время как входная щель анализатора находится при нулевом потенциале. Поэтому кроме воздействия рентгеновского излучения образец подвергается бомбардировке вторичными электронами фильтрующей фольги, ускоренными до различных энергий, зависящих от интервала значений энергии связи фотоэлектронов в регистрируемом спектре. Специально проведенные сравнительные измерения при противоположных знаках потенциала на держателе образца (серия 6, [33]) показали, что вторичные электроны не влияют заметным образом на скорость убывания фтора. Это означает, что взаимодействие ПВДФ с AlKα-фотонами играет доминирующую роль в процессе карбонизации. Это также доказывает, что при постоянной мощности рентгеновского источника полная кумулятивная длительность экспозиции является хорошим физическим параметром для описания кинетики дефторирования [38] вне зависимости от вариаций энергии вторичных электронов при сканировании различных интервалов энергии связи. Давление остаточных газов в вакуумной камере спектрометра ЭС ИФМ-4 при измерениях не превышало 10–9 Торр. Образцы, подвергаемые карбонизации, представляли собой прямоугольные пленки размером 10 × 8 мм. Через энергоанализатор спектрометра проходили электроны, рожденные в области 8 × 2 мм.
В экспериментах серии 3 получено 93 спектра С1s-электронов при энергии пропускания 70 эВ для проведения компонентного анализа. На рис. 5 представлены примеры этих спектров на различных стадиях радиационной карбонизации серии 3. Параллельно в этой же серии, как и во всех остальных проведенных на спектрометре ЭС ИФМ-4, при энергии пропускания 320 эВ регистрировали спектры С1s и F1s в широких энергетических интервалах, включающих сателлиты неупругих потерь фотоэлектронов, полосы СKVV, а также спектры валентных электронов, в том числе линии F2s. Спектр С1s при энергии пропускания 320 эВ регистрировали до и после каждого другого спектра для измерения относительной атомной концентрации F/С, мониторинга мощности рентгеновского источника, электростатического заряда поверхности и неоднократно наблюдавшихся изменений ее морфологии (например, [17, 21]).
Рис. 5.
Эволюция С1s-спектров пленки ПВДФ Kynar при увеличении продолжительности радиационного воздействия AlKα-излучения (серия 3).
Для всех серий экспериментов по изучению кинетики радиационного дефторирования ПВДФ Kynar характерны однотипная систематическая эволюция формы, заключающаяся в перераспределении интенсивности между главными особенностями, и энергетический сдвиг спектральных особенностей в сторону меньших значений энергии связей. Этот факт, как и авторы [13, 33], связываем с уменьшением электростатического заряда поверхности при ее дефторировании. При проведении серий 2–8 регистрировали также спектры F1s и/или F2s фотоэлектронов, интенсивность которых систематически уменьшалась при увеличении экспозиции.
Для экспериментальных С1s-спектров, полученных в сериях 1–3, проведен компонентный анализ формы, основанный на их представлении в виде суперпозиции функций Гаусса. Для определения ширины и количества компонент использована простая и достаточно очевидная идеология: каждый гауссиан соответствует пику эмиссии остовных электронов из атома углерода, принадлежащего некоторой функциональной группе в определенном химическом окружении; ширина всех гауссианов одинакова, так как ширина одиночного спектрального пика зависит только от естественной ширины возбуждающей Kα-линии и разрешения анализатора при данных условиях регистрации спектров, она равна ширине модельного гауссиана, наилучшим образом описывающего форму пика С1s эмиссии из СF2-группы в спектре, полученном в самом начале каждого эксперимента; число модельных компонент должно быть минимальным, но достаточным для описания основных особенностей экспериментального спектра.
Сначала проанализируем результаты, полученные в экспериментах с использованием MgKα-излучения (серии 1 [36] и 2 [37]). На рис. 6 представлен пример разложения на семь компонент экспериментального C1s-спектра, полученного спустя 159 мин с начала измерений первой серии [36]. Ширина модельных пиков на половине максимальной высоты при анализе результатов измерений первой и второй серий составляла 1.4 эВ. Краткая авторская интерпретация происхождения каждого компонента и энергетические положения их максимумов представлены в табл. 3. Нулевая энергия связи привязана к пику С1s эмиссии из СF2-группы. Авторская трактовка некоторых спектральных особенностей не представляется однозначной. Начнем с обсуждения пика 4. Его энергетическое положение относительно пика 6 достаточно близко к величине первичного химического сдвига, вызываемого атомом фтора (2.9 эВ [35]). Это позволяет предположить, что пик 4 возникает за счет эмиссии С1s-электронов CF-групп, окруженных атомами углерода, не связанными с фтором. Это могут быть CF-группы либо полиеновой цепи, либо участвующие в межцепочечной сшивке (схема 2 ). Пик 5 может быть результатом эмиссии С1s-электронов CF-групп, участвующих в межцепочечной сшивке (схема 3 ). Его энергетическое положение определяется комбинацией первичного и вторичного химических сдвигов. Такая интерпретация пика 5 присутствует в [36] в качестве альтернативы основной гипотезе.
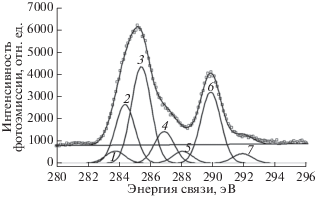
Таблица 3.
Энергетические положения максимумов модельных гауссианов и интерпретация соответствующих спектральных особенностей (серия 1) (адаптировано из [36])
| Номер пика | Энергетическое положение относительно СF2, эВ | Интерпретация |
|---|---|---|
| 1 | –6.2 | Поверхностные загрязнения |
| 2 | –5.4 | =С= или –С≡ |
| 3 | –4.5 | СН2 или СН (ПВДФ) |
| 4 | –3.1 | $\mathop {{\text{CF}}}\limits^\centerdot $ радикал |
| 5 | –2.0 | CF |
| 6 | 0 | CF2 |
| 7 | +1…+2 | CF2–CF2 (дефекты “голова к голове”) |
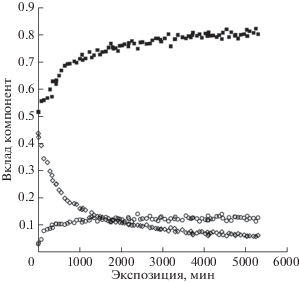
Исключительно интересным и удивительным фактом представляется то, что площади под пиками 4 и 5 оставались пропорциональны друг другу на протяжении всего эксперимента, причем коэффициент пропорциональности был близок к двум (рис. 7). Это наблюдение подтвердилось при проведении второй серии измерений (рис. 8). Если считать межцепочечные сшивки, осуществляемые по схемам 2 и 3 , равновероятными, то в рамках данной модели карбонизации ПВДФ количество СF-групп во фторзамещенных полиеновых цепях приблизительно такое же, как в каждой из сшивок двух возможных типов.
Рис. 7.
Вариации вклада компонент 4 (треугольники), 5 (квадраты), 6 (кружки) в суммарную интегральную интенсивность (площадь) С1s-спектра при проведении измерений серии 1. Адаптировано из [36].
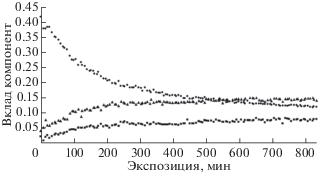
Рис. 8.
Вариации вклада компонент 4 (треугольники), 5 (квадраты), 6 (кружки) в суммарную интегральную интенсивность (площадь после вычитания фона методом Ширли) С1s-спектра при проведении измерений серии 2.
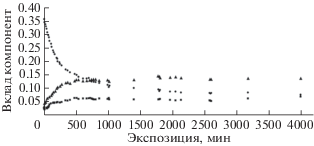
Отметим также основные отличия результатов [13] и [36, 37]. Во-первых, если в [13] интенсивность спектральной особенности, возникновение которой обусловлено эмиссией из атомов углерода, не связанных с фтором, не изменяется, то в [36, 37] суммарная площадь под пиками 1, 2 и 3 в течение приблизительно 400 мин увеличивается, несмотря на уменьшение пиков 1 и 3, а затем стабилизируется. Во-вторых, при увеличении продолжительности облучения уменьшение пика 6 и возрастание пиков 4 и 5 происходит в [36, 37] нелинейно (рис. 7, 8). Площади под пиками 4 и 5 увеличиваются только в течение первых 600 мин, причем медленнее, чем уменьшается пик 6, а затем стабилизируются. Данный факт свидетельствует о том, что одновременно с распадом СF2 → СF происходит отщепление фтора и от СF-групп. Такое поведение кинетических зависимостей соответствует известной модели двухстадийного дегидрофторирования ПВДФ [32]:
Схема 4 .
При низких дозах облучения формируются фрагменты фторзамещенных полиеновых структур. При увеличении дозы облучения и дальнейшем удалении фтора и водорода они преобразуются в кумуленовые или полииновые. Стабилизация площадей под пиками 4 и 5 после 600 мин облучения означает выравнивание скоростей дегидрофторирования на обеих стадиях процесса, поскольку в рамках данной модели единственным результатом распада СF2-группы является образование фторметиновой группы СF в основном либо радикальном состоянии.
Перейдем к описанию результатов компонентного анализа С1s-спектров при изучении кинетики дефторирования пленки ПВДФ Kynar под воздействием AlKα-излучения (серия 3, [14]). Энергетическое разрешение при регистрации было хуже, чем в сериях 1 и 2, поэтому ширина модельных кривых Гаусса на половине их максимальной высоты составляла 2 эВ. Это позволило достаточно точно описать форму экспериментальных спектров, используя не семь, как в сериях 1 и 2, а всего пять кривых Гаусса, причем все состояния фторметиновых групп могли быть описаны одним гауссианом. Предварительно спектры были сглажены для подавления статистического шума. Фоновую составляющую вычитали из спектра методом Ширли [35]. Примеры разложения на компоненты С1s-спектров, полученных при экспозициях 284 и 2184 мин, представлены на рис. 9.
Рис. 9.
Компонентный анализ С1s-спектров пленки ПВДФ Kynar, полученных в серии 3 при экспозициях 284 (а) и 2184 мин (б). Интенсивность эмиссии нормирована на высоту главного максимума спектра. Символы в виде черных ромбиков – экспериментальные результаты после сглаживания полиномами. Сплошная линия и остальные символы соответствуют пяти модельным гауссианам. Адаптировано из [14].
Пять модельных гауссовых кривых одинаковой ширины и положения, фиксированные вблизи –6.1, –5.1, –4.5 и –2.6 эВ относительно компоненты CF2 с наибольшей энергией связи, оказались достаточны для хорошего соответствия экспериментальным данным. Первые три из перечисленных модельных кривых соответствуют спектральным особенностям, возникающим вследствие фотоэмиссии остовных электронов атомов углерода, непосредственно не связанных с фтором, но соседствующих с различным числом групп CF2 и CF. В частности, положение третьей кривой позволяет связать ее с эмиссией из атома углерода СН2-группы, окруженной двумя группами CF2. Из рис. 9 видно, что интенсивности третьей и пятой компонент в каждом из двух спектров попарно близки. Это позволяет предположить, что при облучении пленки фтор и водород отщепляются преимущественно в равных количествах, что соответствует схеме 4 [32]. Особенность, описываемая четвертой кривой Гаусса, может быть соотнесена с эмиссией C1s-электронов фторметиновых групп. Небольшое, но отчетливо видимое вверху рисунка несоответствие между экспериментом и результатом моделирования в интервале энергии связи 291–293 эВ, скорее всего, вызвано вкладом структурных дефектов типа “голова к голове” и “хвост к хвосту”, вызывающих дополнительные химические сдвиги для C1s-электронов, эмитируемых из двух соседних CF2-групп.
Результаты компонентного анализа серии 3 представлены на рис. 10. Верхние черные квадраты соответствуют вкладу атомов углерода, не связанных с фтором. Их содержание повышается от 50 до 80% в течение всего времени воздействия AlKα-излучения. Серии пустых ромбиков и кружков соответствуют уменьшению содержания CF2 и увеличению содержания CF-групп. В каждой серии символов прослеживается явная тенденция к насыщению.
Рис. 10.
Кинетика изменения содержания атомов углерода: не связанных с фтором (◼); связанных с двумя (◇) атомами; с одним (⚪) атомом фтора. Адаптировано из [14].
Таким образом, несмотря на различия энергии MgKα- и AlKα-фотонов при проведении серий экспериментов 1–3 по радиационной карбонизации пленки ПВДФ Kynar, а также способов регистрации и обработки экспериментальных спектров фотоэмиссии остовных электронов углерода при анализе их тонкой структуры, получены качественно согласующиеся кинетические характеристики эволюции фторсодержащих функциональных групп. Количественные различия между результатами, полученными в [14, 36, 37] и [13], могут объясняться различной устойчивостью исследованных образцов к радиационной деградации, но, вероятнее всего, различной мощностью рентгеновских источников, применяемых для возбуждения спектров и/или различиями методик регистрации и обработки данных.
2. ПРИМЕНЕНИЕ РФЭС ДЛЯ АНАЛИЗА ХИМИЧЕСКОГО СОСТАВА ПОВЕРХНОСТИ ПВДФ И ПРОДУКТОВ ЕГО РАДИАЦИОННОЙ КАРБОНИЗАЦИИ
Как правило, исследователей, применяющих метод РФЭС, в первую очередь интересует химический состав изучаемой поверхности. Вследствие простоты химической структуры ПВДФ можно регистрировать неперекрывающиеся фотоэлектронные линии и оже-полосы, характерные для фтора и углерода, что существенно облегчает процесс обработки данных с целью количественного химического анализа и изучения тонкой структуры спектров. Напротив, довольно быстрое дефторирование поверхности полимера при экспозиции усложняет получение надежных кинетических результатов, позволяющих оценить, как мгновенное химическое и структурное состояние поверхности, так и ее эволюцию при облучении в зависимости от времени или дозы. Поэтому возникла необходимость выработки определенной тактики регистрации спектров.
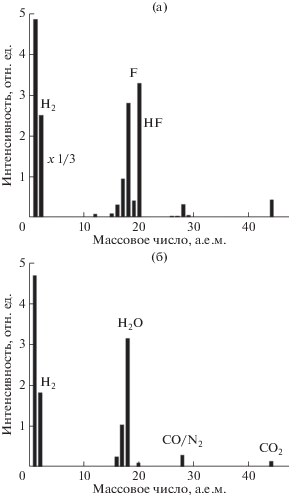
Она основана на том, что исследования радиационной модификации ПВДФ, проведенные одновременно с регистрацией масс-спектров [7, 16, 18], показали отсутствие углеродсодержащих молекул в продуктах молекулярной эмиссии. В частности, в [16] исследована деградация пленки ПВДФ под действием рентгеновского синхротронного излучения в широком интервале значений энергии фотонов (0.5–2.0 кэВ). Масс-спектр, зарегистрированный при неработающем рентгеновском источнике, обнаруживает молекулы H2O, CO (или N2) и CO2. В масс-спектрах, полученных при включенном рентгеновском источнике, кроме перечисленных молекул, были зафиксированы H2, HF, F2 в соотношении, соответственно, 20 : 10 : 2. Отсутствовала эмиссия групп, содержащих атомы углерода (C, CH, CH2, CF, CF2). Результаты этого эксперимента представлены на рис. 11. Они свидетельствуют о том, что разрыв связей в главной цепочке полимера не происходит и что количество углерода в образце при облучении фотонами не меняется. Поэтому интегральная интенсивность (площадь под линией спектра после вычитания фона) С1s-электронов пропорциональна количеству атомов углерода в анализируемом методом РФЭС приповерхностном слое ПВДФ в любой момент времени продолжительного эксперимента и является естественным внутренним эталоном для нормировки интенсивности остальных спектральных линий и полос.
Рис. 11.
Масс-спектры продуктов молекулярной эмиссии из пленки ПВДФ при экспозиции (а) и в отсутствие синхротронного излучения (б). Адаптировано из [16].
Тем не менее это обстоятельство полностью проблемы количественного химического анализа поверхности не решает, даже при наличии исчерпывающих сведений о функции пропускания используемого спектрометра [39] и элементной чувствительности [40]. Толщина анализируемого слоя в методе РФЭС зависит от рода исследуемого вещества и кинетической энергии фотоэлектронов и имеет порядок нескольких межатомных расстояний. Кинетическая энергия разных групп эмитированных электронов и соответствующая глубина анализа увеличиваются в следующей последовательности: CKVV, FKLL, F1s, C1s, F2s, С2s. Интегральная интенсивность линий F1s и F2s, а также полосы FKLL пропорциональна количеству атомов фтора в приповерхностных слоях, толщины которых определяются значениями кинетической энергии соответствующих фотоэлектронов и оже-электронов. Несовпадение глубины анализа для С1s и этих трех групп электронов позволяет говорить лишь о пропорциональности отношений интегральных интенсивностей F1s/С1s, F2s/С1s и FKLL/С1s средней относительной атомной концентрации F/С в трех приповерхностных слоях различной толщины.
Таким образом, для корректного измерения концентраций необходимо учитывать эффективную длину пробега соответствующих групп фотоэлектронов в веществе мишени, характеризующую толщину поверхностного слоя, доступную для анализа. Исходя из общих рассуждений, основанных на законе сохранения энергии, можно было бы предположить, что эффективная длина пробега должна быть пропорциональна кинетической энергии фотоэлектронов. Тем не менее, во многих монографиях указывается, что данная зависимость выражается степенной функцией с положительным показателем. Разные авторы приводят различные значения показателя степени, причем существенно меньше единицы, например 0.5 в [35].
Для решения поставленной задачи в общем случае требуется измерение концентрации с использованием далеко отстоящих друг от друга по шкале энергии нескольких линий одного элемента. Для ПВДФ это могут быть только линии F1s и F2s. Эти группы фотоэлектронов значительно отличаются кинетической энергией и, следовательно, эффективной длиной пробега. Поэтому их интенсивность будет нести информацию о количестве атомов фтора в двух поверхностных слоях различной толщины. Если градиент концентрации фтора по глубине отсутствует или невелик, что должно быть характерно для необлученного (исходного) ПВДФ, а также для случая малых экспозиций, то относительные концентрации F/С, определенные в результате одновременного измерения интегральных интенсивностей линий F1s, F2s и С1s, должны быть одинаковы. Таким образом, сравнение отношений F1s/С1s и F2s/С1s может позволить определить вид зависимости глубины анализа от кинетической энергии электронов. Несмотря на техническую неосуществимость одновременной регистрации этих линий, что несколько осложняет данную задачу, в ряде исследований она была успешно решена (например, [41]). Для пленки ПВДФ Kynar искомый показатель степени оказался очень близок к единице (0.98 ± 0.05). Это означает, что эффективная длина пробега прямо пропорциональна кинетической энергии электронов, по меньшей мере, в интервале 800–1450 эВ. Отметим, что для измерения F/C не требуются абсолютные численные значения эффективной длины пробега различных групп электронов. Достаточно знать лишь их отношения, факторы элементной чувствительности и функцию пропускания спектрометра.
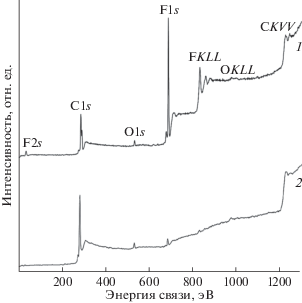
Установленный в [41] факт пропорциональности глубины анализа и кинетической энергии эмитированных электронов был использован в [21, 22] для изучения кинетики уменьшения относительной атомной концентрации F/C в приповерхностных слоях различной толщины при облучении пленки ПВДФ Kynar в течение 7200 мин (серия 4). Электронная эмиссия и радиационная карбонизация поверхности происходили под воздействием немонохроматического AlKα-излучения, энергия пропускания магнитного анализатора составляла 320 эВ. На рис. 12 изображены обзорные РФЭ-спектры исходной и облученной в этом эксперименте при максимальной экспозиции пленки ПВДФ Kynar. Небольшой не изменяющийся при экспозиции вклад электронов кислорода объясняется следами пластификатора, добавляемого в жидкий полимер при производстве пленки методом выдувной экструзии. На рисунке видно существенное уменьшение интенсивности спектральных особенностей, связанных с наличием фтора.
Рис. 12.
Обзорные спектры исходной (1) и облученной в течение 7200 мин (2) пленки ПВДФ Kynar. Спектральные особенности идентифицированы соответствующими символами атомных оболочек.
Исследования [21, 22] интересны также и попыткой проведения компонентного анализа С1s-спектров. Вклад фторметиновых групп визуально не наблюдался вследствие невысокого разрешения прибора при используемой энергии пропускания. Этот факт, как и недооцененное авторами влияние вторичных химических сдвигов на энергию связи С1s-электронов СН2-групп, вызывает сомнение в точности результатов компонентного анализа. Тем не менее компонентный анализ позволяет провести независимую оценку величины F/С при различных экспозициях и сравнить их с данными, полученными из отношений F1s/C1s и F2s/C1s.
Полученные результаты представлены на рис. 13. Из рисунка следует, что при малых экспозициях величины относительной атомной концентрации F/C, измеренные тремя независимыми способами, достаточно близки, однако по мере дефторирования все больше различаются, свидетельствуя о возникновении и увеличении градиента концентрации фтора по глубине. Причиной возникновения градиента, вероятно, является уменьшение количества дефторирующих пленку ПВДФ фотонов по мере проникновения излучения вглубь вещества. Поэтому средняя концентрация остаточного фтора тем больше, тем больше толщина анализируемого слоя.
Рис. 13.
Зависимости относительной атомной концентрации фтора от продолжительности облучения пленки ПВДФ Kynar. Символы ▲, ◻, ◆ демонстрируют кинетику уменьшения величин F/C, полученных, соответственно, из результатов измерений отношений F1s/C1s, F2s/C1s и компонентного анализа. Адаптировано из [21, 22].
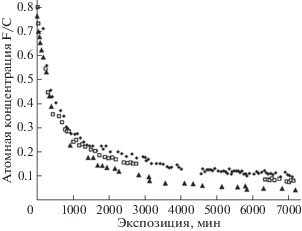
Из данных, представленных на рис. 13, следуют, по меньшей мере, три важных вывода. Во-первых, близость величин F/C, измеренных при малых экспозициях из отношений интегральных интенсивностей F1s/C1s и F2s/C1s, подтверждает пропорциональность эффективной длины пробега и, следовательно, глубины анализируемого слоя ПВДФ кинетической энергии эмитированных электронов. Во-вторых, оказалось, что даже весьма приблизительный компонентный анализ дает при малых экспозициях почти такую же величину F/C. В-третьих, при длительных экспозициях величина F/C, измеренная из отношения интегральных интенсивностей F1s/C1s и характеризующая среднюю концентрацию фтора в наиболее близком к поверхности слое вещества, меньше, чем величина F/C, измеренная из F2s/C1s. Это полностью соответствует упомянутым выше классическим бугеровским представлениям о поглощении излучения веществом. Отметим, однако, что компонентный анализ, для которого необходима регистрация только одного, а именно C1s-спектра, при длительных экспозициях дает наибольшее значение F/C. Этот факт не укладывается в предыдущие логические построения, поскольку кинетическая энергия С1s-электронов при фотоэффекте больше, чем F1s, но меньше, чем F2s-электронов.
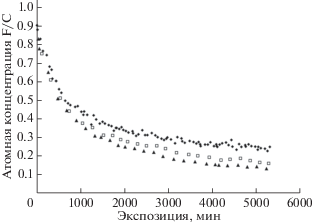
Связано ли это несоответствие с неточностями компонентного анализа в [21, 22] либо является закономерностью? Экспериментальные спектры электронов С1s, F1s и F2s, полученные при долговременных измерениях серии 3, и результаты компонентного анализа С1s-спектров, специально измеренных с лучшим разрешением при энергии пропускания 70 эВ [14], позволяют проверить воспроизводимость этого результата. Основные характеристики оборудования и условия проведения данного эксперимента описаны выше. На рис. 14 представлена кинетика уменьшения величины F/С, измеренной теми же тремя способами. Использованы те же обозначения, что на рис. 13. Из сравнения рис. 13 и 14 следует качественное согласие результатов измерений серии 3 и [21, 22], проявляется кажущееся нелогичным поведение кинетической кривой, полученной из компонентного анализа.
Рис. 14.
Зависимости относительной атомной концентрации фтора от продолжительности облучения пленки ПВДФ Kynar (серия 3). Символы ▲, ◻, ◆ демонстрируют кинетику уменьшения величин F/C, полученных, соответственно, из результатов измерений отношений F1s/C1s, F2s/C1s и компонентного анализа.
Возможное объяснение этого явления приведено в [21, 22]. При измерении кинетики уменьшения содержания фтора из отношения интегральных интенсивностей F1s/C1s и F2s/C1s учитывают, что отношения эффективных длин свободного пробега F1s, F2s и C1s фотоэлектронов различны. Однако рассчитать изменение эффективной длины пробега в процессе дефторирования практически нельзя, возможна лишь качественная оценка влияния этого эффекта на результат измерения. На самом деле модификация плотности образца неоднородна по глубине. F1s-электроны эмитируют из наиболее близкого к поверхности слоя полимера, который имеет наименьшую плотность вследствие более сильного дефторирования. Поэтому их длина свободного пробега увеличивается при облучении быстрее, чем F2s-электронов. Эффективная длина пробега C1s-электронов увеличивается с промежуточной скоростью. Совокупность этих эффектов приводит к переоценке содержания фтора, измеряемого из отношения интенсивностей F1s/С1s и, наоборот, к его недооценке при измерении, основанном на отношении F2s/С1s. Эти отклонения от истинного среднего содержания фтора в двух анализируемых приповерхностных слоях различной толщины нарастают с увеличением экспозиции.
В справедливости такого достаточно разумного объяснения можно было бы убедиться, если бы при увеличении экспозиции наблюдался монотонный рост интенсивности С1s-спектра. Однако в процессе эксперимента обнаруживаются лишь немонотонные колебания этой величины с отклонениями от среднего значения порядка 15% в большую и меньшую стороны. Эти колебания могут быть связаны с различными факторами. Конкурирующее влияние на плотность вещества оказывает уменьшение расстояний между атомами углерода в цепочке за счет образования кратных связей. Этот же эффект приводит к изменению морфологии поверхности [17, 21]: после 7200 мин облучения первоначально прозрачная и абсолютно плоская пленка становится неравномерно коричневой и гофрированной. Наиболее интенсивно окрашенный край образца даже скрутился в виде свитка и треснул перпендикулярно оси скручивания приблизительно посредине. Вогнутая поверхность скрутки была обращена в сторону источника излучения, что свидетельствует об образовании на ней более коротких кратных связей углерод–углерод. Гофрировка способна существенно влиять на интенсивность С1s-спектра за счет затенения отдельных участков пленки, а также за счет воспрепятствования попаданию фотоэлектронов на входную щель анализатора. Нельзя исключить и постепенное изменение мощности излучения при включении рентгеновского источника за счет разогрева алюминиевого анода.
3. ПРИМЕНЕНИЕ РФЭС И ОЖЕ-СПЕКТРОСКОПИИ ДЛЯ ИЗУЧЕНИЯ МОДИФИКАЦИИ ВАЛЕНТНОГО СОСТОЯНИЯ УГЛЕРОДА ПРИ РАДИАЦИОННОЙ КАРБОНИЗАЦИИ ПВДФ
Дефторирование ПВДФ под воздействием излучения, регистрируемое в многочисленных РФЭС-экспериментах, сопровождается, как показано в [16], эмиссией водорода и фтористого водорода без разрыва связей углерод–углерод в цепочечном скелете полимера. Это позволяет обозначить обсуждаемый процесс терминами “радиационное дегидрофторирование”, “радиационная деградация” или “радиационная карбонизация”. Радиационная карбонизация наиболее приемлема с химической точки зрения, поскольку отражает увеличение доли углерода в облучаемых образцах. Кратные связи, образующиеся в дегидрофторированных фрагментах цепей, и межцепочечные сшивки изменяют характерный для ПВДФ sp3-тип гибридизации валентных электронов атомов углерода. В результате реальный облученный полимер может оказаться сложной комбинацией углеродистых структур [16]. Так как именно тип гибридизации определяет структуру и многие физико-химические свойства углеродсодержащих объектов [42], очень важно с точки зрения и теории, и практики разрабатывать методы и методики идентификации и мониторинга состояния валентной оболочки атома углерода. Некоторые перспективы в этом плане имеет анализ спектров электронной эмиссии [32].
Надежная идентификация возможна при сравнении со спектрами, форма которых характерна для определенного типа гибридизации. Такими, например, являются С1s- и оже-спектры сильно ориентированного пиролитического графита (СОПГ) – чистого углерода с sp2-типом гибридизации валентных электронов. В общем случае необходим комплексный анализ формы спектров эмиссии всех групп электронов углерода, в том числе и менее интенсивной валентной полосы.
На рис. 15 представлены оригинальные С1s-спектры СОПГ, полиэтилена и пленки ПВДФ, карбонизированной AlKα-излучением в течение 9000 мин (серия 5) в спектрометре ЭС ИФМ-4. Для краткости этот материал условно назовем карбиноидом. Относительную атомную концентрацию остаточного фтора F/C на поверхности этого вещества измеряли из отношения площадей под пиками F1s/C1s и F2s/C1s. Она составила, соответственно, 0.039 и 0.070. Это означает, что в пределах анализируемого приповерхностного слоя пленки не более 7% атомов углерода могут быть связаны с одним атомом фтора и не более 3.5% – с двумя. Так как углеродный скелет полимера не претерпевает разрывов [16], межцепочечные сшивки с формированием тетраэдрической конфигурации вряд ли возможны. Иными словами, оказывается, что гибридизация валентных электронов не менее 96% атомов углерода отлична от sp3.
Рис. 15.
С1s-спектры СОПГ (◆), полиэтилена (+) и карбонизированной AlKα-излучением в течение 9000 мин пленки ПВДФ (карбиноида – ◻).
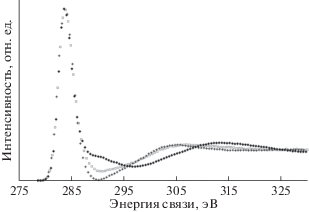
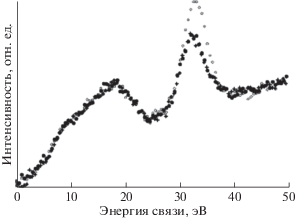
Для удобства сравнения спектры выравнены по положению главного максимума и нормированы на его высоту. Ширина на половине высоты главного пика во всех трех спектрах одинакова, что свидетельствует об однородности электростатического заряда поверхностей полиэтилена и карбиноида. Из рисунка видно, что наибольшие отличия спектра СОПГ наблюдаются в области сателлита неупругих энергетических потерь. Они связаны с наличием в графите подвижных π-электронов, характерных для sp2-гибридизированного состояния. В то же время С1s-спектры карбиноида и полиэтилена очень схожи, а наблюдаемые между ними отличия определенно могут быть связаны с вкладом остаточных дифторметиленовых и фторметиновых групп вследствие неполноты дегидрофторирования ПВДФ.
Таким образом, если бы интерпретировали данные, имея только С1s-спектры, то пришлось бы предположить, что гибридное состояние валентных электронов атомов карбонизированного образца и полиэтилена одно и то же – sp3. Однако такое предположение противоречит вкладу CF-групп в тонкую структуру С1s-спектров. Отличие доминирующего типа гибридизации карбиноида от sp3 выявляется при анализе формы спектров валентных электронов этих же трех углеродистых объектов, представленных на рис. 16. На этом рисунке спектр карбиноида радикально отличается по форме от спектра полиэтилена, но весьма напоминает спектр графита. Небольшие отличия спектров карбиноида и графита можно вполне объяснить вкладом в первый из них фотоэлектронов F2p и F2s [43], но это, как следует из последующего рассмотрения, отнюдь не означает, что валентные электроны атомов карбиноида находятся в sp2-гибридизированном состоянии.
Рис. 16.
Спектры валентных электронов СОПГ (◆), полиэтилена (+) и карбонизированной в течение 9000 мин AlKα-излучением пленки ПВДФ (карбиноида – ◻).
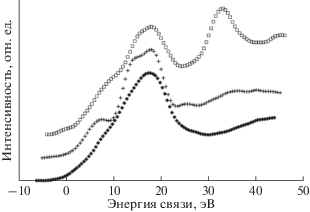
Форма спектра валентных электронов исходного ПВДФ, к сожалению, не позволяет судить о типе гибридизации 2s- и 2p-электронов углерода вследствие существенного вклада эмиссии из F2p-состояний, а также F2s-фотоэлектронов, возбужденных фотонами AlKα3,4. При радиационной карбонизации этот вклад постепенно убывает, как показано на рис. 17.
Рис. 17.
Модификация формы спектров валентных электронов пленки ПВДФ Kynar под воздействием AlKα-излучения различной продолжительности. В интервале значений энергии связи 0–25 эВ для лучшей визуализации различий спектры представлены также и в масштабе, увеличенном по вертикали.
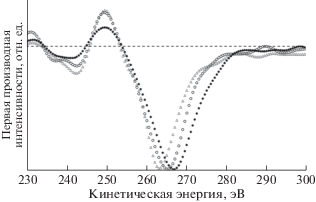
Перейдем к рассмотрению результатов оже-спектроскопии. Оже-спектр углерода является самосверткой валентных состояний р-симметрии, форма которой осложнена многоэлектронными эффектами [44, 45]. Данные экспериментов по оже-спектроскопии обычно принято представлять в виде первой производной интенсивности эмиссии по кинетической энергии, чтобы лучше выявлять нюансы довольно бесструктурного профиля спектра. В литературе существует и другой подход, основанный на сопоставлении интегрального экспериментального спектра с самосвертками парциальных валентных состояний симметрии s и р. К сожалению, в большом количестве таких исследований (например, [46, 47]) игнорируется физическая невозможность самосвертки π*π-состояний в случае графита, совершенно очевидная вследствие локальной природы оже-эффекта и наличия единственного π-электрона на атом [48]. Тем не менее достоверно показано, что спектры оже-эмиссии атомов углерода (CKVV) имеют специфические особенности, позволяющие различать sp2- и sp3-гибридизированные состояния [46, 47, 49].
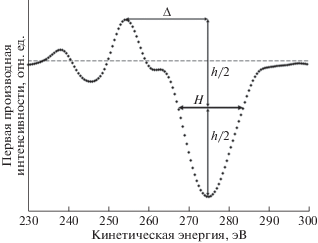
На рис. 18 к первым производным оже-спектров углерода СОПГ, полиэтилена и карбиноида добавлена производная СKVV-спектра исходного ПВДФ Kynar. Для удобства сравнения спектры совмещены по энергетическому положению положительного экстремума, а нормированы на величину амплитуды отрицательного экстремума. На рис. 18 хорошо заметны два радикально различающихся параметра формы первых производных CKVV-спектров СОПГ и остальных углеродистых материалов: энергетический зазор между положительным и отрицательным экстремумами и ширина отрицательной особенности, доминирующей в дифференциальном оже-спектре. Оба параметра явно больше в случае графита. Первый из них предложен в качестве характеристики типа гибридизации в [49]: он оказался для алмаза, полиэтилена [49] и исходного ПВДФ равен ~13.5, а для СОПГ – ~20 эВ [17, 49]. Для характеристики ширины доминирующей отрицательной особенности авторы [17, 21] предложили использовать длину горизонтального отрезка, заключенного между ее ветвями на половине амплитуды интенсивности спектра. На рис. 19 представлена схема измерения обоих параметров (соответственно, Δ и Н).
Рис. 18.
Первые производные оже-спектров СОПГ (◆), полиэтилена (+), исходной пленки ПВДФ (Δ) и карбонизированной пленки ПВДФ, облученной в течение 9000 мин AlKα-фотонами (карбиноида – ◻). Штриховой линией обозначена нулевая линия.
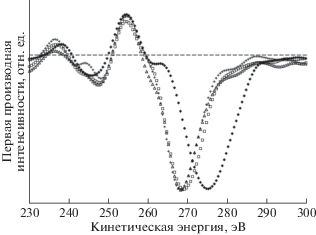
Рис. 19.
Схема измерения параметров Δ и Н дифференциальных оже-спектров на примере спектра СОПГ. Штриховой линией обозначена нулевая линия.
Все приведенные на рис. 15–18 данные получены при энергии пропускания 320 эВ. Их комплексный анализ свидетельствует о возможности идентификации графитоподобного (спектры С1s и CKVV) и алмазоподобного (спектр валентной полосы) характера упорядочения углерода, однако не позволяет сделать однозначный вывод о гибридном состоянии валентных электронов в атомах материала, который предварительно назван карбиноидом. Спектры электронной эмиссии этого вещества не имеют явных особенностей, характерных для sp2- и sp3-гибридизированных состояний углерода. Поэтому можно с некоторой долей осторожности предположить существование в нем атомов углерода, упорядоченных в цепочки. В этой связи название “карбиноид” вполне уместно.
Для решения проблемы идентификации строения вещества, синтезированного на поверхности пленки ПВДФ Kynar при длительном облучении фотонами мягкого рентгеновского облучения, авторы [17, 21] предложили интересную идею, основанную на общепризнанном представлении о более высокой термодинамической устойчивости при обычных условиях форм углерода sp2-типа [42]: если новый синтезированный углеродистый материал с заранее неизвестными типами гибридизации электронов подвергнуть бомбардировке ионами, то, вероятнее всего, некоторая часть атомов перейдет в более устойчивое и легче идентифицируемое sp2-состояние.
При проведении экспериментов серии 4 пленки ПВДФ Kynar карбонизировали AlKα-излучением в течение 7200 мин в спектрометре ЭС ИФМ-4 [17, 21, 22]. Относительную атомную концентрацию остаточного фтора F/C на поверхности пленки определяли с использованием отношения площадей под пиками F1s/C1s и F2s/C1s. Она составляла, соответственно, 0.043 и 0.078 после завершения облучения фотонами.
Переход валентных оболочек атомов углерода в новое гибридное состояние приводит к изменению параметров Δ и Н дифференциальных оже-спектров. При экспозиции пленки в течение 1000–1200 мин оба параметра монотонно возрастают, соответственно, от 13.7 до 15.1 и от 11 до 12.6 эВ, а при дальнейшем облучении практически не меняются. Данные изменения параметров, по-видимому, отражают уменьшение количества атомов углерода в sp3-состоянии.
Затем облученная фотонами поверхность образца подвергалась дополнительному облучению расфокусированным пучком ионов Ar+ энергией ~1 кэВ и плотностью тока 6 мА/м2. Четыре последовательные дозы бомбардировки составляли 0.2, 1.1, 3.9 и 9.6 × 1016 см–2. Относительная атомная концентрация остаточного фтора на поверхности пленки после максимальной дозы бомбардировки, измеренная из отношений F1s/C1s и F2s/C1s, стала еще меньше (соответственно, 0.025 и 0.05).
Ионная бомбардировка приводит к изменению формы спектров электронной эмиссии. На рис. 20 представлены первые производные CKVV-спектров карбонизированного образца, облученного фотонами в течение 6000 мин, а затем после дальнейшего облучения до 7200 мин, подвергнутого ионной бомбардировке дозами 1.1, 3.9 и 9.6 × × 1016 см–2. Для сравнения также приведен спектр СОПГ. При увеличении дозы до 3.9 × 1016 см–2 параметры Δ и Н возрастают сходным образом до 17.6 и 14.8 эВ соответственно и остаются приблизительно такими же при дозе 9.6 ×1016 см–2, не достигая значений ~20 и ~16 эВ, характерных для СОПГ.
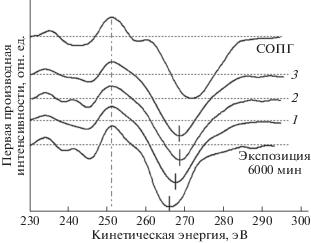
Непосредственное сравнение С1s-спектров этого образца до и после ионной бомбардировки оказалось затруднительным. Вследствие изменения конструкции держателя образца для обеспечения доступа ионов к исследуемой поверхности пленка стала заряжаться неоднородно, что приводило к уширению спектральных линий. Тем не менее, поскольку данное уширение оставалось постоянным и не зависело от дозы, оказалось возможным проанализировать модификацию формы сателлитов С1s-линии при увеличении дозы. Результаты такого анализа графически представлены на рис. 21. Как и в случае оже-спектров, заметны небольшие, но явные признаки увеличения концентрации атомов углерода с sp2-типом гибридизации валентных состояний: увеличение интенсивности спектра в области так называемого π-плазмона и сдвиг максимума широкого π + σ-плазмона в сторону больших значений энергии связи. Отметим, что первый из них проявляется даже несмотря на уменьшающуюся при бомбардировке концентрацию остаточных дифторметиленовых и фторметиновых групп, С1s-электроны которых дают вклад в эмиссию при тех же значениях энергии связи. Поскольку, как отмечалось выше, признаки sp3-гибридизации отсутствуют в спектрах валентных электронов карбонизированного фотонами вещества, увеличение числа атомов углерода в sp2-состоянии при бомбардировке ионами, вероятнее всего, происходит за счет атомов в состоянии sp, т.е. характерных для одномерного характера упорядочения.
Рис. 21.
Спектры сателлитов С1s-линий образца пленки ПВДФ Kynar, карбонизированной фотонами в течение 7200 мин, после ионной бомбардировки до доз 1.1 (⚪) и 9.6 × 1016 см–2 (⚫). Для удобства визуального сравнения спектры предварительно выравнены по энергетическому положению главных максимумов С1s-линий (не показаны).
Очевидные свидетельства различного гибридного состояния атомов углерода после радиационной карбонизации AlKα-фотонами (серия 3) и ионами аргона (серии 7, 8, табл. 2) также получены в [33]. Две независимые серии бомбардировок ионами проведены со ступенчатым нарастанием дозы в одинаковом дозовом интервале, но с разным количеством ступеней, соответственно, восемь и десять, и различной общей продолжительностью экспериментов. Максимальная кумулятивная доза 4.5 × 1015 см–2 была одинакова в обеих сериях. РФЭС и CKVV-спектры измеряли в промежутках между бомбардировками так же, как в серии 3. Проведение независимых измерений в сериях 7 и 8 было связано с необходимостью надежного выявления принципиальных отличий результатов двух воздействий разных типов на полимерную пленку. Измерения показали, что дефторирование поверхности исследуемых пленок вызывают оба воздействия, но валентные состояния атомов углерода в карбонизированных слоях различны.
На рис. 22 представлены спектры валентных электронов, полученных почти в самом конце экспериментов серий 3 (экспозиция 5276 мин), а также 7 и 8 (максимальная доза). Относительные атомные концентрации фтора F/C, измеренные из отношений площадей F1s/С1s и F2s/С1s равны, соответственно, 0.135 и 0.164 (серия 3), 0.068 и 0.105 (серия 7), 0.071 и 0.105 (серия 8). Визуальное сравнение интенсивностей F2s-пиков (~33 эВ, рис. 22) показывает, что содержание остаточного фтора действительно больше в случае воздействия фотонов, а в двух сериях ионной бомбардировки практически одинаково.
Рис. 22.
Спектры валентных электронов, полученных при завершении измерений серий 3 (⚪), 7 (+) и 8 (⚫).
Спектры, снятые при завершении серий 7 и 8, очень похожи по форме, и это характерно не только для валентных полос. Несмотря на различия условий проведения экспериментов этих серий, формы спектров эмиссии одинаковых групп электронов при максимальной дозе бомбардировки практически идентичны. Поэтому, чтобы не усложнять рис. 22 и 23, на них приведены спектры эмиссии, соответственно, электронов С1s и CKVV, полученных при завершении серий 3 и 8. Кроме того, на рис. 24 добавлена первая производная по кинетической энергии CKVV-спектра исходного ПВДФ Kynar. Все спектры (рис. 22–24) получены при энергии пропускания анализатора 320 эВ. На рис. 23 лучше, чем на рис. 21, проявляются спектральные особенности, характерные для sp2-состояния атомов углерода: в случае ионной бомбардировки наблюдается как увеличение интенсивности C1s-спектра в области π-плазмона несмотря на меньшую концентрацию остаточных групп CF2 и CF, так и сдвиг максимума сателлита C1s-линии в сторону больших значений энергии связи. Спектральные данные об оже-эмиссии углерода (рис. 24) также свидетельствуют об увеличении содержания sp2-гибридизированных атомов после ионной бомбардировки и хорошо согласуются с результатами [17, 21, 22], детально описанными выше. Действительно, карбонизация пленки AlKα-фотонами приводит к некоторому росту параметров Δ и Н первых производных спектров, а воздействие ионной бомбардировки на их увеличение оказывается еще более существенным [33].
Рис. 23.
Спектры C1s-электронов, полученные при завершении измерений серий 3 (⚪) и 8 (⚫). На вставке области сателлитов представлены в увеличенном виде для наглядности.
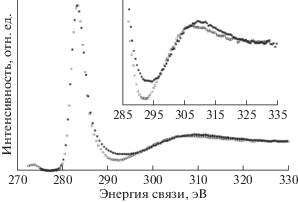
Рис. 24.
Первые производные CKVV-спектров, полученные при завершении измерений серий 3 (⚪) и 8 (⚫), а также исходного ПВДФ Kynar (Δ). Штриховой линией обозначена нулевая линия.
Таким образом, анализ всей совокупности результатов, полученных в [17, 21, 22, 33], приводит к выводу о формировании на поверхности ПВДФ при воздействии фотонов мягкого рентгеновского излучения одномерных углеродных структур. В настоящее время возможна лишь косвенная и весьма приблизительная количественная оценка их содержания в доступном для анализа приповерхностном слое, основанная на измерении концентрации остаточного фтора. Тем не менее само существование таких цепочек, состоящих только из углерода, можно считать принципиально доказанным, поскольку ионная бомбардировка приводит к частичному переходу атомов в термодинамически более устойчивое sp2-гибридизированное состояние. Наиболее вероятным механизмом такого перехода представляются межцепочечные сшивки, инициируемые ударным воздействием ионов.
4. МОДЕЛИ РАДИАЦИОННОЙ КАРБОНИЗАЦИИ ПВДФ
4.1. Неоднородность распределения остаточного фтора по глубине
Как уже отмечалось выше, концентрации фтора, измеренные с использованием отношений площадей под спектрами F1s/C1s и F2s/C1s, при воздействии на пленку ПВДФ фотонов мягкого рентгеновского излучения при малых экспозициях близки. По мере увеличения экспозиции они монотонно убывают с различной скоростью. Это свидетельствует о возникновении и увеличении неоднородности распределения остаточного фтора по глубине анализируемого слоя, что может быть связано с поглощением фотонов веществом полимера.
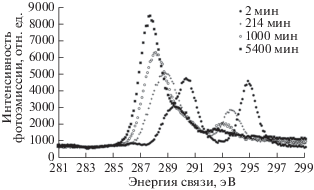
В ряде исследований [33, 50–52 ] было обнаружено еще одно свидетельство данного явления: изменение соотношения между интенсивностями F1s-линии и ее сателлита неупругих потерь. На рис. 25 представлены спектры F1s-электронов пленки ПВДФ Kynar на различных стадиях радиационной карбонизации. Для удобства визуального сравнения спектры выравнены по энергетическому положению и высоте главных максимумов. Из рисунка видно, что интенсивность сателлита по отношению к интенсивности F1s-линии возрастает с увеличением экспозиции, т.е. с уменьшением содержания остаточного фтора.
Рис. 25.
Спектры F1s-электронов пленки ПВДФ Kynar на различных стадиях радиационной карбонизации при облучении AlKα-фотонами.
Этот эффект, как показывает анализ данных многочисленных серий экспериментов, носит монотонный характер и обусловлен тем, что чем глубже от поверхности находится эмитирующий атом, тем больше вероятность неупругого взаимодействия фотоэлектронов с электронами твердого тела в процессе транспорта к поверхности. Равномерное уменьшение концентрации фтора, например, в два раза, в доступном для анализа слое глубиной L приведет к уменьшению вдвое интегральной интенсивности как F1s-пика, так и сателлита. Теперь предположим, что верхняя часть этого поверхностного слоя глубиной L/2 не содержит атомов фтора, а в более глубокой оставшейся части (также L/2) концентрация фтора не изменилась по сравнению с исходной пленкой. В этом случае интегральная интенсивность всего F1s-спектра уменьшится также в два раза, но интенсивность самого F1s-пика уменьшится больше, чем в два раза, а интенсивность сателлита – меньше, чем вдвое, вследствие увеличения вероятности неупругих потерь. Поэтому отношение интенсивности сателлита к интенсивности линии будет возрастать.
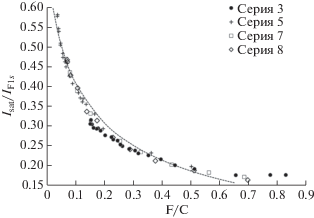
Раздельное измерение интегральных интенсивностей (площадей под пиками) F1s-линии и ее сателлита вряд ли может дать надежные результаты, что связано с неоднозначностью процедуры выделения этих составляющих из спектров, форма которых, как видно из рис. 25, постоянно меняется. Более определенную качественную информацию об эволюции распределения атомов фтора по глубине в процессе поверхностной карбонизации полимера дает зависимость отношения интенсивности сателлита в точке на фиксированном энергетическом расстоянии (25 эВ) от положения максимума F1s к величине этого максимума (Isat/IF1s) от концентрации F/C. Эта зависимость представлена на рис. 26. Чтобы не перегружать рисунок, взяты данные, полученные при измерениях лишь 3, 5, 7 и 8 серий. Однако полученная зависимость оказалась универсальной не только для кинетики радиационной карбонизации ПВДФ Kynar, но и множества других многократно исследованных пленок ПВДФ. Относительная атомная концентрация F/C измерена с использованием отношения площадей под пиками F1s/С1s. Хотя уменьшение содержания остаточного фтора по мере карбонизации имеет очевидную тенденцию к насыщению (рис. 7, 8, 10, 13, 14), оно приводит, как видно из рис. 26, к все более резкому увеличению относительной интенсивности сателлита F1s-линии.
Рис. 26.
Зависимость отношения Isat/IF1s интенсивности F1s-спектра в точке сателлита (25 эВ выше положения главного максимума) к величине главного максимума от концентрации остаточного фтора F/С. Точки – эксперимент, штриховая линия – результат модельного расчета.
Штриховая линия на рис. 26 демонстрирует результаты полуколичественного моделирования обсуждаемой зависимости. Учитывая экспоненциальный характер поглощения излучения веществом, можно предположить, что относительная атомная концентрация остаточного фтора ${{n}_{{\text{F}}}}\left( x \right)$ в слое вещества бесконечно малой толщины на глубине х выражается следующим соотношением [51, 52 ] :
Средняя относительная атомная концентрация остаточного фтора в анализируемом слое толщиной L
Коэффициент поглощения AlKα-излучения веществом ПВДФ можно приближенно оценить, усреднив известные данные для политетрафторэтилена и полиэтилена (2.2 × 105 м–1 [51]). Однако такая величина оказывается слишком малой для объяснения разницы значений F/C, измеряемых с использованием отношений интенсивностей F1s/С1s и F2s/С1s. В частности, для объяснения данных, полученных при завершении измерений серии 3, необходима величина χ = 6 × 107 м–1. Предположение о решающей роли вторичных электронов фильтрующей фольги в формировании концентрационного профиля остаточного фтора по глубине, высказанное в [51], опровергается результатами специально проведенного более позднего эксперимента [33]. Вполне возможно, что величина χ определяется поглощением характеристических FKα-фотонов, энергии которых достаточно для фотоионизации атомов углерода, либо иными вторичными процессами, также инициирующими элиминирование НF. В модельном полуколичественном расчете зависимости отношения Isat/IF1s от содержания остаточного фтора, результат которого представлен штриховой линией на рис. 26, для упрощения вычислений параметр χ предполагали равным 6 × 107 м–1 и постоянным, поскольку вариации параметра χ в достаточно широких пределах не оказывают воздействия на общий характер зависимости.
Если выделить слой вещества бесконечно малой толщины dx, находящийся на расстоянии x от поверхности, то число фотоэлектронов dN(x), вылетевших из данного слоя в направлении входной апертуры анализатора, пропорционально $A{{n}_{{\text{F}}}}\left( x \right)Sdx,$ где А – безразмерный коэффициент, зависящий от интенсивности фотонов, их энергии и, соответственно, вероятности фотоионизации F1s-оболочки, а S – площадь поверхности образца, с которой происходит эмиссия регистрируемых спектрометром электронов.
Интегрируя данное выражение, получаем, что общее число F1s-электронов $~N$, вылетевших из приповерхностного слоя толщиной L, пропорционально $ALSc$ + 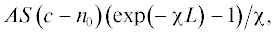 или после сокращения на площадь анализируемого слоя и постоянную А:
или после сокращения на площадь анализируемого слоя и постоянную А:
Число фотоэлектронов $~N{\kern 1pt} '\left( x \right),$ вылетевших из того же слоя dx и потерявших при этом энергию, пропорционально $\alpha xA{{n}_{{\text{F}}}}\left( x \right)Sdx,$ где α – коэффициент, равный вероятности потери фотоэлектроном энергии при прохождении слоя вещества единичной толщины. Интегрируя данное выражение по толщине слоя $L$, из которого могут вылететь остовные электроны фтора, получаем после аналогичного сокращения:
Экспериментальные значения отношения Isat/IF1s сравнивали с расчетными величинами ${{N{\kern 1pt} '} \mathord{\left/ {\vphantom {{N{\kern 1pt} '} {\left( {N - N{\kern 1pt} '} \right)}}} \right. \kern-0em} {\left( {N - N{\kern 1pt} '} \right)}}.$ При определенных постоянных значениях α = 2.5 × 108 м–1, $\chi $ = 6 × 107 м–1 и K = = 1.5 × 10–8 м данная простейшая математическая модель неплохо согласуется с экспериментальной зависимостью (рис. 26). Расчетные значения $L$ увеличиваются от 0.83 до 3.6 нм при уменьшении параметра n0 от 1 до 0.02. Таким образом, простая математическая модель, предполагающая монотонный экспоненциальный характер убывания содержания остаточного фтора с глубиной при радиационном дегидрофторировании, дает достаточно разумное согласие с экспериментальными данными. Тем не менее вопрос об аномально большой величине поглощения дегидрофторирующего излучения веществом пленки остается открытым.
Также вызывает удивление и требует дополнительного изучения факт, что характер распределения фтора по глубине не зависит от того, фотоны (серии 3, 5) или ионы (серии 7, 8) используются для радиационной карбонизации. Из рис. 26 видно, что все экспериментальные точки с большой точностью укладываются на одну кривую, отражающую усиление неоднородности распределения атомов фтора по глубине в процессе дефторирования приповерхностного слоя пленки ПВДФ. Само по себе более высокое содержание остаточного фтора в более удаленных от облучаемой или бомбардируемой поверхности слоях вещества вполне предсказуемо. Однако данные, представленные и обсужденные в предыдущих разделах, убедительно свидетельствуют о различном электронном строении и, следовательно, различной структурной организации углерода в слоях пленок, карбонизируемых фотонами и ионами. Из общих соображений ясно, что должна быть различна даже плотность вещества, из которого происходит фотоэмиссия, а значит и эффективная длина свободного пробега фотоэлектронов, т.е. толщина слоя, доступного для анализа методами РФЭС и оже-спектроскопии.
4.2. Кинетика распада групп CF2 и CF при воздействии излучения
Как следует из предыдущей части данного обзора, с середины 1980-х годов были проведены многочисленные исследования карбонизации ПВДФ в результате облучения фотонами. Тем не менее микроскопический механизм элиминирования фтора до сих пор полностью не изучен. Кинетические исследования могут дать ценную информацию по этой проблеме. Например, в [38] на основе данных РФЭС была впервые предложена простая математическая модель кинетики одновременного удаления фтора и водорода с поверхности ПВДФ. Она основана на предположении, что удаление фтора из группы CF2 (CF) возможно лишь в том случае, если она соседствует с группой CН2 (CН). Именно поэтому уменьшение числа групп CF2 (CF) за бесконечно малый промежуток времени пропорционально квадрату их текущего общего содержания.
Это предположение, однако, может быть не совсем точным. Даже для идеальной структуры ПВДФ из-за случайного характера взаимодействия фотонов с полимерным остовом на любой стадии карбонизации существует возможность образования изолированных групп CF2, CF, CН2 и CН, например,
Таким образом, несмотря на хорошее согласие с экспериментальными данными, модель [38] нуждалась в дальнейшем уточнении, поскольку она не учитывала практической невозможности полного удаления фтора. Кроме того, общая продолжительность воздействия AlKα-фотонов в [38] составляла около 1000 мин, а относительное содержание F/C остаточных атомов фтора по окончании эксперимента – около 0.4. Последующие измерения серии 3 [14] позволили провести более глубокую деградацию ПВДФ и проверить применимость предложенной в [38] модели при меньших значениях F/C. Кроме того, лучшее спектральное разрешение позволило авторам [14] провести деконволюцию пиков C1s на отдельные компоненты, отличающиеся различными химическими сдвигами. Такая обработка, детально описанная выше, дала экспериментальную возможность выявить эволюцию не только групп CF2, но и CF (рис. 10).
Изолированные группы CF2 и CF останутся в пленке при продолжительном облучении, поскольку в ближайшем окружении нет атомов водорода. Содержание этих групп N∞ вряд ли может существенно меняться в различных экспериментах, проведенных в одинаковых условиях, из-за очень большого количества атомов на поверхности облученного образца, а также из-за статистического усреднения. На этот параметр может влиять различная мощность источников излучения за счет изменения скорости образования изолированных фторсодержащих групп. Таким образом, основное уравнение исходной модели [38] должно быть переписано в таком виде, чтобы включить этот новый параметр (индексы 1 и 2 будут далее использоваться для обозначения параметров, относящихся, соответственно, к группам CF2 и CF):
(1)
$\frac{{{\text{C}}{{{\text{F}}}_{2}}}}{{\text{C}}} = {{N}_{1}} + {{N}_{{1\infty }}} = \frac{{{{N}_{{1(0)}}}}}{{1 + {{k}_{1}}{{N}_{{1(0)}}}{\kern 1pt} t}} + {{N}_{{1\infty }}},$На рис. 27 приведены результаты, демонстрирующие кинетику деградации CF2-групп. Они получены из компонентного анализа спектров C1s, измеренных в экспериментальных сериях 1, 2 и 3 (маркеры), и расчета по уравнению (1) (штриховые и пунктирная линии). Из рисунка видно, что при подборе для каждой серии определенной совокупности параметров N1∞, N1(0) и k1 (табл. 4) можно добиться хорошего согласия экспериментальных и расчетных данных.
Рис. 27.
Сравнение экспериментальных (точки) и расчетных зависимостей (линии) относительной концентрации CF2-групп от продолжительности экспозиции в сериях 1, 2 и 3.
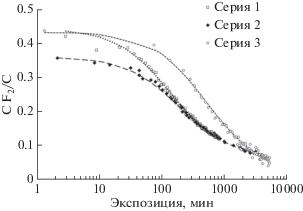
Таблица 4.
Параметры уравнения (1), при которых достигается наилучшее соответствие экспериментальным результатам серий 1, 2 и 3
| № серии | N1(0) | k1, мин–1 | N1∞ |
|---|---|---|---|
| 1 | 0.380 | 0.019 | 0.063 |
| 2 | 0.300 | 0.017 | 0.060 |
| 3 | 0.412 | 0.0047 | 0.024 |
Проведем краткий анализ результатов, представленных в табл. 4. Отличия параметров N1(0) в разных экспериментах друг от друга и значения 0.5, ожидаемого из структурной формулы ПВДФ, скорее всего, связаны с неодинаковой продолжительностью времени между включением рентгеновского источника (фактическим началом облучения образца) и регистрацией первого в каждой серии С1s-спектра. Это тем более вероятно, если учесть, что в начале измерений при высокой концентрации фтора дефторирование происходит с максимальной скоростью (рис. 7, 8, 10, 13, 14). В сериях 1 и 2 условия экспериментов были одинаковы, поэтому значения параметров k1 и N1∞ близки. Меньшая мощность источника в экспериментах серии 3 обусловила существенно меньшие скорости как собственно дефторирования, так и образования изолированных CF2-групп. Последний фактор демонстрирует принципиальную возможность за счет уменьшения мощности источника глубокого, хотя и более длительного дефторирования полимера. В [38] параметр k1 равен 0.0043, а существование изолированных групп не учитывали, и, следовательно, параметр N1∞ не вводили. Отметим, что в [38] и при измерениях серии 3 [14] эксперимент проводился в одинаковых условиях, что и обусловило близкие значения k1.
Тем не мене, очевидно, что чем меньше в расчетном уравнении варьируемых параметров, тем надежнее окажутся результаты сопоставления с экспериментом. Двухпараметрическое уравнение можно получить, если учесть простым вычитанием текущее содержание не остаточных CF2-групп, а тех, которые уже деградировали в промежуток времени от нуля (начало сканирования первого в серии спектра C1s) до t:
(2)
$\begin{gathered} N_{{\text{1}}}^{'} = {{N}_{{1(0)}}} - {{N}_{1}} = {{N}_{{1(0)}}} - \frac{{{{N}_{{1(0)}}}}}{{1 + {{k}_{1}}{{N}_{{1(0)}}}t}} = \\ = {{N}_{{1(0)}}}\left( {\frac{{{{k}_{1}}{{N}_{{1(0)}}}t}}{{1 + {{k}_{1}}{{N}_{{1(0)}}}t}}} \right). \\ \end{gathered} $(3)
$N_{{\text{1}}}^{'}\left( t \right) = N_{{\text{1}}}^{'}\left( {t = 0} \right) + {{N}_{{1(0)}}}\left( {\frac{{{{k}_{1}}{{N}_{{1(0)}}}t}}{{1 + {{k}_{1}}{{N}_{{1(0)}}}t}}} \right).$Рис. 28.
Зависимости текущего содержания деградированных групп CF2 (◇), CF (◼) и остаточных групп CF (⚪) от продолжительности облучения AlKα-фотонами в серии 3. Адаптировано из [14].

Приведенные выше соображения и принятые в [38] допущения применимы и к эволюции CF-групп. Таким образом, соответствующее уравнение должно иметь форму, очень похожую на уравнение (3):
(4)
$N_{{\text{2}}}^{'}\left( t \right) = N_{{\text{2}}}^{'}\left( {t = 0} \right) + {{N}_{{2(0)}}}\left( {\frac{{{{k}_{2}}{{N}_{{2(0)}}}t}}{{1 + {{k}_{2}}{{N}_{{2(0)}}}t}}} \right).$Хорошее согласие расчетных и экспериментальных данных подтверждает справедливость основных допущений, сделанных ранее [38]: деградация групп CF2 (CH2) и CF (CH) при воздействии мягкого рентгеновского излучения подчиняется дифференциальным уравнениям второго порядка, что позволяет предположить двухступенчатый процесс с равными текущими количествами метиленовых и дифторметиленовых, а также метиновых и фторметиновых групп. Длительная экспозиция пленки ПВДФ и повышение разрешения спектров позволили уточнить модель деградации [14]. Даже очень продолжительное облучение фотонами не может обеспечить полного дегидрофторирования поверхности ПВДФ вследствие образования изолированных групп CF2 (CF) и CH2 (CH), которые никогда не смогут деградировать в условиях, применяемых в конкретном исследовании. Сравнительный анализ экспериментальных данных серии 3 и модельного расчета дает значения относительной концентрации изолированных групп CF2 (CH2) и CF (CH), соответственно, 2.4 и 14.1%. Константы скорости деградации k1 и k2 групп CF2 и CF различны, но одного и того же порядка величины.
ЗАКЛЮЧЕНИЕ
Как следует из представленного обзора, изучение явления радиационной карбонизации ПВДФ имеет довольно продолжительную историю. Накопленные экспериментальные данные позволили исследовать кинетику изменения не только общего количества фтора в модифицируемой излучением пленке, но и вариации содержания дифторэтиленовых и фторметиновых групп, а также разработать математическую модель процесса. Анализ тонкой структуры спектров электронной эмиссии выявил различия состояний атомов углерода в производных ПВДФ, карбонизированных в результате различных радиационных воздействий. Оказалось, что бомбардировка ионами аргона и облучение фотонами мягкого рентгеновского излучения формируют на поверхности полимерной пленки углеродные структуры, отличающиеся типом гибридизации валентных электронов. В последнем случае весьма вероятным представляется формирование цепочечных (карбиноидных) фрагментов. Обнаружение этих различий, как и усиливающейся по мере карбонизации неоднородности распределения остаточного фтора по глубине, может иметь практическое применение в плане создания слоевых углеродистых гетероструктур.
Следует признать, что математическая модель радиационной карбонизации ПВДФ может описывать процесс дегидрофторирования лишь схематически, в довольно общем виде, в то время как детальный механизм все еще остается неясным. В обсуждаемой модели вероятности k1 и k2 можно рассматривать как произведения индивидуальных вероятностей независимых событий, отражающих взаимодействие фотона с группами CF2 и CH2 или с CF и CH. Знание этих четырех индивидуальных вероятностей важно для более детального понимания радиационной карбонизации поверхности ПВДФ. Можно сформулировать еще целый ряд вопросов, представляющих несомненный научный интерес. Как выглядит детальный механизм единичного акта элиминирования, и какова при этом принципиальная роль фото- или ударной ионизации? Происходит ли элиминирование атомов фтора и водорода одновременно или ступенчато с очень короткой задержкой в фемтосекундной области? Существуют ли какие-либо различия в характере диссипации энергии и, следовательно, в механизме элиминирования, если фотон поглощается атомом углерода метиленовой (CH2) или дифторметиленовой (CF2) группы, а в последнем случае атомом углерода или фтора?
Сформулированные проблемы показывают, что изучение явления радиационной карбонизации ПВДФ далеко от завершения и требует дальнейших экспериментальных и теоретических исследований.
Список литературы
Энциклопедия полимеров. Т. 1. Гл. ред. Каргин В.А. М.: Советская энциклопедия, 1972. 1224 с.
Русаков В.П. Производство полимеров. М.: Высшая школа, 1988. 280 с.
Кочервинский В.В. // Успехи химии. 1996. Т. 65. № 10. С. 936.
Ferroelectric Polymers – Chemistry, Physics and Applications. Ed. Nalva H.S. N.Y.: Marcel Dekker Inc., 1995. 912 p.
Kawai H. // Jpn. J. Appl. Phys. 1969. V. 8. № 7. P. 975. https://doi.org/10.1143/JJAP.8.975
Борисенок В.А., Симаков В.Г., Куропаткин В.Г., Брагунец В.А., Волгин В.А., Ромаев В.Н., Тукмаков В.В., Кручинин В.А., Лебедева А.А., Гончарова Д.Р., Жерноклетов М.В. // Приборы и техника эксперимента. 2008. № 4. С. 113.
Okudaira K.K., Yamane H., Ito K., Imamura M., Hasegawa S., Ueno N. // Surf. Rev. Lett. 2002. V. 9. № 1. P. 335. https://doi.org/10.1142/S0218625X02002294
McNeill C.R., Asadi K., Watts B., Blom P.W.M., de Leeuw D.M. // Small. 2010. V. 6. № 4. P. 508. https://doi.org/10.1002/smll.200901719
Верховская К.А., Плаксеев А.А., Гаврилова Н.Д., Гайнутдинов Р.В., Лотонов А.М., Лысова О.А., Юдин С.Г. // Физика твердого тела. 2009. Т. 51. Вып. 10. С. 2055.
Zuo X., Yu S., Xu X., Bao R., Xu J., Qu W. // J. Membr. Sci. 2009. V. 328. № 1–2. P. 23. https://doi.org/10.1016/j.memsci.2008.08.012
Mariappan V.K., Krishnamoorthy K., Pazhamalai P., Kim S.-J. // Mater. Adv. 2020. V. 1. № 6. P. 1644. https://doi.org/10.1039/D0MA00324G
Buntov E.A., Zatsepin A.F., Guseva M.B., Ponosov Yu.S. // Carbon. 2017. V. 117. P. 271. https://doi.org/10.1016/j.carbon.2017.03.010
Duca M.D., Plosceanu C.L., Pop T. // J. Appl. Polym. Sci. 1998. V. 67. № 13. P. 2125. https://doi.org/10.1002/(SICI)1097-4628(19980328)67: 13<2125::AID-APP2>3.0.CO;2-G
Sidelnikova A.L., Andreichuk V.P., Pesin L.A., Evsyukov S., Gribov I.V., Moskvina N.A., Kuznetsov V.L. // Polym. Degrad. Stab. 2014. V. 110. P. 308. https://doi.org/10.1016/j.polymdegradstab.2014.09.009
Евсюков С.Е., Кудрявцев Ю.П., Коршак Ю.В. // Успехи химии. 1991. Т. 60. № 4. С. 764.
Morikawa E., Choi J., Manohara H.M., Ishii H., Seki K., Okudaira K.K., Ueno N. // J. Appl. Phys. 2000. V. 87. № 8. P. 4010. https://doi.org/10.1063/1.372447
Чеботарев С.С., Песин Л.А., Грибов И.В., Москвина Н.А., Кузнецов В.Л., Евсюков С.Е. // Поверхность. Рентген., синхротр. и нейтрон. исслед. 2008. № 7. С. 49.
Le Moel A., Duraud J.P., Balanzat E. // Nucl. Instrum. Methods Phys. Res. B. 1986. V. 18. № 1–6. P. 59. https://doi.org/10.1016/S0168-583X(86)80012-X
Adem E.H., Bean S.J., Demanet C.M., Le Moel A., Duraud J.P. // Nucl. Instrum. Methods Phys. Res. B. 1988. V. 32. № 1–4. P. 182. https://doi.org/10.1016/0168-583X(88)90206-6
Brzhezinskaya M.M., Morilova V.M., Baitinger E.M., Pesin L.A., Evsyukov S.E. // Polym. Degrad. Stab. 2014. V. 99. P. 176. https://doi.org/10.1016/j.polymdegradstab.2013.11.009
Chebotaryov S.S., Volegov A.A., Pesin L.A., Evsyukov S.E., Moskvina N.A., Gribov I.V., Kuznetsov V.L. // Physica E. 2007. V. 36. № 2. P. 184. https://doi.org/10.1016/j.physe.2006.10.011
Chebotaryov S.S., Baitinger E.M., Volegov A.A., Margamov I.G., Gribov I.V., Moskvina N.A., Kuznetsov V.L., Evsyukov S.E., Pesin L.A. // Rad. Phys. Chem. 2006. V. 75. № 11. P. 2024. https://doi.org/10.1016/j.radphyschem.2005.12.051
Pesin L.A., Morilova V.M., Zherebtsov D.A., Evsyukov S.E. // Polym. Degrad. Stab. 2013. V. 98. № 2. P. 666. https://doi.org/10.1016/j.polymdegradstab.2012.11.007
Le Moel A., Duraud J.P., Lecomte C., Valin M.T., Henriot M., Le Gressus C., Darnez C., Balanzat E., Demanet C.M. // Nucl. Instrum. Methods Phys. Res. B. 1988. V. 32. № 1–4. P. 115. https://doi.org/10.1016/0168-583X(88)90192-926
Ross G.J., Watts J.F., Hill M.P., Morrissey P. // Polymer. 2000. V. 41. P. 1685. https://doi.org/10.1016/S0032-3861(99)00343-2
Ross G.J., Watts J.F., Hill M.P., Morrissey P. // Polymer. 2001. V. 42. P. 403. https://doi.org/10.1016/S0032-3861(00)00328-1
Кудрявцев Ю.П., Евсюков С.Е., Бабаев В.Г. // Изв. Акад. наук. Сер. хим. 1992. № 5. С. 1223.
Margamov I.G., Pesin L.A., Kudryavtsev Yu.P., Evsyukov S.E. // Appl. Surf. Sci. 1999. V. 148. № 3–4. P. 183. https://doi.org/10.1016/S0169-4332(99)00154-3
Mavrinskaya N.A., Pesin L.A., Baumgarten M., Mavrinskiy A.V., Baitinger E.M., Evsyukov S.E. // Magnetic Resonance in Solids EJ. 2008. V. 10. № 1. P. 31.
Живулин В.Е., Москвина Н.А., Грибов И.В., Андрейчук В.П., Морилова В.М., Хабибуллина Р.В., Песин Л.А. // Поверхность. Рентген., синхротр. и нейтрон. исслед. 2017. № 9. С. 38. https://doi.org/10.7868/S0207352817090062
Живулин В.Е., Чернов В.М., Осипов А.А., Штенберг М.В., Евсюков С.Е., Песин Л.А. // Физика твердого тела. 2017. Т. 59. Вып. 7. С. 1387.
Carbyne and Carbynoid Structures. Ed. Heimann R.B., Kavan L., Evsyukov S. Dordrecht: Kluwer Academic Publishers, 1999. 446 p.
Песин Л.А., Чеботарев С.С., Кувшинов А.М., Беспаль И.И., Грибов И.В., Москвина Н.А., Кузнецов В.Л., Евсюков С.Е., Вязовцев А.В., Кравец Н.С. // Поверхность. Рентген., синхротр. и нейтрон. исслед. 2010. № 3. С. 37.
Beamson G., Briggs D. High Resolution XPS of Organic Polymers: The Scienta ESCA 300 Database. N.Y.: Wiley, 1992. 295 p.
Анализ поверхности методами оже- и рентгеновской фотоэлектронной спектроскопии. Ред. Бриггс Д., Сих М.П. М.: Мир, 1987. 600 с.
Kuvshinov A.M., Pesin L.A., Chebotaryov S.S., Kuznetsov M.V., Evsyukov S.E., Sapozhnikova T., Mirzoev A.A. // Polymer Degradation and Stability. 2008. V. 93. № 10. P. 1952. https://doi.org/10.1016/j.polymdegradstab.2008.06.015
Кувшинов А.М., Песин Л.А., Кузнецов М.В., Грибов И.В., Москвина Н.А, Кузнецов В.Л., Евсюков С.Е. // Вестн. ЮУрГУ. Сер. Математика. Механика. Физика. 2009. Вып. 1. № 22(155). С. 72.
Voinkova I.V., Ginchitskii N.N., Gribov I.V., Klebanov I.I., Kuznetsov V.L., Moskvina N.A., Pesin L.A., Evsyukov S.E. // Polymer Degradation and Stability. 2005. V. 89. № 3. P. 471. https://doi.org/10.1016/j.polymdegradstab.2005.01.027
Соколов О.Б., Кузнецов В.Л. Развитие экспериментальных возможностей метода электронной спектроскопии с использованием магнитного энергоанализатора. Учебное пособие. Челябинск: ЧПИ, 1990. 56 с.
Elliott I., Doyle C., Andrade F.D. // J. Electron Spectr. Relat. Phenom. 1983. V. 28. P. 303. https://doi.org/10.1016/0368-2048(83)80010-3
Pesin L.A., Gribov I.V., Kuznetsov V.L., Evsyukov S.E., Moskvina N.A., Margamov I.G. // Chem. Phys. Lett. 2003. V. 372. № 5–6. P. 825. https://doi.org/10.1016/S0009-2614(03)00522-0
Шулепов С.В. Физика углеродных материалов. Челябинск: Металлургия, 1990. 336 с.
Сарайкин А.Г., Чеботарев С.С., Евсюков С.Е., Москвина Н.А., Грибов И.В., Кузнецов В.Л., Песин Л.А. // Изв. Челяб. науч. центра. 2003. Вып. 1(18). С. 27.
Houston J.E., Rogers J.W., Rye R.R., Hatson J.E., Ramaker D.E. // Phys. Rev. B. 1986. V. 34. № 2. P. 1215. https://doi.org/10.1103/PhysRevB.34.1215
Песин Л.А., Байтингер Е.М., Ковалев И.Н., Евсюков С.Е., Кудрявцев Ю.П. // Журн. структ. химии. 1999. Т. 40. № 3. С. 493.
Speranza G., Laidani N., Galliari L., Anderle M. // Diamond and Related Mater. 1999. V. 8. № 2–5. P. 517. https://doi.org/10.1016/S0925-9635(98)00287-8
Дементьев А.П., Маслаков К.И. // Физика твердого тела. 2004. Т. 46. Bып. 4. С. 662.
Pesin L.A., Baitinger E.M. // Carbon. 2002. V. 40. № 3. P. 295. https://doi.org/10.1016/S0008-6223(01)00130-0
Galuska A.A., Madden H.H., Allred R.E. // J. Electron Spectroscopy and Related Phenomena. 1988. V. 12. P. 293. https://doi.org/10.1016/0169-4332(88)90012-8
Грибов И.В., Песин Л.А., Евсюков С.Е., Курбанова А.В., Маргамов И.Г., Воинкова И.В., Кузнецов В.Л., Москвина Н.А. // Химическая физика. 2005. Т. 24. № 3. С. 38.
Воинкова И.В., Песин Л.А., Волегов А.А., Евсюков С.Е., Грибов И.В., Кузнецов В.Л., Москвина Н.А. // Поверхность. Рентген., синхротр. и нейтрон. исслед. 2007. № 8. С. 20.
Песин Л.А., Андрейчук В.П., Морилова В.М., Грибов И.В., Москвина Н.А., Кузнецов В.Л., Евсюков С.Е., Корякова О.В., Мокрушин А.Д., Егоров Е.В. // Поверхность. Рентген., синхротр. и нейтрон. исслед. 2013. № 5. С. 51. https://doi.org/10.7868/S0207352813050119
Дополнительные материалы отсутствуют.
Инструменты
Поверхность. Рентгеновские, синхротронные и нейтронные исследования