

Поверхность. Рентгеновские, синхротронные и нейтронные исследования, 2022, № 11, стр. 9-14
Особенности моделирования XANES-спектров для центров Cu(II) в плоско-квадратной координации в Cu-обменных цеолитах
И. А. Панкин a, *, А. В. Солдатов a
a Международный исследовательский институт интеллектуальных материалов,
Южный федеральный университет
344090 Ростов-на-Дону, Россия
* E-mail: pankin@sfedu.ru
Поступила в редакцию 28.04.2022
После доработки 05.05.2022
Принята к публикации 05.05.2022
- EDN: TBJMEW
- DOI: 10.31857/S102809602211019X
Аннотация
Понимание взаимосвязи структуры активных центров катализаторов на основе Cu-обменных цеолитов и их производительности является важным шагом на пути получения катализаторов с оптимизированными свойствами. Одной из основных экспериментальных методик, используемых для характеризации металлических центров в Cu-обменных цеолитах, является XANES-спектроскопия, которая позволяет получать информацию об изменении заряда и структуры активных центров при активации и в условиях протекания химических реакций. Рассмотрены особенности моделирования спектров XANES для модельных конфигураций центров Cu(II) в плоско-квадратной координации. Показано, что для воспроизведения формы экспериментальных кривых важно учитывать вклады двух конкурирующих каналов поглощения, обусловленных так называемыми эффектами “shake-down”, в которых атомы меди имеют электронные конфигурации 3d10L и 3d9 в возбужденном состоянии. Данный метод позволяет получить наиболее репрезентативные теоретические XANES-спектры для центров Cu(II) и в дальнейшем может быть использован для анализа структуры медных центров в Cu-обменных цеолитах различных топологий.
ВВЕДЕНИЕ
Cu-обменные цеолиты могут быть использованы в качестве эффективных катализаторов для ряда важнейших химических реакций, в числе которых селективное каталитическое восстановление вредоносных оксидов азота в присутствии молекул аммиака (так называемая NH3-SCR реакция (SCR – Selective Catalytic Reduction)) [1] и реакция прямой конверсии метан–метанол (так называемая MTM реакция (МТМ – Methane-to-Methanol)) [2]. Было показано, что для эффективной работы Cu-обменные катализаторы требуют предварительной высокотемпературной активации. Варьирование условий активации, а также параметры самого каркаса, например, соотношение Si : Al, могут приводить к формированию центров Cu с различной структурной топологией и зарядовым состоянием [3–5].
Важным инструментов для исследования зарядового состояния и структурной топологии центров меди в Cu-обменных цеолитах является спектроскопия рентгеновского поглощения в ближней (XANES – X-ray Absorption Near Edge Structure) и протяженной (EXAFS – Extended X-ray Absorption Fine Structure) области спектра. В частности, XANES-спектроскопия позволяет получать информацию об усредненном зарядовом состоянии центров меди и об их структурной топологии. Данная информация также может быть получена непосредственно в процессе протекания химических реакций (в режимах in situ/operando). В недавних работах [4–6] с помощью статистических методов анализа массива экспериментальных XANES-спектров (таких как метод MCR–ALS (Multivariate Curve Resolution–Alternating Least Square) [7, 8]) было показано, что при высокотемпературной активации Cu-обменные цеолиты топологии шабазит (далее Cu-CHA) характеризуются совокупностью центров меди различных типов, которые вносят вклад в усредненный экспериментальный сигнал, что в свою очередь существенно усложняет теоретическую интерпретацию экспериментальных кривых. Однако применение таких методов, как MCR–ALS, позволяет выделить XANES-спектры, соответствующие так называемым “чистым компонентам”, т.е. отдельно взятым топологиям центров меди, что актуализирует вопрос применения теоретического моделирования XANES-спектров для структурного анализа.
В настоящей работе рассмотрены особенности моделирования спектров XANES в рамках метода конечных разностей с использованием полного потенциала (программный комплекс FDMNES [9, 10]) для центров меди Cu(II) в плоско-квадратной координации на примере модельных конфигураций мобильного тетра-амино-комплекса m-Cu(NH3)4 и каркас-координированного комплекса Z2Cu@6MR. Данные конфигурации могут быть сформированы в Cu-CHA в условиях NH3-SCR-реакции (в низкотемпературном режиме) и при высокотемпературной активации в атмосфере кислорода соответственно.
Продемонстрирована важность учета двух возможных каналов поглощения, в которых атомы меди имеют заполненную и незаполненную 3d-оболочку (в возбужденном состоянии), для корректного описания экспериментальных кривых четырехкоординированных комплексов Cu(II) в квадратно-плоской координации.
МЕТОДЫ
Получение структурных моделей
Структурные модели конфигураций мобильного m-Cu(NH3)4 и каркас-координированного Z2Cu@6MR были получены с помощью оптимизации атомной структуры исходных моделей в рамках метода PAW-псевдопотенциалов (PAW – Projector Augmented Wave), реализованного в программном комплексе VASP [11]. Размер базиса плоских волн был ограничен энергией 450 эВ. Минимизацию энергии в ходе оптимизации структурных параметров проводили в рамках метода сопряженных градиентов, минимизацию энергии в рамках самосогласованного расчета электронной плотности – с помощью алгоритма Дэвидсона. Критерий сходимости составил 10–6 эВ. В соответствии с результатами систематического исследования применения различных функционалов для описания связей Cu–N и Cu–O в Cu-обменных цеолитах [12] был использован функционал Perdew–Burke–Ernzerhof с поправкой Хаббарда (параметр U = 6 в соответствии с [13]).
Для каркас-координированного комплекса Z2Cu@6MR была рассмотрена ячейка CHA с 36 “тетраэдрическими” позициями (2 атома Al, 34 атома Si и 72 атома O). Исходная модель каркаса была взята из базы данных структур цеолитов (www.iza-structure.org). Атом меди помещали приблизительно в центр плоскости шестичленного кольца (6MR) с двумя атомами Si, замещенными атомами Al. Для мобильного комплекса m-Cu(NH3)4 оптимизация была выполнена в квадратной ячейке со стороной 10 Å с целью исключения возможных взаимодействий молекул, расположенных в соседних ячейках. Исходные структурные параметры были заданы согласно [14, 15].
Моделирование XANES-спектров
Моделирование XANES-спектров за K-краем Сu было выполнено в рамках метода конечных разностей и полного потенциала с использованием программного комплекса FDMNES [9, 16]. Спектр рассчитывали из первых принципов в рамках одноэлектронного приближения в прямом пространстве для сферического атомного кластера, в центре которого расположен поглощающий атом. Решение дискретизированного уравнения Шредингера получено в конечном количестве узловых точек, где межатомный потенциал определяется суперпозицией самосогласованных атомных потенциалов. Энергетически зависимую часть обменно-корреляционного потенциала определяли в рамках приближения локальной плотности LDA (Local Density Approximation) и формализма Хедина–Лангквиста [9].
В работе было применено постоянное гауссово и энергозависимое лоренцево уширение в рамках модели арктангенса. С целью учета уширения, вызванного конечным временем жизни вакансии 1s-уровня Cu, значение соответствующего параметра функции “размазки” gamma hole было задано на уровне 1.55 эВ в соответствии с табулированными значениями [17]. Параметры функции “размазки” в рамках модели арктангенса были оптимизированы ранее при моделировании XANES-спектров оксидов меди [18].
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Несмотря на большой потенциал применения XANES-спектроскопии для анализа локальной атомной и электронной структуры Cu-центров в медных цеолитах, далеко не всегда для интерпретации экспериментальных данных и, как результат, структурного анализа применяется моделирование XANES-спектров. Так, в [19, 20] проводили моделирование для некоторых каркас-координированных и мобильных водно- или аммиак-координированных комплексов меди (формируемых в Cu-CHA при различных условиях) в рамках метода псевдопотенциалов с использованием программного кода CASTEP. В [21] моделирование XANES-спектров за K-краем Cu применяли для уточнения результатов рентгеноструктурного и EXAFS-анализа с целью уточнения параметров атомной структуры ионов меди в окрестности шестичленного кольца Cu-CHA. В ряде работ [3, 4] моделирование XANES-спектров для каркас-взаимодействующих конфигураций ионов меди ZCu(I), ZCu(II) и ZCu(II)OH было выполнено в рамках метода молекулярных орбиталей в программном комплексе ADF. Ранее в работе [18] методом конечных разностей с использованием полного потенциала (программный код FDMNES) в рамках стандартного одноэлектронного приближения было проведено моделирование спектров XANES ряда каркас-взаимодействующих Cu-оксидных комплексов, инкорпорированных в каркасы цеолитов топологии шабазит CHA и морденит (MOR). Также метод конечных разностей был использован для моделирования спектров мобильного ди-амино-комплекса меди Cu(I)[NH3]2 [22] и уточнения структуры центров Cu, формируемых в Cu-обменном мордените, при твердотельном ионном обмене H–MOR и CuCl2 [23].
Тем не менее важным вопросом для дискуссий остается предсказательная точность различных методов моделирования XANES-спектров. В частности, известно, что определенные трудности возникают при моделировании XANES-спектров для комплексов Cu(II) c квадратно-плоской координацией ионов меди. В работе [24] для более точного описания экспериментальной кривой мобильного тетра-амино-комплекса было учтено влияние нескольких слоев молекул воды в качестве растворителя. Моделирование проводили в рамках метода полного многократного рассеяния в среде MXAN. Авторы пришли к выводу, что спектр амино-комплекса меди в водном растворе может быть наиболее точно описан только при учете пятикоординированной квадратно-пирамидальной модели с использованием дополнительного лиганда NH3 в первой координационной сфере меди и молекул воды в качестве растворителя во второй и третьей координационных сферах. Однако в [25] было показано, что корректное описание экспериментальной кривой для тетра-амино-комплекса без учета дополнительного лиганда или молекул растворителя можно получить в рамках теории полного многократного рассеяния (использовали коды FEFF и CONTINUUM) с учетом вкладов двух конкурирующих каналов поглощения в рамках так называемого эффекта “shake-down”. Было показано, что теоретический спектр, получаемый в результате суперпозиции расчетных спектров для двух различных каналов возбуждения 3d10L (где L указывает на формирование вакансии на p-орбитали лиганда за счет переноса электрона с лиганда на 3d-уровень меди – так называемый LMCT-переход (Ligand-to-Metal Charge Transfer)) и 3d9, позволяет воспроизвести форму основного максимума (пика WL (White Line)) экспериментального сигнала, представленного двумя обособленными максимумами, разность в энергетической локализации которых составляет ~5.9 эВ (рис. 1). Суперпозицию учитывали в пропорции 2/3 вклада 3d 10L- и 1/3 вклада 3d 9-каналов возбуждения. Возникновение двух каналов поглощения было также описано в [26, 27] при интерпретации поляризованных XANES-спектров некоторых соединений меди.
Рис. 1.
Результаты моделирования XANES-спектров за K-краем Cu (1–3) для структурных моделей m-Cu(NH3)4 (a) и Z2Cu@6MR (b) в сравнении с экспериментальными кривыми (4): 1 – расчеты в рамках “одноканального” процесса; 2, 3 – расчеты в рамках “двухканального” процесса с учетом конфигураций 3d9(4s) и 3d9(4p) соответственно; 4 – оцифрованы экспериментальные кривые для комплекса Cu(NH3)4 [15] и O2-активированного Cu-CHA с высоким содержанием Al в каркасе (Si : Al = 5) [28]. Теоретические кривые для комплекса m-Cu(NH3)4 сдвинуты по шкале энергии на +1.57 эВ с целью лучшего согласования с экспериментом. Теоретические кривые, полученные для модели Z2Cu@6MR, представлены без сдвига.
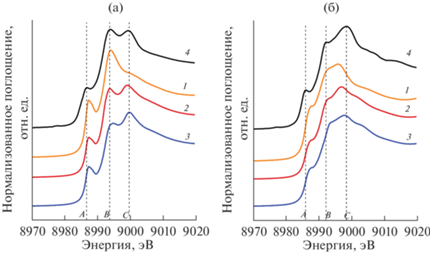
В настоящей работе был применен аналогичный подход для моделирования XANES-спектров комплексов Cu (II) в квадратно-плоской координации в рамках метода конечных разностей с использованием полного потенциала. На рис. 1 продемонстрированы результаты моделирования XANES-спектров для структурных моделей мобильного тетра-аминокомплекса m-Cu(NH3)4 и каркас-координированного центра Z2Cu@6MR. Полученные в результате геометрической оптимизации структурные модели, а также длины связей Cu–N и Cu–O в первой координационной сфере атомов Cu показаны на рис. 2. В случае m-Cu(NH3)4 можно видеть, что стандартный расчет в рамках “одноканального” подхода (рис. 1а, кривая 1) позволяет корректно предсказать лишь наличие одной из двух особенностей (пик B) в области основного максимума (пика WL) экспериментального спектра, расщепленного на два пика (B и C). Интенсивность краевого максимума (пик A) также оказывается несколько переоцененной по сравнению с экспериментальной кривой. Из рис. 1a можно видеть, что учет “двухканального” процесса позволяет более аккуратно описать форму основного максимума экспериментальной кривой (пики B и C), а также получить более корректное значение интенсивности краевого пика A. Важно отметить, что в рамках “двухканального” процесса не учитывали создание вакансии на p‑оболочке лигандов, т.е. рассматривали атом меди, содержащий все 29 электронов (соответствующий нейтральному атому), а формирование 3d 9-канала поглощения обуславливалось переносом одного d-электрона на 4s- и 4p-оболочку атома меди (рис. 1б, кривые 2 и 3). Энергетический сдвиг при суммировании компонент каналов поглощения 3d 10 и 3d 9 определяли непосредственно из расчета как разницу в значениях параметра “epsii”.
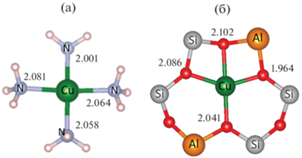
Рис. 2.
Структурные модели комплексов m-Cu(NH3)4 (а) и Z2Cu@6MR (б), полученные в результате оптимизации структурных параметров. Для Z2Cu@6MR показаны атомы каркаса только в составе шестичленного кольца, остальная часть каркаса цеолита не приведена. Длины связей Cu–N и Cu–O указаны в Å; обозначены все атомы за исключением H и O.
Важно отметить, что в рамках “двухканального” процесса с использованием как 3d 9(4s)-, так и 3d9(4p)-конфигураций удается довольно точно определить разность в энергетическом положении пиков B и C, в то время как конфигурация 3d 9(4s) более корректно описывает относительные интенсивности пиков, что обеспечивает более точное описание экспериментальной кривой и делает данный метод расчета предпочтительным. Стоит заметить, что в случае как “одноканального”, так и “двухканального” процессов энергетическая разница в положении пиков A и B остается несколько недооцененной.
Переходя к обсуждению результатов моделирования для конфигурации Z2Cu, стоит отметить, что экспериментальная кривая (оцифрованные данные [28]) образца Cu-CHA (Si : Al = 5), характеризуемого содержанием преимущественно парных центров 2Al, после активации в присутствии O2 демонстрирует существенно большую разницу в интенсивностях пиков B и C в области основного максимума поглощения, а структурная модель Z2Cu@6MR, описывающая положение иона Cu(II) в плоскости шестичленного кольца в окрестности центров 2Al, обладает заметно меньшей симметрией и однородностью длин связей Cu–O по сравнению со связями Cu–N в структуре мобильного комплекса Cu(NH3)4 (рис. 2). При моделировании спектров Z2Cu@6MR были также рассмотрены стандартный “одноканальный” и “двухканальный” подходы с учетом конфигураций 3d 9(4s) и 3d 9(4p).
В случае Z2Cu@6MR можно видеть, что моделирование в рамках “одноканального” процесса (рис. 1б, кривая 1) позволяет получить более корректный (по сравнению с тетра-амино-комплексом) теоретический спектр, что может свидетельствовать о менее выраженном эффекте “shake-down” для центров Cu(II) в данной конфигурации. Действительно, в рамках “одноканального” подхода удается воспроизвести обе особенности основного максимума экспериментального спектра (пики B и C) и даже корректно воспроизвести тенденцию в распределении их интенсивности. Однако можно заметить, что энергетическое положение пика C оказывается существенно недооцененным по сравнению с экспериментальной кривой, а интенсивность краевого пика “A” (аналогично комплексу m-Cu(NH3)4), напротив, переоцененной. В рамках “двухканального” подхода удается более корректно описать интенсивность пика A и в случае 3d 9(4p)-конфигурации (рис. 1б, кривая 3) получить более корректное положение пика C. Однако стоит заметить, что оно сопровождается переоценкой энергетической локализации пика B (по отношению к экспериментальной кривой). Аналогично моделированию комплекса m-Cu(NH3)4 для конфигурации 3d 9(4s) (рис. 1б, кривая 2) удается получить наиболее корректное соотношение интенсивностей максимумов B и C по сравнению с результатами моделирования в рамках “одноканального” процесса и конфигурации 3d 9(4p) в рамках “двухканального” процесса. Аналогично m-Cu(NH3)4 во всех методах моделирования также наблюдается недооценка энергетической разности в положении пиков A и B относительно экспериментальной кривой. Также стоит отметить, что в рамках “двухканального” процесса недостатком является отсутствие заметного уменьшения поглощения в области справа от максимума C.
В целом стоит отметить, что рассмотрение “двухканального” подхода обеспечивает лучшее согласие теоретических и экспериментальных кривых для модельных систем, где ионы Cu(II) имеют плоско-квадратную координацию. Важно подчеркнуть, что ввиду более приемлемого результата, полученного в рамках стандартного “одноканального” подхода, в случае модели Z2Cu@6MR с меньшей симметрией в локальном окружении атома меди стоит ожидать, что так называемый эффект “shake-down” при формировании спектра рентгеновского поглощения будет менее выражен. Учет двух каналов возбуждения при моделировании XANES-спектров для центров Cu(II) обеспечивает более точный результат и в дальнейшем может быть использован для моделирования и интерпретации экспериментальных XANES-спектров Cu-обменных цеолитов, где центры меди представлены конфигурациями Cu(II), в том числе в составе димеров и тримеров.
ВЫВОДЫ
По результатам моделирования спектров XANES за K-краем Cu методом конечных разностей для комплексов меди Cu(II) в плоско-квадратной координации было показано, что учет двух каналов поглощения, возникающих в результате так называемого эффекта “shake-down”, позволяет более корректно описать экспериментальные спектры тетра-амино-комплекса и Cu-CHA с большим соотношение Si : Al в каркасе цеолита после O2-активации. В результате моделирования спектров XANES в рамках стандартного “одноканального” процесса для Z2Cu@6MR комплекса показано, что в этом случае так называемый эффект “shake-down”, вероятно, менее выражен. При рассмотрении “двухканального” процесса учет 3d 9-конфигурации с переносом d-электрона на 4s-уровень меди обеспечивает более корректное описание интенсивности пиков B и C в области основного максимума поглощения. Ожидается, что представленный в работе подход может быть использован для более надежного структурного анализа различных центров Cu(II), формируемых в Cu-обменных цеолитах в условиях O2-активации и в процессе протекания реакций, на основе интерпретации экспериментальных XANES-спектров.
Список литературы
Groothaert M.H., van Bokhoven J.A., Battiston A.A., Weckhuysen B.M., Schoonheydt R.A. // J. Am. Chem. Soc. 2003. V. 125. № 25. P. 7629.https://doi.org/10.1021/ja029684w
Groothaert M.H., Smeets P.J., Sels B.F., Jacobs P.A., Schoonheydt R.A. // J. Am. Chem. Soc. 2005. V. 127. № 5. P. 7629.https://doi.org/10.1021/ja047158u
Borfecchia E., Lomachenko K.A., Giordanino F., Falsig H., Beato P., Soldatov A.V., Bordiga S., Lamberti C. // Chem. Sci. 2015. V. 6. № 1. P. 548.https://doi.org/10.1039/c4sc02907k
Martini A., Borfecchia E., Lomachenko K.A., Pankin I.A., Negri C., Berlier G., Beato P., Falsig H., Bordiga S., Lamberti C. // Chem. Sci. 2017. V. 8. № 10. P. 6836.https://doi.org/10.1039/C7SC02266B
Martini A., Alladio E., Borfecchia E. // Top. Catal. 2018. V. 61. № 14. P. 1396. https://doi.org/10.1007/s11244-018-1036-9
Usberti N., Gramigni F., Nasello N.D., Iacobone U., Selleri T., Hu W., Liu S., Gao X., Nova I., Tronconi E. // Appl. Catal. B. 2020. V. 279. P. 119397. https://doi.org/10.1016/j.apcatb.2020.119397
de Juan A., Tauler R. // Anal. Chim. Acta. 2003. V. 500. № 1. P. 195.https://doi.org/10.1016/S0003-2670(03)00724-4
de Juan A., Jaumot J., Tauler R. // Anal. Methods. 2014. V. 6. № 14. P. 4964.https://doi.org/10.1039/C4AY00571F
Joly Y. //Phys. Rev. B. 2001. V. 63. № 12. P. 125120.https://doi.org/10.1088/1742-6596/190/1/012007
Joly Y., Bunau O., Lorenzo J.E., Galera R.M., Grenier S., Thompson B. // 14th International Conference on X-ray Absorption Fine Structure / Eds. DiCicco A., Filipponi A., Iop Publishing Ltd, Bristol, 2009.
Kresse G., Furthmuller J. // Comput. Mater. Sci. 1996. V. 6. № 1. P. 15.https://doi.org/10.1016/0927-0256(96)00008-0
Chen L., Janssens T.V.W., Grönbeck H. // Phys. Chem. Chem. Phys. 2019. V. 21. № 21. P. 10923.https://doi.org/10.1039/c9cp01576k
Isseroff L.Y., Carter E.A. // Phys. Rev. B. 2012. V. 85. № 23. P. 23514.https://doi.org/10.1103/PhysRevB.85.235142
McEwen J.S., Anggara T., Schneider W.F., Kispersky V.F., Miller J.T., Delgass W.N., Ribeiro F.H. // Catal. Today. 2012. V. 184. № 1. P. 129.https://doi.org/10.1016/j.cattod.2011.11.037
JanssensT.V.W., Falsig H., Lundegaard L.F., Vennestrøm P.N.R., Rasmussen S.B., Moses P.G., Giordanino F., Borfecchia E., Lomachenko K.A., Lamberti C., Bordiga S., Godiksen A., Mossin S., Beato P. // ACS Catal. 2015. V. 5. № 5. P. 2832. https://doi.org/10.1021/cs501673g
Guda S.A., Guda A.A., Soldatov M.A., Lomachenko K.A., Bugaev A.L., Lamberti C., Gawelda W., Bressler C., Smolentsev G., Soldatov A.V., Joly Y. // J. Chem. Theory Comput. 2015. V. 11. № 9. P. 4512.https://doi.org/10.1021/acs.jctc.5b00327
Krause M.O., Oliver J.H. // J. Phys. Chem. Ref. Data. 1979. V. 8. № 2. P. 329.https://doi.org/10.1063/1.555595
Pankin I.A., Martini A., Lomachenko K.A., Soldatov A.V., Bordiga S., Borfecchia E. // Catal. Today. 2020. V. 345. P. 125.https://doi.org/10.1016/j.cattod.2019.09.032
Zhang R., Szanyi J., Gao F., McEwen J.-S. // Catal. Sci. Technol. 2016. V. 6. № 15. P. 5812. https://doi.org/10.1016/j.cattod.2016.01.025
Zhang R., McEwen J.-S. // J. Phys. Chem. Lett. 2018. V. 9. № 11. P. 3035. https://doi.org/10.1021/acs.jpclett.8b00675
Deka U., Juhin A., Eilertsen E.A., Emerich H., Green M.A., Korhonen S.T., Weckhuysen B.M., Beale A.M. // J. Phys. Chem. C . 2021. V. 116. № 7. P. 4809. https://doi.org/10.1021/jp212450d
Lomachenko K.A., Borfecchia E., Bordiga S., Soldatov A.V., Beato P., Lamberti C. // J. Phys. Conf. Ser. 2016. V. 712. P. 012041.https://doi.org/10.1088/1742-6596/712/1/012041
Srabionyan V.V., Sukharina G.B., Kurzina T.I., Durymanov V.A., Ermakova A.M., Avakyan L.A., Alayon E.M.C., Nachtegaal M., van Bokhoven J.A., Bugaev L.A. // J. Phys. Chem. C. 2021. V. 125. № 46. 25867. https://doi.org/10.1021/acs.jpcc.1c08240
Frank P., Benfatto M., Hedman B., Hodgson K.O. // Inorg. Chem. 2008. V. 47. № 10. P. 4126.https://doi.org/10.1021/ic7021243
Chaboy J., Muñoz-Páez A., Carrera F., Merkling P., Marcos E.S. // Phys. Rev. B . 2005. V. 71. № 13. P. 134208. https://doi.org/10.1103/PhysRevB.71.134208
Kosugi N., Yokoyama T., Kuroda H. // in EXAFS and Near Edge Structure III (Springer, Berlin, Heidelberg, 1984). P. 55–57. https://doi.org/10.1007/978-3-642-46522-2_15
Kosugi N., Yokoyama T., Asakura K., Kuroda H. // Chem. Phys. 1984. V. 91. № 2. P. 249.https://doi.org/10.1016/0301-0104(84)80058-0
Paolucci C., Parekh A.A., Khurana I., Di Iorio J.R., Li H., Albarracin Caballero J.D., Shih A.J., Anggara T., Delgass W.N., Miller J.T., Ribeiro F.H., Gounder R., Schneider W.F. // J. Am. Chem. Soc. 2016. V. 138. № 18. P. 6028. https://doi.org/10.1021/jacs.6b02651
Дополнительные материалы отсутствуют.
Инструменты
Поверхность. Рентгеновские, синхротронные и нейтронные исследования