

Поверхность. Рентгеновские, синхротронные и нейтронные исследования, 2022, № 5, стр. 51-66
Моделирование электрических свойств самоорганизованных островковых пленок полярных молекул C60F18 на химически неактивных поверхностях
А. В. Горячевский a, *, Л. П. Суханов a, b, **, А. М. Лебедев a, Н. Ю. Свечников a, К. А. Меньшиков a, Р. Г. Чумаков a, В. Г. Станкевич a
a Национальный исследовательский центр “Курчатовский институт”
123182 Москва, Россия
b Московский физико-технический институт
141700 Долгопрудный, Московская область, Россия
* E-mail: goryachevsky94@gmail.com
** E-mail: sukhanov1955@mail.ru
Поступила в редакцию 29.06.2021
После доработки 27.08.2021
Принята к публикации 30.08.2021
- EDN: KSHDCD
- DOI: 10.31857/S1028096022050107
Аннотация
Изучены электрические свойства молекул фторида фуллерена C60F18, обладающих высоким электрическим дипольным моментом, при их физической адсорбции на различных поверхностях с целью исследования возможности создания переходных слоев с заданными физико-химическими характеристиками и управления их свойствами. Были получены пространственные карты электростатического потенциала и напряженности электрического поля отдельной молекулы. Методами теории функционала плотности промоделировано распределение электростатического потенциала однослойных и многослойных, плотноупакованных и разреженных, а также упорядоченных и разупорядоченных ансамблей молекул фторфуллерена C60F18, наблюдавшихся ранее в экспериментах, и их воздействие на подложку. Расчеты подтвердили экспериментально установленное расщепление F1s-уровней в электрическом поле, направленном вдоль оси симметрии молекулы (линейный эффект Штарка). По характеру взаимодействия молекул адсорбата с подложкой и друг с другом, влиянию коллективных электростатических эффектов на структуру монослоя и сдвигу остовных электронных уровней сделаны выводы о присутствии локальных электрических полей над островковой пленкой и в приповерхностной области подложки, процессе роста пленки. Результаты настоящего исследования могут быть использованы для интерпретации распределений электростатического потенциала, полученных методом сканирующей микроскопии квантовой точки.
ВВЕДЕНИЕ
В последние два десятилетия возрос исследовательский интерес к процессам адсорбции молекул на различных поверхностях. На подложке возможно образование самоорганизованных монослоев полярных органических молекул, т.е. молекулярных ансамблей, формирующих двумерные упорядоченные домены разной протяженности. Они представляют собой новый класс перспективных наноматериалов с широким спектром всевозможных применений в нелинейной оптике, биосенсорах, биосовместимых материалах и особенно в молекулярной электронике (например, [1]). В этой связи макромолекулы типа C60F18, обладающие высоким электрическим дипольным моментом от 10 до 11 Д [2], могут в значительной мере изменять электронные свойства переходных слоев молекула–металл.
Последовательный теоретический анализ систем с самоорганизованными монослоями на поверхности подложки возможен на уровне расчетов их электронного строения методами теории функционала плотности (ТФП) [3] с оптимизацией геометрии и наложением периодических граничных условий (например, [4–13]).
Для изучения процессов адсорбции и самоорганизации полярных молекул C60F18 на поверхности разных подложек прежде всего требуется информация об электронных и электрических свойствах самой молекулы, включая ее электрический дипольный момент, распределение электростатического потенциала, напряженности электрического поля и электронной плотности. До недавнего времени такая информация была либо ограниченной, либо совсем отсутствовала [14, 15]. Например, разброс рассчитанных значений электрического дипольного момента молекулы составлял от 12.4 до 15.7 Д. Чтобы восполнить пробел, мы впервые [2] рассчитали эти свойства с контролируемой точностью с помощью различных приближений ТФП [3]. Было показано, что известное приближение точечного диполя, используемое в электростатических моделях описания электрических характеристик самоорганизованных монослоев полярных органических молекул на поверхности подложки (например, [8, 16]), выполняется лишь с точностью 30% только на расстояниях, вдвое больших размера молекулы. Это приближение не применимо к описанию обнаруженных в [17] методом сканирующей туннельной микроскопии гексагональных плотноупакованных слоев молекул C60F18 на поверхности Au(111), когда межмолекулярное расстояние практически совпадает с размером молекулы.
В отличие от бесконечных периодических структур, рассмотренных в [8], экспериментально установлен [18] принципиально иной тип самоорганизации пленки C60F18, а именно непериодическое покрытие поверхности подложки молекулами в виде трехмерных островков с упорядочением молекул внутри них. Такой тип самоорганизации пленки согласуется с ее островковым ростом по механизму Вольмера–Вебера [19, 20], когда энергия взаимодействия молекул с подложкой много меньше энергии межмолекулярного взаимодействия. В свете вышеизложенного при моделировании распределения электрического поля ансамбля молекул типа C60F18 необходим был оригинальный подход к теоретическому исследованию, отличный от описанных выше в литературе.
Предлагаемый подход приобретает особую актуальность в связи с развитием сканирующей микроскопии квантовой точки (СМКТ) – нового бесконтактного метода измерения распределения электростатического потенциала вдоль поверхности с субнанометровым разрешением [21–26]. СМКТ включает в себя присоединение одной органической молекулы – квантовой точки – к кончику иглы атомно-силового микроскопа. Таким образом, квантовая точка становится зондом вместо кончика иглы прибора, поднимая тем самым боковое разрешение до субнанометровых масштабов. Существует множество областей применения СМКТ, например, в исследовании больших биомолекулярных структур. Новая микроскопия нуждается в теоретическом дополнении исследования электрических свойств макромолекул, которые пока еще редко рассчитывают методами квантовой химии. Поскольку метод СМКТ измеряет распределение электростатического потенциала, значительная часть усилий будет сосредоточена на построении пространственных карт распределения электростатического потенциала молекулярных кластеров (C60F18)n.
Настоящая работа посвящена моделированию электрических свойств молекул фторида фуллерена C60F18, обладающих высоким дипольным моментом и формирующих самоорганизованные пленки при их адсорбции на различных химически неактивных поверхностях, с целью исследования возможности создания переходных слоев с заданными физико-химическими характеристиками и управления их свойствами. Молекулы в самоорганизованных пленках проявляют новые свойства, которых нет у одиночных молекул. Данные свойства, обнаруженные в эксперименте, могут быть использованы при изготовлении электронных приборов.
ДЕТАЛИ РАСЧЕТНОЙ МЕТОДИКИ
При учете специфического характера самоорганизации молекул C60F18 на поверхности в виде островков, а также результатов [2] в настоящей работе предложен оригинальный подход к теоретическому исследованию электрических свойств островковых пленок молекул C60F18. Применяемое в литературе приближение точечного диполя для описания электрических характеристик самоорганизованных монослоев полярных органических молекул на поверхности подложки (например, [8, 16]) – лишь частный случай предлагаемого подхода. Электростатический потенциал φ(r) молекулы (в атомных единицах) определяется электронной плотностью ρ(r) и зарядами ядер Za:
(1)
$\varphi ({\mathbf{r}}) = - \int {\frac{{\rho ({\mathbf{r}}{\kern 1pt} '){\kern 1pt} d{\mathbf{r}}{\kern 1pt} '}}{{\left| {{\mathbf{r}} - {\mathbf{r}}{\kern 1pt} '} \right|}}} + \sum\limits_a {\frac{{{{Z}_{a}}}}{{\left| {{\mathbf{r}} - {{{\mathbf{r}}}_{a}}} \right|}}} {\kern 1pt} ,$Полученную экспериментально геометрию молекулы C60F18 [14, 15, 28] приводили к конфигурации, обладающей симметрией C3v, путем усреднения по всем конфигурациям, применяя все преобразования симметрии группы C3v к исходной геометрии с использованием языка Python [29] и модуля NumPy [30], который позволяет проводить матричные вычисления.
Квантово-химическое моделирование молекулы осуществляли в приближении ТФП [3] с помощью программного пакета GAUSSIAN09 [31]. Использовали функционал плотности B3LYP (трехпараметрический обменный функционал Беке [32, 33] с корреляционным функционалом Ли–Янга–Парра [34]) с применением базисного набора Попла 6-31G* [31]. Базисный набор выбирали на основании сравнения теоретических и экспериментальных данных для модельной системы FCH3 [2]. Расчеты проводили в отсутствие и в присутствии электрического поля, направленного вдоль оси симметрии третьего порядка молекулы.
На основе расчетов в отсутствие внешнего поля было построено трехмерное распределение электростатического потенциала и напряженности электрического поля отдельной молекулы. Для построения поля отдельной молекулы использовали результаты квантово-химического моделирования. С помощью программы GAUSSIAN09 были вычислены электрический дипольный момент и напряженность поля внутри куба размером 50 × 50 × 50 Å с центром, совпадающим с электростатическим центром молекулы, и с шагом 0.5 Å. Электростатический центр – это точка M трехмерного пространства, для которой справедливо следующее равенство [31]:
(2)
$\left( {\sum\limits_a {{{Z}_{a}}} } \right)~{{{\mathbf{r}}}_{M}} = \sum\limits_a {{{Z}_{a}}{{{\mathbf{r}}}_{a}}} ,$(3)
$\varphi = \frac{{\left( {{\mathbf{\mu }},{\mathbf{r}}} \right)}}{{{{r}^{3}}}},\,\,\,\,{\mathbf{E}} = \frac{{3\left( {{\mathbf{\mu }},{\mathbf{n}}} \right){\mathbf{n}} - {\mathbf{\mu }}}}{{{{r}^{3}}}},$Для определения электростатического потенциала между узлами сетки использовали трилинейную интерполяцию [35]. Суть ее заключается в следующем. Если известны значения функции в вершинах единичного куба, то значения функции внутри него можно вычислить по формуле:
(4)
$\begin{gathered} f\left( {x,y,z} \right) = \left( {1 - x} \right)\left( {1 - y} \right)\left( {1 - z} \right)f\left( {0,0,0} \right) + \\ + \,\,\left( {1 - x} \right)\left( {1 - y} \right)z{\kern 1pt} f\left( {0,0,1} \right) + \\ + \,\,\left( {1 - x} \right)y\left( {1 - z} \right)f\left( {0,1,0} \right) + \\ + \,\,\left( {1 - x} \right)yz{\kern 1pt} f\left( {0,1,1} \right) + x\left( {1 - y} \right)\left( {1 - z} \right)f\left( {1,0,0} \right) + \\ + \,\,x\left( {1 - y} \right)z{\kern 1pt} f\left( {1,0,1} \right) + \\ + \,\,xy\left( {1 - z} \right)f\left( {1,1,0} \right) + xyz{\kern 1pt} f\left( {1,1,1} \right). \\ \end{gathered} $Электростатический потенциал островковых структур рассчитывали по принципу суперпозиции на основании информации об электростатическом потенциале отдельной молекулы с помощью технологии программирования CUDA (Compute Unified Device Architecture) [36] и языков Python [29] и C++ [37]. Технология CUDA позволяет проводить параллельные вычисления на персональном компьютере с установленной на нем видеокартой. Так как требуемая вычислительная задача имеет высокую степень распараллеливания, то с помощью CUDA можно существенно снизить время ее выполнения. Карты распределения электростатического потенциала строили с помощью программы Gnuplot [38], которая позволяет автоматизировать процесс построения карт электрических свойств.
ОСНОВНЫЕ РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Перед проведением численного эксперимента необходимо оценить, как ведет себя электрическое поле кластера (C60F18)n при увеличении его размеров n. Для этого была разработана теоретическая схема, позволяющая оценить поле больших кластеров и сравнить его с полем бесконечных структур.
Насыщение электрических свойств при увеличении размеров кластера
Проведем теоретические оценки насыщения электрических свойств кластера при увеличении количества молекул в нем. Рассмотрим три случая – одномерный, двумерный и трехмерный.
В одномерном случае рассмотрим бесконечную цепочку диполей, выстроенных в одну линию на равных расстояниях друг от друга. Их дипольные моменты перпендикулярны данной линии и сонаправлены. Тогда на каждый диполь будет действовать электрическое поле остальных диполей, которое равно:
(5)
$E = - 2\sum\limits_{m\,{\kern 1pt} = \,{\kern 1pt} 1}^\infty {\frac{{{{\mu }_{m}}}}{{r_{m}^{3}}}} = - 2\frac{\mu }{{{{R}^{3}}}}\sum\limits_{m{\kern 1pt} \, = {\kern 1pt} \,1}^\infty {\frac{1}{{{{m}^{3}}}}} = - \frac{{2\mu }}{{{{R}^{3}}}}\zeta (3),$(6)
$\begin{gathered} \Delta E = - 2\frac{\mu }{{{{R}^{3}}}}\sum\limits_{m\,{\kern 1pt} = \,{\kern 1pt} n\, + \,{\kern 1pt} 1}^\infty {\frac{1}{{{{m}^{3}}}}} \sim - 2\frac{\mu }{{{{R}^{3}}}}\int\limits_{n\,{\kern 1pt} + \,{\kern 1pt} 1}^\infty {\frac{{dx}}{{{{x}^{3}}}}} = \\ = - \frac{\mu }{{{{R}^{3}}}}\frac{1}{{{{{\left( {n + 1} \right)}}^{2}}}}, \\ \end{gathered} $В двумерном случае рассмотрим квадратную решетку из полярных молекул, дипольные моменты которых сонаправлены и перпендикулярны плоскости решетки. Тогда на каждый диполь будет действовать электрическое поле остальных диполей, которое равно:
(7)
$E = \sum\limits_{i{\kern 1pt} = {\kern 1pt} - \infty }^\infty {\sum\limits_{j{\kern 1pt} = {\kern 1pt} - \infty }^\infty { - \frac{\mu }{{r_{{ij}}^{3}}}} } = - \frac{\mu }{{{{R}^{3}}}}\sum\limits_{\begin{array}{*{20}{c}} {i,j{\kern 1pt} = {\kern 1pt} - \infty } \\ {{{i}^{2}} + {{j}^{2}}{\kern 1pt} \ne {\kern 1pt} 0} \end{array}}^\infty {\frac{1}{{{{{({{i}^{2}} + {{j}^{2}})}}^{{{3 \mathord{\left/ {\vphantom {3 2}} \right. \kern-0em} 2}}}}}}} = - \frac{\mu }{{{{R}^{3}}}}k,$(9)
$2\pi \int\limits_{n\surd 2}^\infty {dr\frac{r}{{{{r}^{3}}}}} < Q\left( n \right) < 2\pi \int\limits_n^\infty {dr\frac{r}{{{{r}^{3}}}}} ,\,\,\,\,~\frac{{\pi \surd 2}}{n} < Q\left( n \right) < \frac{{2\pi }}{n}{\kern 1pt} .$Наконец, исследуем трехмерный случай. Рассмотрим кубическую решетку диполей с постоянной решетки a. Введем систему координат с началом отсчета в одной из молекул, а оси координат направим вдоль базисных векторов решетки. Выберем систему, состоящую из всех диполей, расположенных в полупространстве z ≤ 0. Пусть дипольный момент каждой молекулы направлен вдоль оси z.
Рассмотрим два случая – внутри и вне массива диполей. В обоих случаях в силу осевой симметрии x- и y-компоненты напряженности электрического поля равны нулю, так что осталось вычислить z-компоненту. Тогда, пользуясь формулой (3), получим значение z-компоненты напряженности электрического поля:
(10)
${{E}_{z}} = \frac{{3\left( {{\mathbf{\mu }},{\mathbf{n}}} \right){{n}_{z}} - {{\mu }_{z}}}}{{{{r}^{3}}}} = \frac{{\mu (3n_{z}^{2} - 1)}}{{{{r}^{3}}}}.$(11)
$\begin{gathered} {{E}_{z}} \approx \iint\limits_{\begin{array}{*{20}{c}} {z\,{\kern 1pt} \leqslant \,{\kern 1pt} 0,} \\ {l{\kern 1pt} \, \leqslant \,{\kern 1pt} r\,{\kern 1pt} \leqslant \,{\kern 1pt} L} \end{array}} {\frac{{dxdydz}}{{{{a}^{3}}}}\frac{{\mu (3n_{z}^{2} - 1)}}{{{{r}^{3}}}}} = \\ = \int\limits_0^{2\pi } {d\varphi } \int\limits_{\frac{\pi }{2}}^\pi {d\theta {\text{sin}}\theta } \int\limits_l^L {\frac{{{{r}^{2}}dr}}{{{{a}^{3}}}}\frac{{\mu (3{\text{co}}{{{\text{s}}}^{2}}\theta - 1)}}{{{{r}^{3}}}}} = \\ = \frac{{2\pi \mu }}{{{{a}^{3}}}}\int\limits_{\frac{\pi }{2}}^\pi {d\left( { - {\text{cos}}\theta } \right)} {\kern 1pt} (3{\text{co}}{{{\text{s}}}^{2}}\theta - 1)\int\limits_l^L {\frac{{dr}}{r}} = \\ = \frac{{2\pi \mu }}{{{{a}^{3}}}}{\text{ln}}\frac{L}{l}\int\limits_0^1 {dt(3{{t}^{2}} - 1)} = 0. \\ \end{gathered} $Далее будут описаны результаты смоделированного электростатического потенциала для различных кластеров – монослойных и многослойных, плотноупакованных и разреженных, упорядоченных и разупорядоченных. Все эти кластеры небольшие, в них порядка нескольких десятков молекул. Данный размер интересен тем, что именно с таких кластеров начинается рост островковой пленки [18], и крайне необходимо вычислить электростатический потенциал структур в зоне ее роста. Острова формируются из одиночных молекул. Отдельные молекулы адсорбируются на поверхности и создают на ней электростатический потенциал. Далее на нее осаждаются другие молекулы, испытывая притяжение за счет неоднородностей потенциала, вызванных отдельными малыми фрагментами островковой пленки на подложке. Электростатический потенциал реальных молекул не выражается в аналитическом виде, поэтому наряду с теоретическими оценками потенциала бесконечных структур целесообразно провести компьютерный расчет электрических свойств малых кластеров.
Трехмерное пространственное распределение электростатического потенциала и напряженности электрического поля отдельной молекулы C60F18
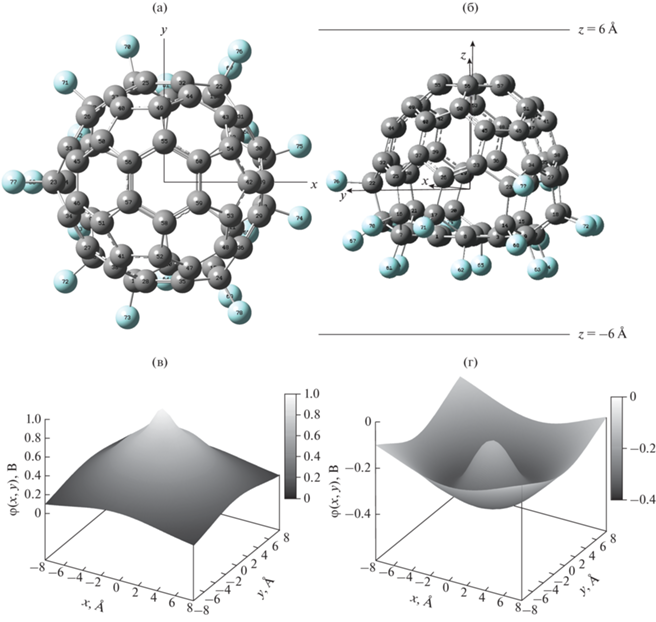
Ранее был проведен квантово-химический расчет электронной структуры молекулы C60F18 [2]. Геометрия молекулы была взята из результатов эксперимента по рентгеновской дифракции на кристаллах фторфуллерена C60F18 [28]. В настоящей работе она была приведена к симметрии C3v путем усреднения по всем элементам данной группы симметрии. В [2] была показана анизотропия поля вблизи и внутри каркаса молекулы C60F18, которая выражается в несимметричности поля при отражении относительно плоскости yz (рис. 1а) и неэквивалентности осей x и y. Это указало на необходимость расчета полей в широкой области пространства и с более детальным разрешением, чем это было сделано в [2]. В настоящей работе построено трехмерное распределение электростатического потенциала. На рис. 1 показано распределение электростатического потенциала в плоскостях, перпендикулярных вектору электрического дипольного момента молекулы (рис. 1б) и расположенных на 6 Å выше (рис. 1в) и ниже (рис. 1г) электростатического центра ядерной подсистемы молекулы. Распределение на далеких расстояниях с большой точностью соответствует полю точечного диполя [2], а при приближении к молекуле начинают проявляться отклонения от дипольного приближения (рис. 1в, 1г). Ввиду указанных особенностей дипольное приближение было использовано вдали от молекулы при оптимизации расчета поля островковых структур. Вблизи же молекулярного каркаса в распределении потенциала присутствуют особенности, связанные со структурой расположения атомов в молекуле. Распределение электростатического потенциала, наводимого в области подложки при адсорбции молекулы на поверхности атомами фтора, немонотонно и создает для электронов подложки сложный потенциальный рельеф, похожий на мексиканскую шляпу (рис. 1г). Адсорбированная молекула формирует в подложке соответствующее электростатическое изображение, которое является потенциальной ямой для электронов. Это может быть использовано для деформирования распределения электронной плотности на границе раздела и формирования на ней областей с разным типом проводимости за счет адсорбции островковых структур и возникновения эффекта поля. Также подобные самоорганизующиеся адсорбированные пленки можно использовать в качестве переходных слоев, выравнивающих зонные структуры двух материалов для обеспечения транспорта зарядов между ними [39, 40].
Рис. 1.
Молекула фторфуллерена C60F18 в кубе размером 16 × 16 × 16 Å: а – пространственная структура, вид сверху; б – вид сбоку на структуру, стрелкой показан вектор электрического дипольного момента молекулы, горизонтальными линиями отмечены расстояния, на которых вычисляли электростатический потенциал; в, г – двумерные срезы пространственной карты электростатического потенциала в плоскости z = 6, –6 Å соответственно.
Двумерные срезы трехмерной карты электрических свойств кластеров молекул фторфуллерена: использование дипольного приближения и квантово-химического расчета электрического поля
В результате моделирования были получены двумерные срезы трехмерной карты электростатического потенциала кластеров молекул фторфуллерена. Проведен расчет кластеров разных размеров и конфигураций. Рассматривали монослойные и многослойные, плотноупакованные и разреженные кластеры.
Следует пояснить, что электростатические модели применимы только в тех областях пространства, где нет сильного перекрывания распределений электронов взаимодействующих молекул [27]. Внутри молекулы имеется существенная электронная плотность, что позволяет говорить о взаимодействии электронов соседних молекул, проявляющемся в самоорганизации молекулярных островов. Здесь и далее рассматриваются распределения электрических свойств, рассчитанные на существенном удалении от углеродно-фторного каркаса молекулы, потому что в этой области корректно применять электростатические модели и рассматривать суперпозицию электрических полей. Однако для исследования взаимодействия соседних молекул важно знать распределение этих характеристик и внутри молекулярного каркаса, чтобы оценить вклад во взаимодействие молекул за рамками дипольного приближения. Такая оценка, использующая квантово-химические расчеты для отдельной молекулы, корректна, потому что молекулы при адсорбции не испытывают деформаций и не меняют свою внутреннюю структуру, что подтверждается экспериментально [18]. В физсорбированном ансамбле не происходят химические реакции молекул друг с другом и с подложкой, что позволяет использовать данные расчетов электронной структуры и структуры поля вблизи и внутри отдельной молекулы. Поэтому представленные ниже результаты описывают электрические поля вблизи и внутри островковых и доменных структур, и рассчитанные электронные свойства самой молекулы могут быть использованы для интерпретации экспериментальных результатов и определения напряженности электрического поля в пленке.
На рис. 2а показана структура монослойного кластера, состоящего из 19 молекул фторфуллерена, в котором расстояния между центрами молекул равны 12 Å, что чуть больше удвоенного бокового радиуса молекулы r = 5.3 Å [2]. Данное расстояние было выбрано с учетом исследований пленок методом сканирующей туннельной микроскопии в [17], где наблюдалась их гексагональная плотноупакованная структура на поверхности Au(111), существованию которой можно дать следующее объяснение. Отталкивательный участок кривой U(R) потенциала межмолекулярных взаимодействий проходит через нулевое значение. В узком диапазоне расстояний R между центрами молекул значение U(R) близко к нулю, что при наличии стабилизирующих взаимодействий молекул с их электрическими образами [8] благодаря подложке золота обеспечивает устойчивость системы в целом.
Рис. 2.
Монослойный плотноупакованный кластер из 19 молекул фторфуллерена C60F18 в кубе размером 80 × 80 × 80 Å: а – пространственное расположение молекул в кластере, вид сверху (крестиками показаны положения электростатических центров молекул, вектор электрического дипольного момента каждой молекулы перпендикулярен плоскости рисунка и направлен из рисунка наружу); б – вид сбоку (горизонтальными линиями отмечены расстояния, на которых вычисляли электростатический потенциал); в–з – двумерные срезы пространственной карты электростатического потенциала в плоскости z = 15, –15, 8, –8, 5.5, –5 Å соответственно.
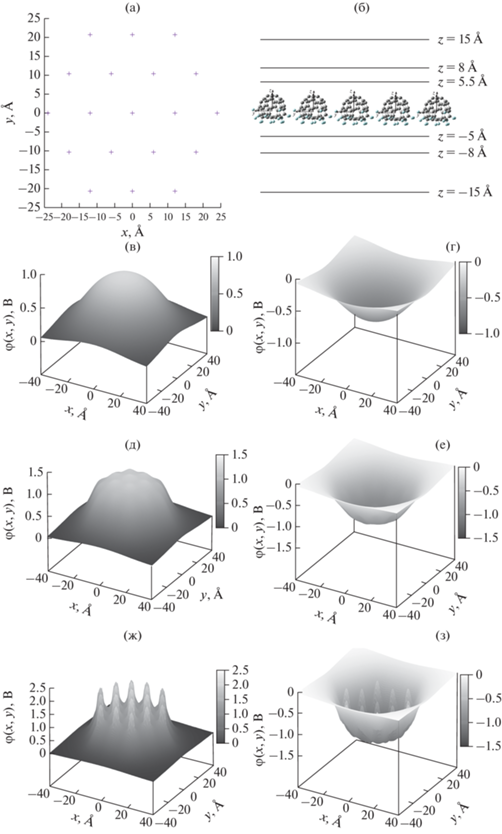
Электростатические центры всех молекул лежат в плоскости рис. 2а и отмечены крестиками, а векторы дипольного момента молекул перпендикулярны плоскости рисунка и выходят из нее. На рис. 2б представлена структура кластера (вид сбоку на одну из сторон шестиугольного кластера). Горизонтальными линиями обозначены плоскости, в которых проводился расчет электрических свойств. Здесь было выбрано расстояние 15 Å от монослоя, сравнимое с расстоянием между молекулами в кластере и почти в два раза превышающее размер молекулы в направлении оси z. На рис. 2в, 2г представлены двумерные графики электростатического потенциала в зависимости от координат x и y. Распределение похоже на распределение в случае отдельной молекулы (рис. 1в, 1г) с той лишь разницей, что здесь не наблюдается немонотонность потенциала, характерная для поля отдельной молекулы. По сути, электростатический потенциал отвечает полю объединенного диполя, распределенному в плоскости молекулярного кластера. При приближении к монослою начиная с расстояния 8 Å проявляются осцилляции электростатического потенциала кластера, вызванные вкладами отдельных молекул (рис. 2д, 2е).
Вблизи монослоя отчетливо видна структура поля отдельных молекул, которые формируют сложный немонотонный потенциальный рельеф (рис. 2ж, 2з). Присутствуют сильные осцилляции поля, что существенно отличается от распределения электростатического потенциала вдали от монослоя (рис. 2в–2е). Дальнейшее погружение в зону высокой электронной плотности приводит к неограниченному росту электростатического потенциала. Область неограниченно растущего электростатического потенциала (острые пики) расположена внутри и на расстояниях не более 2 Å вокруг каркаса молекулы C60F18. Эта область лежит в пределах $ - 5 \leqslant z \leqslant 5.5\,{\text{{\AA}}}.$ Острые пики электростатического потенциала вызваны вкладами потенциала ядер молекулы (1) и проявляются на масштабах 1 Å, что существенно меньше размера самой молекулы (~10 Å). Электростатический потенциал в этой области пространства не может быть измерен зондовыми методами, например СМКТ [21–26]. Следовательно, его нельзя сравнить с экспериментом, так как радиус закругления кончика иглы зондового микроскопа составляет от единиц до десятков нанометров, а размер квантовой точки – порядка нанометра [25]. Также в данной области пространства отсутствуют атомы подложки и другие адсорбированные молекулы, не принадлежащие данному кластеру. Поэтому в рамках приближения неперекрывающихся распределений электронов моделирование электростатического потенциала в точках $ - 5 \leqslant z \leqslant 5.5~\,{\text{{\AA}}}$ может дать качественную информацию о взаимодействии молекул в кластере. Зная расположение ядер и распределение электронной плотности внутри молекулы, можно оценить электростатический потенциал внутри молекулярного кластера.
В качестве следующей модельной системы был взят трехслойный кластер, состоящий из 50 молекул (рис. 3а), так как острова, наблюдаемые при малом покрытии поверхности высокоориентированного пиролитического графита (ВОПГ) молекулами C60F18, не превышают 100 нм в поперечнике и состоят из двух–трех монослоев [18]. Первый и третий слои кластера (темные крестики) состоят из 19 молекул и повторяют их расположение на рис. 2а. Второй слой (светлые крестики) состоит из 12 молекул, которые расположены между молекулами первого и третьего слоев. Выбрано расстояние 8 Å между соседними слоями, что слегка больше 7 Å, т.е. размера молекулы вдоль ее оси симметрии. На рис. 3б показана схема кластера (вид сбоку на одну из сторон шестиугольника). Горизонтальными линиями, проходящими через молекулы, показаны их электростатические центры, взятые за начало координат на рис. 1б. Горизонтальными линиями вне молекул показаны точки, в которых рассчитывали электростатический потенциал. Они находятся на том же расстоянии 15 Å от электростатических центров молекул C60F18, что и на рис. 2б. На рис. 3в, г показаны карты электростатического потенциала на данных расстояниях от молекул, которые аналогичны распределениям на рис. 2в, 2г с той лишь разницей, что величины электростатического потенциала почти в два раза больше. Это объясняется аддитивностью потенциала – вкладом всех трех слоев кластера. Более глубокие слои дают меньший вклад в потенциал, чем поверхностный слой, потому что в этих слоях меньше молекул и они располагаются дальше от точки наблюдения, что отражается в меньших значениях потенциала.
Рис. 3.
Многослойный плотноупакованный кластер из 50 молекул фторфуллерена C60F18 в кубе размером 80 × 80 × 80 Å: а – пространственное расположение молекул в кластере, вид сверху (темными крестиками показаны положения электростатических центров молекул в первом и третьем слоях, светлыми – во втором слое, вектор электрического дипольного момента каждой молекулы перпендикулярен плоскости рисунка и направлен из рисунка наружу); б – вид сбоку (горизонтальными линиями отмечены расстояния, на которых вычисляли электростатический потенциал); в–е – двумерные срезы пространственной карты электростатического потенциала в плоскости z = 15, –15, 5.5, –5 Å соответственно.

На рис. 3д, 3е показаны значения электростатического потенциала для того же кластера, но здесь точки наблюдения располагаются гораздо ближе к молекулам (на расстояниях 5.5 и 5 Å сверху и снизу кластера, соответственно, как и на рис. 2ж, 2з). Электростатический потенциал аналогичен потенциалу на рис. 2ж, 2з, хотя осцилляции, отвечающие отдельным молекулам, искажены добавкой потенциала более глубоких слоев кластера. Можно сказать, что электростатический потенциал нижележащих слоев деформирует потенциал верхнего монослоя молекул.
По результатам моделирования электростатического потенциала можно заключить, что осцилляции потенциала кластера заметно проявляются, начиная с расстояний 8 Å от плоскостей, проходящих через электростатические центры молекул фторфуллерена. Поле вдали островковых структур достаточно гладкое и не имеет немонотонностей. Электростатический потенциал многослойных структур существенно зависит от количества слоев и возрастает при добавлении новых слоев. Этот факт может иметь значение при воздействии электрического поля на подложку и влиять на форму и размеры островов на поверхности, а также на свойства отдельной молекулы в пленке, что будет показано в следующих разделах.
На рис. 4а показана структура монослойного разреженного кластера, состоящего из 19 молекул фторфуллерена (вид сбоку на одну из сторон шестиугольного кластера), в котором расстояния между центрами молекул равны 20 Å. Это расстояние было выбрано с учетом данных рентгенодифракционных исследований о структуре молекулярных кристаллов фторфуллерена [28]. На рисунке горизонтальными линиями обозначены плоскости, в которых проводился расчет электрических свойств. Было выбрано расстояние 15 Å от монослоя (рис. 2, плотноупакованный кластер), которое сравнимо с расстоянием между молекулами в данном кластере и почти вдвое больше размера молекулы в направлении оси z. На рис. 4в, 4г представлены двумерные графики электростатического потенциала в зависимости от координат x и y.
Рис. 4.
Монослойный разреженный кластер из 19 молекул фторфуллерена C60F18 в кубе размером 160 × 160 × 160 Å: а – пространственное расположение молекул в кластере, вид сверху (крестиками показаны положения электростатических центров молекул, вектор электрического дипольного момента каждой молекулы перпендикулярен плоскости рисунка и направлен из рисунка наружу); б – вид сбоку (горизонтальными линиями отмечены расстояния, на которых вычисляли электростатический потенциал); в–з – двумерные срезы пространственной карты электростатического потенциала в плоскости z = 15, –15, 8, –8, 5.5, –5 Å соответственно.

На рис. 4д, 4е изображен потенциал вблизи монослоя в точках, указанных на рис. 4б. Видно, что при приближении к монослою на расстояние 8 Å усиливаются осцилляции электростатического потенциала отдельных молекул. На рис. 4ж, 4з изображен потенциал вблизи монослоя в точках, указанных на рис. 4б. Вблизи монослоя отчетливо проявляется структура поля отдельных молекул, которые формируют сложный немонотонный рельеф потенциала. На рис. 4ж, 4з присутствуют сильные осцилляции поля, что существенно отличается от распределения электростатического потенциала вдали от монослоя (рис. 4в–4е). Дальнейшее приближение к зоне высокой электронной плотности приводит к неограниченному росту потенциала за счет ядер молекулы.
Сравнивая данные о разреженных кластерах с данными о плотноупакованных кластерах, можно заключить, что электростатический потенциал разреженного кластера начинает осциллировать на более далеких расстояниях, чем потенциал плотноупакованного кластера (рис. 2в, 2г, 4в, 4г). Кроме того, значения электростатического потенциала вдали от кластера существенно зависят от плотности упаковки молекул в кластере. Чем больше плотность упаковки, тем выше потенциал (рис. 2в, 2г, 4в, 4г). Вблизи кластера потенциал растет с ростом плотности упаковки и зависит преимущественно от расстояния до плоскости наблюдения (рис. 2ж, 2з, 4ж, 4з). В случае разреженных кластеров область неоднородности электростатического потенциала существенно больше, чем в случае плотноупакованных кластеров (рис. 2, 4). Более того, модель разреженного кластера отвечает ситуации, когда на подложке адсорбированы отдельные молекулы, являющиеся зародышами островов, или разрозненные острова. Они эквивалентны распределенным диполям, что было показано при их моделировании. Увеличение расстояний между неоднородностями в данном случае может способствовать адсорбции дополнительных молекул и, как следствие, стимулировать рост островковой пленки. Также это помогает молекулам объединяться и формировать многослойные кластеры.
Электрические свойства разупорядоченных структур
Было изучено влияние разупорядочения молекул в островковых структурах на электрические свойства в пространстве. Исследовали монослойные кластеры, моделирующие плоские пленки, и многослойные кластеры, моделирующие пространственные структуры. Молекулы при моделировании поворачивались случайным образом на малые углы в пределах ~10° как в двумерных, так и трехмерных кластерах. Электрические дипольные моменты молекул становились разупорядоченными, что моделирует расположение молекул в реальных островах на поверхности подложки. В экспериментально наблюдаемых структурах присутствует воздействие электростатического потенциала монослоя, которое приводит к возмущению положений и ориентации молекул. Такое возмущение заведомо нерегулярно в силу конечного размера кластеров, поэтому целесообразно моделировать его при помощи случайного возмущения кластера. Это моделирование не претендует на точность воспроизведения экспериментальных данных в силу того, что точные положения молекул в трехмерном пространстве не могут быть найдены никакими приборами. Можно определить только структуру электронного облака молекулярного адсорбированного кластера, т.е. примерное расположение молекул относительно подложки с точностью не выше нескольких ангстрем [41, 42]. Поэтому в настоящей работе рассмотрены кластеры с молекулами, повернутыми на малые случайные углы. Эти кластеры позволяют понять характер флуктуаций электростатического потенциала, которые неизбежно возникают в реальных структурах. В силу непериодичности таких структур классические дифракционные методы неприменимы для их исследования. В настоящее время существуют фазочувствительные рентгеновские методы [43–48], которые позволяют получить изображение электронной плотности сложных молекулярных структур. Но в связи с трудностями применения такого рода методов к адсорбированным нерегулярным структурам имеет смысл численно промоделировать электростатический потенциал наблюдаемых островков и оценить их влияние друг на друга. Результаты такого расчета говорят о том, что при разупорядочении теряется регулярная структура поля и симметрия, характерная для невозмущенного кластера. Это выражается в изменении амплитуды осцилляций и положений пиков и впадин потенциала. Пики становятся нерегулярными и неидентичными.
Оценки электрического поля, действующего на молекулу в пленке со стороны окружающих молекул
По данным рентгеновской фотоэлектронной спектроскопии пленок фторфуллерена, адсорбированных на ВОПГ [18], помимо основного пика F1s при энергии связи 686.9 эВ выявлен дополнительный пик (8% интенсивности относительно основного пика), сдвинутый на 2 эВ в область больших энергий связи. Для установления вида адсорбции молекул фторфуллерена на подложке были проведены квантово-химические расчеты кластера H3CF–C6H6, моделирующего взаимодействие одиночной связи C–F с графитовой подложкой, которые показали отсутствие химической адсорбции между молекулой и подложкой, а также отсутствие сдвига F1s-орбиталей. Это указывает на коллективные электростатические эффекты, вызванные самоорганизованной адсорбцией дипольных молекул [18].
По данным расчета методом B3LYP в базисе 6-31G* симметричной геометрии молекулы C60F18 было промоделировано действие электрического поля на ее электронную структуру. Поле направляли по оси симметрии молекулы, причем не только вдоль ее дипольного момента, но и против него. Были построены зависимости энергии низших 18 заполненных орбиталей молекулы, отвечающих 1s-орбиталям всех 18 атомов фтора, от напряженности электрического поля (рис. 5). Из рисунка видно, что зависимости укладываются в четыре прямые (линейный эффект Штарка). Каждая из прямых соответствует группе симметрично расположенных атомов фтора с нумерацией согласно рис. 1. По экспериментальному сдвигу уровней оценена напряженность электрического поля, создаваемая молекулами ансамбля в исследуемой системе. Из данных расчетов можно сделать вывод, что расщеплению F1s-уровней молекулы с углублением низшего уровня на ∼2 эВ относительно отмеченного горизонтальной линией на рис. 5 среднего значения в отсутствие поля отвечает напряженность поля ∼1.5 × 108 В/см, направленного против электрического дипольного момента молекулы. Заметим, что по формуле (7) в двумерном случае можно оценить создаваемое молекулами поле по порядку величины:
(12)
$E \sim k\frac{\mu }{{{{R}^{3}}}} \sim 10\frac{{10\,{\text{Д}}}}{{{{{\left( {1\,{\text{нм}}} \right)}}^{3}}}} = 3 \times {{10}^{7}}\,\,\frac{{\text{В}}}{{{\text{см}}}}.$Рис. 5.
Зависимость энергии 18 F1s-орбиталей молекулы C60F18 от напряженности электрического поля Ez, приложенного вдоль оси симметрии третьего порядка молекулы. Вертикальной линией показаны точки с нулевым значением напряженности электрического поля, горизонтальной – средняя энергия F1s-орбиталей молекулы в отсутствие электрического поля. Каждая прямая обозначена номерами соответствующих ей симметрично расположенных атомов фтора (рис. 1).

Сканирующая микроскопия квантовой точки и ее связь с настоящими исследованиями
В связи с появившимся методом СМКТ [21–26], позволяющим измерять электростатический потенциал наноструктур, настоящее исследование представляет интерес как источник теоретических данных, с помощью которого наряду с экспериментальными данными можно проводить сравнительный анализ электрических свойств адсорбированных структур. Метод СМКТ дает возможность измерять с субнанометровым разрешением электростатический потенциал вблизи молекулярного каркаса, где электронная плотность низка и применимо мультипольное разложение электростатического потенциала молекулы. Предлагаемый теоретический подход предназначен для определения электрических свойств именно в этой области пространства, но имеет гораздо более высокое разрешение по сравнению с экспериментом и поэтому может служить в качестве дополнения к экспериментальной методике СМКТ. В совокупности теоретические и экспериментальные методы позволяют получить качественное и количественное распределение электростатического потенциала адсорбированных структур, а также с помощью рассчитанного потенциала молекулы определить ее ориентацию относительно подложки и других молекул в адсорбированном кластере.
ЗАКЛЮЧЕНИЕ
В работе исследованы электрические свойства молекулярных кластеров (C60F18)n, адсорбированных на подложке. Молекулярные структуры создают сложный профиль электростатического потенциала в области подложки и над островковой пленкой, который с недавнего времени можно измерять зондовыми методами [21–26]. Созданное на поверхности подложки электрическое поле изменяет электронную структуру молекулярных кластеров, т.е. энергию орбиталей молекул и электронную плотность. С одной стороны, это отражается в рентгеновских фотоэлектронных спектрах исследуемых систем. С другой стороны, сами структуры создают вокруг себя электрическое поле со сложным профилем. Это поле меняет проводимость приповерхностного слоя подложки и деформирует его зонную структуру, что создает области проводимости различных типов без внесения легирующих примесей в тело подложки.
Была разработана теоретическая схема, позволяющая оценить поле больших кластеров (C60F18)n и сравнить его с полем бесконечных структур. Показано, что насыщение электрических свойств кластера при увеличении количества n молекул в нем существенно зависит от размерности кластерной модели. Теоретические оценки электрических свойств кластеров разных размерностей демонстрируют, что поле дальних молекул в случае изотропного расположения диполей слабо влияет на результирующее поле, и основной вклад в поле вносят локальные поля кластера (C60F18)n. Это указывает на недостаточность дипольного приближения для расчета электростатического потенциала и напряженности электрического поля больших трехмерных кластеров полярных молекул и на необходимость учета реального поля молекулы.
Методом ТФП B3LYP построено трехмерное распределение электростатического потенциала и напряженности электрического поля отдельной молекулы. Распределение на далеких расстояниях с большой точностью соответствует полю точечного диполя, а при приближении к молекуле начинают проявляться отклонения от дипольного приближения. Ввиду указанных особенностей дипольное приближение было использовано вдали от молекулы при оптимизации расчета поля островковых структур. В результате моделирования были получены двумерные срезы трехмерной карты электростатического потенциала кластеров молекул фторфуллерена. Проведен расчет гексагональных кластеров разных размеров и конфигураций: монослойных и многослойных, плотноупакованных и разреженных, а также разупорядоченных структур. Можно заключить, что осцилляции электростатического потенциала кластера проявляются, начиная с расстояний 8 Å от плоскостей, проходящих через электростатические центры молекул фторфуллерена. Поле вдали от островковых структур гладкое и не имеет осцилляций. Электростатический потенциал многослойных структур существенно зависит от количества слоев и возрастает при добавлении новых слоев. Этот факт может иметь значение при воздействии электрического поля на подложку и влиять на форму и размеры островов на поверхности, а также на свойства отдельной молекулы в пленке. Электростатический потенциал разупорядоченных структур теряет свою регулярную структуру и симметрию: проявляются нерегулярность, разные амплитуды осцилляций и положения пиков и впадин потенциала.
С помощью расчетов методом B3LYP в базисе 6-31G* симметричной геометрии молекулы C60F18 было промоделировано действие электрического поля на ее электронную структуру. Были построены зависимости энергии низших 18 заполненных орбиталей молекулы, отвечающих 1s-орбиталям всех 18 атомов фтора, от напряженности электрического поля. По данным квантово-химического расчета кластера было установлено, что расщеплению F1s-уровней молекулы с углублением низшего уровня на ∼2 эВ (С60F18/ВОПГ) отвечает поле с напряженностью ∼1.5 × 108 В/см, направленное против электрического дипольного момента молекулы. Это подтверждает предположение о том, что на молекулу действует поле остальных молекул островковой структуры и поле их изображений.
Теоретически объяснена возможная причина существования гексагональных плотноупакованных слоев молекул C60F18 на поверхности Au(111), обнаруженных методом сканирующей туннельной микроскопии [17], за счет баланса сил межмолекулярных взаимодействий и сил взаимодействия молекул с подложкой в узком диапазоне межмолекулярных расстояний.
Выявленные в работе электрические поля предоставляют уникальную возможность создания наноразмерных потенциальных барьеров и ям. Размерами и формой потенциальных ям можно управлять, меняя расстояния между структурами, создающими поле. Создавая большие по абсолютной величине электрические поля в локальной области пространства, можно влиять на механизмы адсорбции и самоорганизации молекулярных пленок. Информация об электронных и электрических свойствах отдельных молекул позволяет лучше понять причины адсорбции молекулярных ансамблей на различных поверхностях и взаимодействие молекул в ансамблях. Остается открытым вопрос об управлении положением молекул на поверхности. Самоорганизация различных молекулярных структур на поверхности подложки открывает огромные возможности для записи информации с помощью молекулярной электроники.
Список литературы
Battaglini N., Repain V., Lang P., Horowitz G., Rousset S. // Langmuir. 2008. V. 24. № 5. P. 2042. https://doi.org/10.1021/la7031994
Суханов Л.П., Чумаков Р.Г., Горячевский А.В., Лебедев А.М., Меньшиков К.А., Свечников Н.Ю., Станкевич В.Г. // Поверхность. Рентген., синхротр. и нейтрон. исслед. 2018. № 8. С. 30. https://doi.org/10.1134/s0207352818080176
Kohn W., Sham L.J. // Phys. Rev. A. 1965. V. 140. № 4. P. A1133. https://doi.org/10.1103/PhysRev.140.A1133
Rusu P.C., Brocks G. // Phys. Rev. B. 2006. V. 74. P. 073414. https://doi.org/10.1103/PhysRevB.74.073414
Rusu P.C., Brocks G. // J. Phys. Chem. B. 2006. V. 110. P. 22628. https://doi.org/10.1021/jp0642847
Rusu P.C. Charge Transfer and Dipole Formation at Metal-Organic Interfaces: Dissertation to obtain the doctor’s degree at the University of Twente. GILDEPRINT, Enschede, 2007. 123 p.
Rusu P.C., Giovannetti G., Brocks G. // J. Phys. Chem. C. 2007. V. 111. № 39. P. 14448. https://doi.org/10.1021/jp073420k
Kokalj A. // Phys. Rev. B. 2011. V. 84. P. 045418. https://doi.org/10.1103/PhysRevB.84.045418
Egger D.A., Zojer E. // J. Phys. Chem. Lett. 2013. V. 4. P. 3521. https://doi.org/10.1021/jz401721r
Rojas G., Simpson S., Chen X., Kunkel D.A., Nitz J., Xiao J., Dowben P.A. Zurek E., Enders A. // Phys. Chem. Chem. Phys. 2012. V. 14. P. 4971. https://doi.org/10.1039/c2cp40254h
De Renzi V., Rousseau R., Marchetto D., Biagi R., Scandolo S., Del Pennino U. // Phys. Rev. Lett. 2005. V. 95. P. 046804. https://doi.org/10.1103/PhysRevLett.95.046804
Vázquez H., Flores F., Oszwaldowski R., Ortega J., Pérez R., Kahn A. // Appl. Surf. Sci. 2004. V. 234. P. 107. https://doi.org/10.1016/j.apsusc.2004.05.084
Vázquez H., Oszwaldowski R., Pou P., Ortega J., Pérez R., Flores F., Kahn A. // Europhys. Lett. 2004. V. 65. № 6. P. 802. https://doi.org/10.1209/epl/i2003-10131-2
Neretin I.S., Lyssenko K.A., Antipin M.Y., Slovokhotov Y.L. // Russ. Chem. Bull. 2002. V. 51. № 5. P. 754. https://doi.org/10.1023/A:1016012228913
Neretin I.S., Lyssenko K.A., Antipin M.Y., Slovokhotov Y.L., Boltalina O.V., Troshin P.A., Lukonin A.Yu., Sidorov L.N., Taylor R. // Angew. Chemie Int. Ed. 2000. V. 39. № 18. P. 3273. https://doi.org/10.1002/1521-3773(20000915)39:18< 3273::AID-ANIE3273>3.0.CO;2-F
Natan A., Kronik L., Haick H., Tung R.T. // Adv. Mater. 2007. V. 19. P. 4103. https://doi.org/10.1002/adma.200701681
Bairagi K., Bellec A., Chumakov R.G., Menshikov K.A., Lagoute J., Chacon C., Girard Y., Rousset S., Repain V., Lebedev A.M., Sukhanov L.P., Svechnikov N.Yu., Stankevich V.G. // Surf. Sci. 2015. V. 641. P. 248. https://doi.org/10.1016/j.susc.2015.05.020
Горячевский А.В., Суханов Л.П., Лебедев А.М., Меньшиков К.А., Свечников Н.Ю., Чумаков Р.Г., Станкевич В.Г. // Поверхность. Рентген., синхротр. и нейтрон. исслед. 2019. № 10. С. 52. https://doi.org/10.1134/s0207352819100123
Gao Y. // Mater. Sci. Eng. R: Rep. 2010. V. 68. P. 39. https://doi.org/10.1016/j.mser.2010.01.001
Oura K., Lifshits V.G., Saranin A.A., Zotov A.V., Katayama M. Surface Science–An Introduction. Heidelberg–New York: Springer, 2003. 452 p.
Wagner C., Green M.F.B., Maiworm M., Leinen P., Esat T., Ferri N., Friedrich N., Findeisen R., Tkatchenko A., Temirov R., Tautz F.S. // Nat. Mater. 2019. V. 18. P. 853. https://doi.org/10.1038/s41563-019-0382-8
Green M.F.B., Wagner C., Leinen P., Deilmann T., Krüger P., Rohlfing M., Tautz F.S., Temirov R. // Jpn. J. Appl. Phys. 2016. V. 55. P. 08NA04. https://doi.org/10.7567/JJAP.55.08NA04
Wagner C., Fournier N., Tautz F.S., Temirov R. // Phys. Rev. Lett. 2012. V. 109. P. 076102. https://doi.org/10.1103/PhysRevLett.109.076102
Fournier N., Wagner C., Weiss C., Temirov R., Tautz F.S. // Phys. Rev. B. 2011. V. 84. P. 035435. https://doi.org/10.1103/PhysRevB.84.035435
Wagner C., Green M.F., Leinen P., Deilmann T., Krüger P., Rohlfing M., Temirov R., Tautz F.S. // Phys. Rev. Lett. 2015. V. 115. P. 026101. https://doi.org/10.1103/PhysRevLett.115.026101
Wagner C., Tautz F.S. // J. Phys. Condens. Matter. 2019. V. 31. P. 475901. https://doi.org/10.1088/1361-648X/ab2d09
Каплан И.Г. Межмолекулярные взаимодействия. Физическая интерпретация, компьютерные расчеты и модельные потенциалы. М.: БИНОМ, Лаборатория знаний, 2012. 397 с.
Goldt I.V., Boltalina O.V., Sidorov L.N., Kemnitz E., Troyanov S.I. // Solid State Sci. 2002. V. 4. P. 1395. https://doi.org/10.1016/S1293-2558(02)00027-4
Python. https://en.wikipedia.org/wiki/Python_ (programming_ language)
Numpy. https://numpy.org/
Frisch M.J., Trucks G.W., Schlegel H.B. et al. Gaussian 09, Revision A.02. Gaussian, Inc., Wallingford CT, 2009.
Becke A.D. // Phys. Rev. A. 1988. V. 38. № 6. P. 3098. https://doi.org/10.1103/PhysRevA.38.3098
Becke A.D. // J. Chem. Phys. 1993. V. 98. № 2. P. 1372. https://doi.org/10.1063/1.464304
Lee C., Yang W., Parr R.G. // Phys. Rev. B. 1988. V. 37. № 2. P. 785. https://doi.org/10.1103/PhysRevB.37.785
Trilinear Interpolation. URL: https://en.wikipedia. org/wiki/Trilinear_interpolation
CUDA. https://en.wikipedia.org/wiki/CUDA
C++. https://en.wikipedia.org/wiki/C%2B%2B
Williams T., Kelley C., Campbell J. et al. Gnuplot 4.6. Manual. 2012. 238 p.
Taucher T.C., Hehn I., Hofmann O.T., Zharnikov M., Zojer E. // J. Phys. Chem. C. 2016. V. 120. P. 3428. https://doi.org/10.1021/acs.jpcc.5b12387
Abu-Husein T., Schuster S., Egger D.A., Kind M., Santowski T., Wiesner A., Chiechi R., Zojer E., Terfort A., Zharnikov M. // Adv. Funct. Mater. 2015. V. 25. P. 3943. https://doi.org/10.1002/adfm.201500899
Feuchtwang T.E., Cutler P.H., Miskovsky N.M. // Phys. Lett. A. 1983. V. 99. № 4. P. 167. https://doi.org/10.1016/0375-9601(83)90969-6
Tersoff J., Hamann D.R. // Phys. Rev. B. 1985. V. 31. № 2. P. 805. https://doi.org/10.1103/PhysRevB.31.805
Barton J.J. // Phys. Rev. Lett. 1991. V. 67. № 22. P. 3106. https://doi.org/10.1103/PhysRevLett.67.3106
Gog T., Len P.M., Materlik G., Bahr D., Fadley C.S., Sanchez-Hanke C. // Phys. Rev. Lett. 1996. V. 76. № 17. P. 3132. https://doi.org/10.1103/PhysRevLett.76.3132
Lider V.V. // Physics-Uspekhi. 2015. V. 58. № 4. P. 365. https://doi.org/10.3367/ufne.0185.201504d.0393
Tegze M., Faigel G. // Nature. 1996. V. 380. P. 49. https://doi.org/10.1038/380049a0
Tamai A., Seitsonen A.P., Greber T., Osterwalder J. // Phys. Rev. B. 2006. V. 74. P. 085407. https://doi.org/10.1103/PhysRevB.74.085407
Fasel R., Aebi P., Agostino R.G., Naumović D., Osterwalder J., Santaniello A., Schlapbach L. // Phys. Rev. Lett. 1996. V. 76. № 25. P. 4733. https://doi.org/10.1103/PhysRevLett.76.4733
Дополнительные материалы отсутствуют.
Инструменты
Поверхность. Рентгеновские, синхротронные и нейтронные исследования