

Поверхность. Рентгеновские, синхротронные и нейтронные исследования, 2022, № 7, стр. 96-103
Повышение активности низкотемпературного окисления молекул оксида углерода на поверхности металлооксидной системы Au/Al2O3/Mo(110) за счет туннелирования заряда сквозь оксидную пленку
Т. Т. Магкоев *
ФГБОУ ВО “Северо-Осетинский государственный университет им. К.Л. Хетагурова”
362025 Владикавказ, Россия
* E-mail: t_magkoev@mail.ru
Поступила в редакцию 14.12.2021
После доработки 27.01.2022
Принята к публикации 30.01.2022
- EDN: BYPYAE
- DOI: 10.31857/S1028096022070123
Аннотация
Формирование наночастиц золота на поверхности пленки оксида алюминия, образованной на кристалле Мо(110), и окисление молекул оксида углерода на их поверхности исследовано в условиях сверхвысокого вакуума методами электронной оже-спектроскопии, дифракции медленных электронов, инфракрасной абсорбционной спектроскопии, атомно-силовой микроскопии, термопрограммируемой десорбции и измерения работы выхода. Основной акцент сделан на изучении эффективности процесса окисления молекул СО в зависимости от толщины оксидной пленки между Мо и нанесенными частицами Au с целью установления возможного влияния на данный процесс эффекта туннелирования заряда сквозь оксидную диэлектрическую прослойку. Достигнута наибольшая степень идентичности структурных, электронных и адсорбционных свойств систем Au/Al2O3/Mo(110). При различных толщинах пленки оксида алюминия (2, 4, 6, 8 монослоев), показано, что эффективность окисления молекул СО, определяемая как доля молекул СО2 относительно СО, десорбируемых в газовую фазу в процессе измерений спектров термодесорбции, экспоненциально уменьшается с ростом толщины пленки оксида. Принимая во внимание установленный факт, что эффективность окисления СО зависит от величины избыточного заряда, приобретаемого наночастицей золота, можно сделать заключение, что туннелирование заряда сквозь оксидную прослойку повышает эффективность реакции на поверхности соответствующей металлооксидной системы.
ВВЕДЕНИЕ
Металлические наночастицы на оксидах используют в различных областях, одной из которых является гетерогенный катализ, применяемый, в частности, для нейтрализации выбросов двигателей внутреннего сгорания, газовых источников энергии, очистки атмосферы промышленных и жилых помещений и др. [1–3]. Это мотивирует обширные исследования модельных катализаторов на металлической или оксидной подложке для лучшего понимания элементарных стадий, связанных с реакциями на их поверхности [2]. Одним из ключевых факторов, активирующих реакции на поверхности нанесенной металлической частицы, является величина заряда, приобретенного за счет переноса заряда между оксидной подложкой и частицей [3–5]. Например, для частицы Au на подложке из TiO2(110) с помощью теоретических расчетов методом функционала плотности было показано [6], что для обогащенной титаном поверхности перенос электрона происходит от шестикратно координированного атома Ti к атому Au, тогда как для обогащенной кислородом поверхности происходит обратный перенос от атома Au – к поверхностным и внутренним атомам кислорода. Этот процесс тесно связан с каталитическими свойствами системы Au/TiO2. Аналогичная ситуация имеет место для Au, нанесенного MgO, для которого был выявлен [7] частичный перенос заряда с поверхности оксида на кластер золота, что играет существенную роль в активации наноразмерных кластеров золота в качестве катализаторов реакции окисления СО. Позже [8] обнаружили, что оксид магния является эффективным донором электронов для частиц Au за счет переноса заряда от анионных вакансий (F-центров), что также активирует их в качестве катализаторов окисления СО. Каталитическая активация частиц Au, нанесенных на LiF, путем переноса заряда от анионных вакансий также недавно была продемонстрирована в работе [9]. Отрицательно заряженные наночастицы Au, как считается, активнее адсорбируют кислород и активируют связь O–O [10], а также способствуют активации CO [11]. Для металлических частиц, нанесенных на наноструктурированные оксидные слои, выращенные на металлических подложках, имеется дополнительный канал переноса заряда между металлической подложкой и металлическими частицами посредством туннелирования электронов сквозь промежуточный слой оксида [12]. Например, осаждение Cs на тонких пленках оксида алюминия, нанесенных на Мо(110), вызывает довольно большие (0.9–1.1 эВ) положительные сдвиги в кинетической энергии оже-перехода KVV O и энергии связи O 1s, Al 2p [13]. Осаждение алюминия на слой оксида алюминия, выращенный на NiAl(110), приводит к сдвигу энергии связи Al 2p, O 1s и O 2p в сторону более высоких значений на 0.47 ± ± 0.03 эВ [14]. Аналогичный сдвиг в сторону более высокой энергии связи на 0.5 эВ был обнаружен при нанесении V на такую же поверхность оксида алюминия [15]. С учетом того, что Cs, Al и V имеют низкую работу выхода, ожидаемо, что перенос заряда будет происходить от Mo к Cs [16], от NiAl к Al [14] и от NiAl к V [15].
Эти и подобные исследования в основном касаются “статического” состояния частиц на подложке, при котором туннелирование электронов через оксидную прослойку не оказывает заметного влияния на свойства систем рассматриваемого типа. Целью настоящей работы являлось установить характер влияния туннелирования заряда сквозь оксидную пленку на особенности реакции наночастиц металла, нанесенных на ее поверхность. Для нивелирования посторонних процессов, вызванных взаимодействием металла с оксидом и тем самым маскирующих возможный туннельный эффект, выбраны благородный металл (Au) и стабильный оксид (Al2O3), а также простые хорошо изученные реагенты (СО и О2).
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Исследования проведены в модифицированной сверхвысоковакуумной системе VG Escalab MII (давление остаточных газов: 3 × 10–10 мбар), в которой реализованы методы электронной оже-спектроскопии с помощью однокаскадного цилиндрического анализатора с коаксиальной пушкой (Riber); дифракции медленных электронов (Varian) с использованием четырехсеточной электронной оптики; инфракрасной спектроскопии в режиме скользящего падения луча (ИК-спектроскопии); температурно-программируемой десорбции (ТПД) с использованием квадрупольного масс-спектрометра (Hiden Analytical); измерения работы выхода по методу Андерсона и атомно-силовой микроскопия (АСМ) с применением нанозонда Scienta Omicon Nanoprobe, позволяющим реализовывать четырехконтактные измерения наноструктур. Эксперименты методом ИК-спектроскопии были проведены на Фурье-спектрометре Nicolet-Nexus 870 с использованием P-поляризованного света, применение которого обеспечивает чувствительность к колебаниям молекул, перпендикулярных к плоскости поверхности адсорбента. Такие колебания происходят, как правило, в системах, где двухатомные полярные молекулы, такие как СО, адсорбированы вертикально или под наклоном на большинстве металлических и неметаллических подложек [17].
В настоящей работе пленка оксида алюминия контролируемой толщины и структуры формировали на поверхности кристалла Mo, поддерживаемой при повышенной температуре, с помощью хорошо известной процедуры реактивного термического испарения атомов алюминия из ячейки Кнудсена в атмосфере кислорода при парциальном давлении порядка 10–7 мбар [18]. Выбор Mo обусловлен близостью параметров решетки Мо(110) и α-Al2O3(1000) и его тугоплавкостью, что допускает высокотемпературную термическую обработку, необходимую для формирования структурно упорядоченных пленок α-Al2O3. Это позволяет получить эпитаксиальные пленки даже очень малой толщины. Толщину пленки θ оценивали с погрешностью ~15% посредством измерения потока атомов алюминия по изменению частоты кварцевого резонатора и ослаблению интенсивности оже-сигнала перехода MNV (188 эВ) при росте оксидной пленки в соответствии с процедурой [18, 19]. Регистрация частоты выполнена с помощью частотомера QCM-D NanoScience Instruments. Величина потока осаждаемых атомов Al дополнительно была проконтролирована по изменению работы выхода подложки Mo при росте субмонослойной пленки алюминия [20]. Атомы Au наносились на пленки оксида алюминия термическим напылением массивного Au (чистота: 99.9999%) из ячейки Кнудсена на подложки, поддерживаемые при комнатной температуре, с последующим отжигом до 500 К в течение 3 минут для достижения равновесного состояния. Величина покрытия Au определена с помощью кварцевого резонатора и по ослаблению оже-сигнала подложки O KVV (502 эВ). За один монослой условно был принят слой с поверхностной концентрацией атомов Au 1.45 × × 1015 см–2. Молекулы СО и О2 контролируемым образом были поданы в сверхвысоковакуумную камеру из резервуаров, содержащих соответствующие газы высокой степени очистки. Было принято, что экспозиция 1 Л соответствует 10–6 Торр · с. Конструкция экспериментальной установки, в которой кристалл Mo был установлен соосно с кварцевым резонатором, позволяла охлаждать образец до 90 К через присоединенный резервуар, наполняемый жидким азотом, и нагревать до умеренных температур (1000–1500 К) путем пропускания тока через образец и высокотемпературный прогрев до 2700 К электронной бомбардировкой.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Вероятность туннелирования электронов через оксидный диэлектрический слой экспоненциально зависит от его толщины, поэтому измерения эффективности окисления СО на поверхности Au/Al2O3/Mo были проведены при различной толщине межслойного слоя оксида алюминия: в 2, 4, 6 и 8 монослоев. Поскольку реакция на поверхности адсорбента крайне чувствительны к состоянию поверхности, необходимо было обеспечить идентичность структурных, электронных и адсорбционных свойств поверхности пленок оксида алюминия, а также свойств Au/Al2O3. Согласно литературным данным, пленка оксида алюминия толщиной в 2 монослоя обладает свойствами, сходными со свойствами объемного оксида алюминия [18, 19, 21–23]. Более того, с дальнейшим увеличением толщины (более дух монослоев) пленка приобретает диэлектрические свойства [24–26]. Картины дифракции медленных электронов от пленок оксида алюминия толщиной 2, 4, 6 и 8 монослоев практически идентичны и демонстрируют гексагональную симметрию, соответствующую α-Al2O3(1000), подобно наблюдаемой в работах [18, 19, 27] (рис. 1а, 1б). Положение межатомного оже-перехода алюминия, а также его интенсивность по отношению к оже-линии O KVV практически идентичны для пленок оксида алюминия различной толщины. Это указывает на то, что стехиометрия оксидных пленок одинакова.
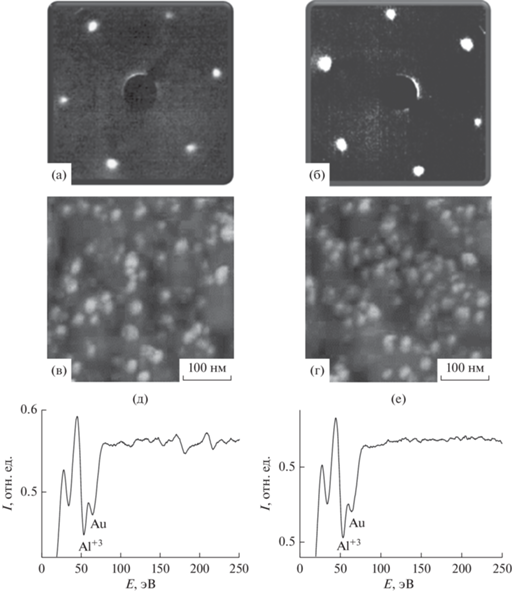
Рис. 1.
Картины дифракции медленных электронов от оксида алюминия толщиной 2 (а) и 8 (б) монослоев на поверхности Мо. АСМ изображения (в, г) поверхностей оксида указанной толщины, покрытых слоем золота толщиной 0.7 монослоя; соответствующие оже-спектры (д, е).
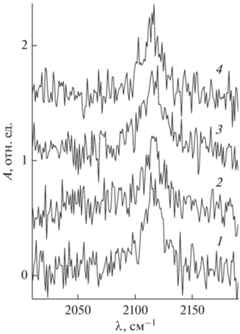
Более точной проверкой состояния поверхности пленок оксида алюминия является изучение адсорбционных свойств тестовых молекул, например, СО [28]. Одним из наиболее чувствительных методов для изучения таких свойств является ИК-спектроскопия из-за высокого разрешения и чувствительности к адсорбированным молекулам, колебательные свойства которых чрезвычайно зависимы от деталей структурного и электронного состояний адсорбента [17]. ИК-спектры в области внутримолекулярных колебаний СО, адсорбированных при насыщающем покрытии (экспозиция 200 Л) на пленках оксида алюминия выбранной толщины, охлажденных до 90 К, приведены на рис. 2. Видно, что спектры подобны, что свидетельствует о том, что состояние поверхности пленки оксида алюминия при всех исследованных толщинах практически одинаково. Поскольку известно, что СО не адсорбируется на регулярных участках оксида алюминия при 90 К [29], наблюдаемая ИК-линия поглощения, вероятно, связана с тем, что молекулы СО преимущественно адсорбируются на дефектах поверхности оксида [30]. Одинаковое энергетическое положение ИК-линии поглощения, ее интенсивность и полуширина позволяют предположить, что природа дефектов и их плотность одинаковы для всех исследованных пленок оксида алюминия. Довольно низкая интенсивность ИК-линии относительно фона указывает на то, что плотность дефектов довольно низкая. Наиболее вероятной природой таких дефектов являются анионные вакансии, которые существуют в определенной степени даже в случае структурно упорядоченных, преимущественно стехиометрических пленок оксида алюминия, выращенных путем реактивного осаждения [31]. Они стимулируют адсорбцию молекул CO на поверхности оксида алюминия за счет переноса заряда с F-центра подложки на антисвязывающую 2π*-орбиталь молекулы СО – процесс незначительный на регулярных центрах идеальной подложки α‑Al2O3(1000) [32].
Рис. 2.
ИК-спектры СО, полученные после экспозиции 200 L, на поверхности пленок оксида алюминия толщиной в 2 (1), 4 (2), 6 (3) и 8 (4) монослоев.
Морфология слоев Au, нанесенных в количестве эквивалентном 0.7 монослоя на пленки оксида алюминия различной толщины, поддерживаемые при 500 K для достижения равновесия, сходна, как видно из соответствующих изображений АСМ (рис. 1в, 1г). Это подтверждается подобием отношения интенсивностей оже-линии Au NVV к Al LVV (рис. 1д, 1е) – величины, характеризующей относительную площадь подложки, покрытой адсорбатом [19, 33]. Эффективный размер частиц Au, как видно из рис. 1в, 1г, составляет порядка 5 нм в латеральном направлении и 5 нм вдоль нормали к поверхности. Согласно теоретическим расчетам [34, 35], Au связывается с α-Al2O3(1000) ионной связью при субмонослойном покрытии, в то время как при покрытии большей толщины, когда образуются трехмерные островки, характер связи металла с оксидом становится более ковалентным (из-за поляризационного эффекта). Особенностью нанесенных на поверхность оксида частиц Au является их, как правило, нейтральный характер на регулярных участках поверхности [12, 36, 37]. Гексагональная симметрия поверхности пленки оксида алюминия, наблюдаемая на картинах дифракции медленных электронов (рис. 1а, 1б), не зарегистрирована на АСМ-изображениях наночастиц золота, нанесенных на поверхность оксида (рис. 1в, 1г). Это следует связать с тем, что для АСМ наблюдаемые поверхности обладали более неравномерным рельефом, следовательно, сканирование проходило в условиях постоянного меняющегося рельефа, что не позволяет достичь атомного разрешения.
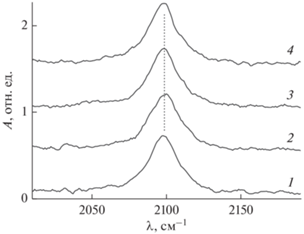
Для более точной проверки, зависит ли состояние частиц Au от толщины пленки оксида алюминия, адсорбция молекул CO, как тестовых частиц, была исследована с помощью ИК-спектроскопии при экспозиции порядка 100 Л. Все зарегистрированные в этих условиях спектры, состоящие из колебательной линии СО, практически идентичны друг другу (рис. 3). С учетом чрезвычайной чувствительности молекулярных колебательных мод к деталям атомной и электронной структуры подложки [17] наблюдаемое сходство ИК-спектров свидетельствует об одинаковом морфологическом и структурном состоянии частиц Au, независимо от толщины пленки оксида алюминия. Поскольку CO практически не адсорбируется на регулярной поверхности Au при используемых условиях [38], наблюдаемая линия поглощения может быть отнесена к молекулам, связанным с низкокоординированными центрами в наноразмерных кластерах Au, а также к межфазной границе раздела металл/оксид, в русле многочисленных интерпретаций, сделанных для этого и других типов частиц Au на оксидных подложках [12, 29, 38–40]. Более низкое значение волнового числа CO на Au (2098 см–1), по сравнению с таковым на поверхности оксида алюминия (2115 см–1), может быть обусловлено более высокой степенью переноса заряда от Au, чем от анионной вакансии оксида, на антисвязывающую 2π*-орбиталь молекулы СО.
Рис. 3.
ИК-спектры СО на поверхности нанесенных на пленку оксида алюминия толщиной 2 (1), 4 (2), 6 (3) и 8 (4) монослоев кластеров золота (с эффективном покрытием, эквивалетным 0.7 монослоя), поддерживаемых при температуре 90 К. Значение волнового числа k максимума линии поглощения во всех случаях составляет 2098 см–1. Экспозиция составляла 100 Л.
Постадсорбция кислорода на поверхности системы CO/Au/Al2O3 при экспозиции 100 Л и температуре 90 K приводит к смещению линии поглощения CO на 5–6 см–1 в более коротковолновую область без заметного изменения ее интенсивности. Такое довольно незначительное изменение волнового числа вряд ли может быть приписано смещению молекулы в другой адсорбционный центр, вызванное адсорбцией кислорода. В последнем случае колебательный сдвиг был бы более значительным [41]. Поскольку известно, что молекулярный кислород диссоциирует на ультрамалых частицах Au [3], наблюдаемый сдвиг ИК-линии можно объяснить снижением заселенности антисвязывающей 2π*-орбитали СО вследствие переноса заряда на 2р-уровень атома кислорода, обладающий более высоким сродством к электрону [42]. Наблюдаемая довольно низкая чувствительность ИК-спектров CO к постадсорбции O2 позволяет предположить, что монооксид углерода и кислород не конкурируют за центр адсорбции на поверхности частицы золота при 90 К. Вместо этого они, вероятно, находятся в соседних адсорбционных центрах, слегка влияя друг на друга, что и проявляется в наблюдаемом незначительном сдвиге ИК-линии поглощения. Согласно расчетам [43], преимущественным адсорбционным центром атомарного кислорода является полость, образованная четырьмя атомами Au, в то время как СО связывается с низкокоординированными атомами кластера Au.
Более выраженный эффект, в конечном итоге приводящий к молекулярному превращению CO + O2 → CO2, появляется при нагревании, стимулирующем миграцию адсорбированных частиц по поверхности и их активацию. Спектры ТПД для CO и CO2, полученные при непрерывном воздействии на систему CO/Au/Al2O3 кислорода, напускаемого в вакуумную камеру до парциального давления 10–6 мбар, приведены на рис. 4а, 4б. Скорость изменения температуры была выбрана равной 1 К/с для обеспечения равновесности процесса, а масс-спектрометр был настроен для одновременной регистрации сигналов с молекулярной массой 28 (CO) и 44 (CO2). Как видно, наряду с десорбцией СО (рис. 4а), образуется также СО2, хотя и в меньшей степени (рис. 4б). Механизм окисления СО на поверхности частиц Au, нанесенных на оксид, является предметом интенсивных исследований в течение последних десятилетий. Считается, что это является результатом сочетания различных параметров, таких как размер частиц, их морфология и структура, природа оксидной подложки и ее стехиометрия, состояние границы раздела металл/оксид, условия реакции и др. [12, 29, 38–40, 44]. Общее мнение в этом отношении состоит в том, что знак и величина заряда частицы Au, контролирующей активацию молекул CO и O2, является ключевым фактором, определяющим характер и скорость реакции [44].
Рис. 4.
ТПД-спектры CO (а) и CO2 (б), полученные при непрерывной экспозиции системы CO/Au/Al2O3 в кислороде посредством заполнения вакуумной камеры до парциального давления 10–6 Торр. На вставке приведены области максимумов ТПД-линий. Толщина пленки оксида алюминия составляла 2 (1), 4 (2), 6 (3) и 8 (4) монослоев.
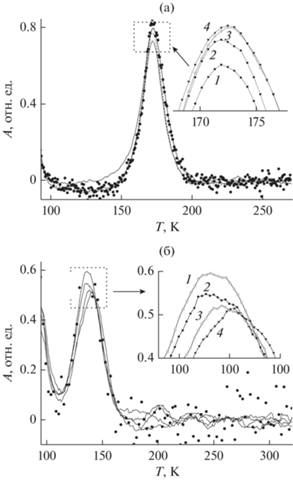
Более детальное рассмотрение ТПД-спектров вблизи максимумов (рис. 4) показывает, что интенсивность сигнала СО растет, а интенсивность сигнала СО2 соответственно, падает с увеличением толщины оксидной пленки. Это означает, что эффективность окисления СО выше для более тонкой межслойной пленки оксида алюминия. Эту эффективность σ можно качественно определить, как отношение интенсивности I ТПД-линий СО2 и СО. В качестве величин интенсивностей были использованы величины максимумов ТПД-линий для каждой толщины оксидного слоя. Зависимость σ от толщины пленки оксида алюминия приведена на рис. 5. Полученная кривая, как видно из рисунка, достаточно хорошо соответствует экспоненциальной зависимости. Принимая во внимание вышеупомянутый факт, что скорость окисления CO на поверхности нанесенных кластеров Au зависит от величины их заряда, наблюдаемую зависимость σ можно расценивать как указание на то, что туннелирование электронов металлической подложки Mo через пленку оксида алюминия в область реакции на поверхности Au/Al2O3 повышает эффективность окисления СО. Реакция происходит при избыточном заряде, который формируется в результате туннельного эффекта сквозь прослойку оксида алюминия. Это согласуется с результатами соответствующих теоретических исследований, свидетельствующих о том, что перезарядка нанесенного кластера Au во время цикла окисления СО существенно влияет на энергетику всех окислительное-восстановительных стадий в каталитических превращениях [45]. Кроме того, согласно ранее опубликованным результатам [46], молекула O2 при адсорбции приобретает отрицательный заряд, а длина связи O–O увеличивается до 1.39–1.47 Å в зависимости от размера кластера Au на поверхности оксидной подложки. Дополнительно к этому на границе раздела металл/оксид происходит перераспределение заряда, вызванное адсорбцией реагентов. Теоретически такая особенность обоснована расчетами [45], согласно которым для реализации процесса реакции на поверхности металлооксидных систем требуется резервуар электронов, которым в настоящей работе является металлическая подложка Мо, являющаяся источником туннелирующих электронов. Данный эффект, однако, не является доминирующим: как видно из рис. 5, повышение эффективности окисления СО не превышает 20%.
Рис. 5.
Зависимость отношения интенсивности ТПД-линий CO2 к CO от толщины пленки оксида алюминия (точками). Непрерывной кривой обозначена экспоненциальная аппроксимация экспериментальной зависимости.

Принимая во внимание возможность туннелирования электронов через оксидный слой, можно ожидать, что заряд частиц Au будет проявляться в изменении частоты ИК-линий поглощения СО с изменением толщины оксидной пленки. Однако это не соответствует действительности: как видно на рис. 3, частота линии СО практически одинакова для всех исследованных толщин пленки оксида алюминия. Это означает, что в ”статическом” режиме туннелирование может быть незначительным. Эффект усиливается в условиях реакции, когда молекулярное окисление требует интенсивной динамической зарядки или перезарядки частиц Au, границы раздела металл/оксид и реагентов.
ЗАКЛЮЧЕНИЕ
Структурные, морфологические и адсорбционные свойства наноразмерных частиц золота на упорядоченной пленке оксида алюминия, выращенной на подложке Mo, не зависят от толщины оксидной пленки, по крайней мере, в диапазоне от 2 до 8 монослоев. В отличие от регулярной поверхности массивного Au, СО легко адсорбируется на нанесенных нанокластерах золота и на границе раздела металл/оксид при температуре подложки 90 К. Состояние адсорбированных молекул СО довольно слабо зависит от последующей адсорбции молекул кислорода, что свидетельствует о том, что эти молекулы занимают различные адсорбционные центры. Нагрев соадсорбированного слоя (СО + О2) приводит к десорбции в газовые фазы как СО, так и СО2. Характерной особенностью этого процесса является то, что доля десорбирующихся молекул СО2 относительно СО экспоненциально уменьшается с увеличением толщины пленки оксида алюминия. Последнее, с учетом известного факта, что эффективность окисления СО зависит от количества избыточного заряда, приобретенного частицей Au, поверхностью раздела металл/оксид и реагентами, является свидетельством того, что туннелирование электронов между реакционной зоной и металлической подложкой через промежуточный оксидный слой стимулирует процесс окисления молекул СО.
Список литературы
Metal Oxides in Heterogeneous Catalysis / Ed. Védrine J.C. Elsevier Inc., 2018. https://doi.org./10.1016/C2016-0-01790-4
Chen S., Xiong F., Huang W. // Surf. Sci. Rep. 2019. V. 74. P. 100471. https://doi.org./10.1016/j.surfrep.2019.100471
Picone A., Riva M., Brambilla A. et al. // Surf. Sci. Rep. 2016. V. 71. P. 32. https://doi.org./10.1016/j.surfrep.2016.01.003
Cai Y., Feng Y.P. // Progr. Surf. Sci. 2016. V. 91. P. 183. https://doi.org/10.1016/j.progsurf.2016.11.001
Honkala K. // Surf. Sci. Rep. 2014. V. 69. P. 366. https://doi.org./10.1016/j.surfrep.2014.09.002
Okazaki K., Morikawa Y., Tanaka S. et al. // Phys. Rev. B. 2014. V. 69. P. 235404. https://doi.org./10.1103/PhysRevB.69.235404
Sanchez A., Abbet S., Heiz U. et al. // J. Phys. Chem. A. 1999. V. 103. P. 9573.
Yan Z., Chinta S., Mohamed A.A. et al. // J. Am. Chem. Soc. 2005. V. 127. P. 1604. https://doi.org./10.1021/ja043652m
Tvauri I.V., Gergieva B.E., Grigorkina G.S. et al. // Solid State Commun. 2015. V. 213–214. P. 42. https://doi.org./10.1016/J.SSC.2015.04.017
Yoon G., Hakkinen H., Landman U. // J. Phys. Chem. A. 2003. V. 107. P. 4066. https://doi.org/10.1021/jp027596s
Lopez N., Norskov J.K., Janssens T.V.W. et al. // J. Catal. 2004. V. 225. P. 86. https://doi.org./10.1016/j.jcat.2006.03.008
Fu Q., Wagner T. // Surf. Sci. Reports. 2007. V. 62. P. 431. https://doi.org/10.1016/j.surfrep.2007.07.001
Heemeier M., Stempel S., Shaikhutdinov S.K. et al. // Surf. Sci. 2003. V. 523. P. 103. https://doi.org./10.1016/S0039-6028(02)02404-4
Libuda J., Frank M., Sandell A. et al. // Surf. Sci. 1997. V. 384. P. 106. https://doi.org/10.1016/S0039-6028(97)00170-2
Baumer M., Biener J., Madix R.J. // Surf. Sci. 1999. V. 432. P. 189. https://doi.org./10.1016/S0039-6028(99)00400-8
Rodriguez J.A., Kuhn M., Hrbek J. // J. Phys. Chem. 1996. V. 100. P. 18240. https://doi.org./10.1021/jp962195w
Hirschmugl C.J. // Surf. Sci. 2002. V. 500. P. 577. https://doi.org./10.1016/B0-12-369397-7/00607-5
Goodman D.W. // J. Vac. Sci. Technol. A. 1996. V. 14. P. 1526. https://doi.org/10.1116/1.580290
Wu M.-C., Goodman D.W. // J. Phys. Chem. 1994. V. 98. P. 9874. https://doi.org/10.1021/j100090a023
Grigorkina G.S., Tvauri I.V., Kaloeva A.G. et al. // Solid State Commun. 2016. V. 233. P. 11. https://doi.org./10.1016/j.ssc.2016.02.010
Jaeger R.M., Kuhlenbeck H., Freund H.-J. et al. // Surf. Sci. 1991. V. 259. P. 235. https://doi.org./10.1016/0039-6028(91)90555-7
Becker C., Kandler J., Raaf H. et al. // J. Vac. Sci. Technol. A. 1998. V. 16. P. 1000. https://doi.org/10.1116/1.581221
Jennison D.R., Verdozzi C., Schultz P.A., Sears M.P. // Phys. Rev. B. 1999. V. 59. P. 15605. https://doi.org./10.1103/PhysRevB.59.R15605
Magkoev T.T., Vladimirov G.G. // J. Phys.: Condens. Matter. 2001. V. 13. P. L655. https://doi.org./10.1088/0953-8984/13/28/101
Magkoev T.T., Christmann K., Moutinho, A.M.C., Murata Y. // Surf. Sci. 2002. V. 515. P. 538. https://doi.org./10.1016/S0039-6028(02)01972-6
Magkoev T.T., Vladimirov G.G., Remar D., Moutinho A.M.C. // Solid State Commun. 2002. V. 122. P. 341. https://doi.org./10.1016/S0038-1098(01)00511-7
Frederick G., Apai G., Rhodin T.N. // Phys. Rev. B. 1991. V. 44. P. 1880. https://doi.org/10.1103/PhysRevB.44.1880
Fierro J.L.G., De La Banda J.F.G. // Catal. Rev. 1986. V. 28. P. 265. https://doi.org./10.1080/01614948608082252
Campbell C.T. // Surf. Sci. Rep. 1997. V. 27. P. 1. https://doi.org./10.1016/S0167-5729(96)00011-8
Zecchina A., Scarano D., Bordiga S. et al. // Catal. Today. 1996. V. 27. P. 403. https://doi.org./10.1023/A:1019089309446
Renaud G., Villette B., Vilfan I., Bourret A. // Phys. Rev. Lett. 1994. V. 73. P. 1825. https://doi.org./10.1103/PhysRevLett.73.1825
Casarin M., Maccato C., Vittadini A. // J. Phys. Chem. B. 2002. V. 106. P. 795. https://doi.org./10.1016/j.susc.2004.05.084
Ossicini S., Memeo R., Ciccacci F. // J. Vac. Sci. Technol. A. 1985. V. 3. P. 387. https://doi.org./10.1116/1.573226
Bogicevic A., Jennison D.R. // Phys. Rev. Lett. 1999. V. 82. P. 4050. https://doi.org./10.1103/PhysRevLett.82.4050
Mattsson A.E., Jennison D.R. // Surf. Sci. 2002. V. 520. P. L611. https://doi.org./10.1016/S0039-6028(02)02209-4
Cao Y., Hu S., Yu M. et al. // Phys. Chem. Chem. Phys. 2016. V. 18. P. 17660. https://doi.org/10.1039/C6CP02324J
Marquez A.M., Graciani J., Sanz J.F. // Theor. Chem. Acc. 2010. V. 126. P. 265. https://doi.org./10.1007/s00214-009-0703-0
Meyer R., Lemire C., Shaikhutdinov Sh. K., Freund H.-J. // Gold Bulletin. 2004. V. 37. P. 72. https://doi.org./10.1007/BF03215519
Grisel R., Weststrate K., Gluhoi A., Nieuwenhuys B.E. // Gold Bulletin. 2002. V. 35. P. 39. https://doi.org./10.1002/chin.200249203
Hussain A., Ferre D.C., Gracia J. et al. // Surf. Sci. 2009. V. 603. P. 2734. https://doi.org./10.1016/J.SUSC.2009.07.023
Davis S.P., Abrams M.C., Brauet J.W. Fourier-transform Spectroscopy / New York, London: Academic Press, 2001. https://doi.org./10.1016/B978-0-12-042510-5.X5000-8
Doyen G., Ertl G. // Surf. Sci. 1974. V. 43. P. 197. https://doi.org/10.1016/0039-6028(74)90228-3
Hussain A., Muller A.J., Nieuwenhuys B.E. et al. // Top Catal. 2011. V. 54. P. 415. https://doi.org./10.1007/s11244-011-9672-3
Wang Y.-G., Yoon Y., Glezakou V.-A. et al. // J. Am. Chem. Soc. 2013. V. 135. P. 10673. https://doi.org./10.1021/ja402063v
Camellone M.F., Kowalski P.M., Marx D. // Phys. Rev. B. 2011. V. 84. P. 035413. https://doi.org./10.1103/PhysRevB.84.035413
Harding C., Habibpour V., Kunz S. et al. // J. Am. Chem. Soc. 2008. V. 131. P. 538. https://doi.org./10.1021/ja804893b
Дополнительные материалы отсутствуют.
Инструменты
Поверхность. Рентгеновские, синхротронные и нейтронные исследования