

Приборы и техника эксперимента, 2021, № 2, стр. 5-18
ГЕТЕРОЭПИТАКСИАЛЬНЫЙ РОСТ АЛМАЗА ИЗ ГАЗОВОЙ ФАЗЫ: ПРОБЛЕМЫ И ПЕРСПЕКТИВЫ (обзор)
С. А. Линник a, *, С. П. Зенкин a, А. В. Гайдайчук a
a Национальный исследовательский Томский политехнический университет
634000 Томск, просп. Ленина, 30, Россия
* E-mail: stepan_lin@mail.ru
Поступила в редакцию 16.07.2020
После доработки 19.08.2020
Принята к публикации 21.08.2020
Аннотация
Рассматриваются современное состояние исследований и новые подходы к решению проблемы гетероэпитаксии пленок алмаза из газовой фазы. Описаны особенности роста алмаза из газовой фазы, процессы нуклеации, текстурирования и основные фундаментальные проблемы и барьеры гетероэпитаксии монокристаллического алмаза. Рассмотрены последние достижения и потенциально перспективные подходы в данной области.
1. ВВЕДЕНИЕ
Алмаз тысячелетиями считался ценнейшим из минералов. Причиной тому, за исключением его редкости, безусловно, являются его уникальные свойства (см. табл. 1), недостижимые для любого другого известного материала. Прежде всего, это экстремально высокая твердость (до 100 ГПа), высочайшая теплопроводность (свыше 2000 Вт/м · К) и практически полная химическая инертность. Помимо этого, алмаз обладает очень высокой радиационной стойкостью [1, 2], низким коэффициентом трения (<0.1) и прозрачностью в очень широком диапазоне длин волн (от дальнего ультрафиолета до дальней инфракрасной области [3, 4], а также для рентгеновского излучения [5]). Несмотря на его высокое удельное сопротивление (1012–1016 Ом · см) и высокую ширину запрещенной зоны (5.45 эВ) данный материал может с успехом применяться в качестве полупроводника в электронике [6] путем создания n- и p-проводимости посредством его легирования атомами бора [7–9], фосфора [10, 11], азота [12, 13], серы [14] и др.
Таблица 1.
| Параметр | Монокристаллический алмаз | CVD-алмаз |
|---|---|---|
| Плотность, г/см3 | 3.515 | 2.8–3.51 |
| Теплоемкость (27°С), Дж/моль · К | 6.195 | 6.12 |
| Теплопроводность (25°С), Вт/м · К | 2200 | 2100 |
| Удельное электрическое сопротивление, Ом · см | 1016 | 1012–1016 |
| Модуль Юнга, ГПа | 820–900 | 910–1250 |
| Коэффициент трения на воздухе | 0.05–0.15 | 0.035–0.3 |
| Твердость по Виккерсу, ГПа | 50–100 | 57–104 |
| Ширина запрещенной зоны, эВ | 5.45 | 5.45 |
2. СИНТЕЗ АЛМАЗА
Попытки искусственного синтеза алмаза долгое время оставались безуспешными, пока в 1939 г. О.И. Лейпунский теоретически не рассчитал термодинамические условия кристаллизации алмаза и впервые определил общий вид диаграммы состояния углерода [21]. Практическая реализация данных условий позволила шведам Э. Лундблату и Х.Р. Лиандеру в 1953 г. провести первый в мире успешный эксперимент по высокотемпературной (2500°С) кристаллизации алмаза под высоким давлением (80 000 атм) [22], положивший начало методу HPHT (high pressure high temperature). Данный метод позволяет проводить синтез алмаза в условиях его термодинамической стабильности и на данный момент позволяет синтезировать монокристаллы массой до 60 карат [23, 24].
Несмотря на высокую производительность и сравнительно низкие энергозатраты, для данного метода характерно одно существенное ограничение – эффективный диаметр сечения даже самого крупного монокристалла не превышает 10–15 мм (и это при том, что стоимость даже такого небольшого монокристалла огромна), что серьезно ограничивает его применение в случаях, когда требуется монокристаллическая поверхность большой площади (десятки квадратных сантиметров) либо, наоборот, очень малой (<1 мкм). К таким областям можно прежде всего отнести микро- и наноэлектронику [25, 26], а также детекторную технику [27].
На рис. 1 представлен общий вид фазовой диаграммы углерода, из которой следует, что алмаз является стабильной аллотропной формой углерода только при существенно высоких температурах и давлениях. При низких давлениях и температурах (в том числе и при нормальных условиях) стабильной формой углерода является графит. Из диаграммы также следует, что синтез алмаза возможен исключительно при давлениях в десятки гигапаскаль (см. области А и В).
Рис. 1.
Классическая фазовая диаграмма состояния углерода (согласно [28, 29]). Области: А отражает диапазон высокотемпературного синтеза алмаза под высоким давлением, B – диапазон каталитического высокотемпературного синтеза под высоким давлением и С – диапазон метастабильного синтеза из газовой фазы.
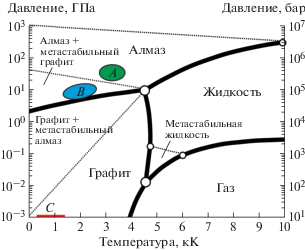
Тем не менее, уже более полувека назад была установлена возможность гомоэпитаксиального роста алмаза из углеводородов при субатмосферных давлениях [30–32], на основе которой впоследствии была разработана полноценная технология газофазного синтеза CVD-алмаза (CVD – chemical vapour deposition) в виде моно- и поликристаллических пленок на гомо- и гетерогенных подложках [16, 17, 33]. Диапазон давлений и температур, характерных для CVD-синтеза, представлен в виде области C на рис. 1. В основе данной технологии лежит конденсация атомарного углерода на поверхности разогретого (преимущественно в диапазоне от 600 до 1100°С) твердого тела, а также различие в скоростях травления алмазной и неалмазных углеродных фаз (в частности, графита [34]). Нарабатываемый в результате различного рода активации (термическая [35, 36], плазменная [37–39], лазерная [40] и др.) газовой смеси водорода и углеводорода (чаще всего метана) атомарный водород стравливает конденсирующийся на поверхности разогретой подложки неалмазный углерод, создавая условия для преимущественной конденсации алмазной фазы (см. рис. 2).
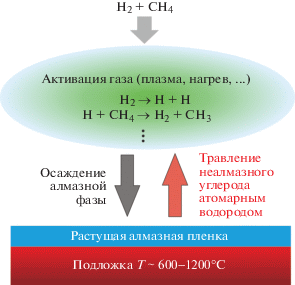
Все способы активации газа для CVD-синтеза алмаза характеризуются существенной вводимой энергией, которая в зависимости от давления варьируется от 50 до 800 Вт/см3. Преимущественно синтез проводится в диапазоне 10–760 Торр (в зависимости от типа активатора и вводимой мощности), так как, с одной стороны, при таких давлениях достигается достаточная для ощутимого роста алмаза концентрация углеродных атомов над подложкой, а с другой, еще генерируется необходимая концентрация атомарного водорода. Температура газа в активируемой области варьируется от 1700 до 5000°С, что является необходимым условием для обеспечения химических реакций.
Химия образования алмаза в газообразной углеводородной среде очень сложна и разнообразна. Можно условно разделить весь процесс на реакции, протекающие в газовой фазе, и реакции у поверхности подложки. В настоящее время общепризнано, что атомы водорода играют ключевую роль в инициации большинства необходимых реакций в газовой фазе [41, 42]. В результате активации газовой смеси водорода и углеводорода создается атмосфера, обогащенная атомарным водородом и разнообразными радикалами (CH3, CH2, CH, C, С2 и др.).
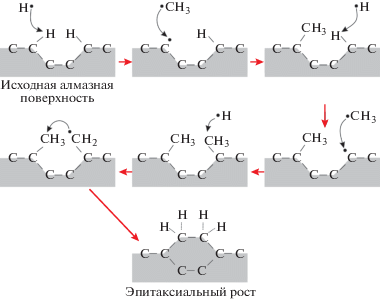
Кроме того, в результате реакции с атомарным водородом могут формироваться более сложные (по сравнению с исходным) углеводороды. На рис. 3 приведена простейшая схема присоединения углеродных атомов в процессе газофазного роста кристалла алмаза, сформированная на основе наиболее распространенной “стандартной” модели, согласно которой ключевую роль при росте алмаза играют метильные радикалы и атомарный водород [43–45]. Водород играет роль посредника, временно связывающего оборванные углеродные связи, уступая впоследствии свое место в решетке новому углеродному атому. Существуют и более сложные модели роста, лучше учитывающие особенности и специфику различных типов CVD-реакторов и более сложные прекурсорные смеси [46–48].
Рис. 3.
Упрощенная схема стандартной модели роста алмаза из газовой фазы на примере метильных радикалов (согласно [45]).
Приведенная выше модель роста хорошо описывает гомоэпитаксиальный рост уже существующей алмазной поверхности аналогично росту кристалла соли в растворе. Понимание же процесса гетероэпитаксии алмаза невозможно без детального рассмотрения процесса его нуклеации на неалмазной поверхности. Существуют две основных теории нуклеации алмазных кристаллитов: гомогенная нуклеация из газовой фазы и наиболее общепринятая теория нуклеации на поверхности.
Теоретическое обоснование возможности гомогенной нуклеации алмазных кристаллитов дано еще в работах [49, 50] на основе классической теории нуклеации. Возможность существования такого вида нуклеации алмаза была подтверждена в ряде работ [51–53], но также был сделан вывод, что подобный механизм в чистом виде физически не способен обеспечить плотность нуклеации, сопоставимую с наблюдаемой на практике.
Для описания нуклеации алмаза на поверхности на данный момент рассматриваются три основных механизма: механизм послойного роста Франка–Ван дер Мерве [54], описывающий строго двумерный рост пленки, механизм трехмерного островкового роста Вольмера–Вебера [55] и смешанный механизм послойного-плюс-островкового роста Странского–Крастанова [56]. Тем не менее, из-за относительно высокого значения поверхностной энергии (около 6 Дж/м2 [57]) алмаз на большинстве подложек имеет тенденцию роста по трехмерному островковому механизму (Вольмера–Вебера).
В общем виде процесс возникновения и роста CVD-алмаза проходит через следующие явно различимые стадии: 1) инкубационный период, 2) образование центров зародышеобразования, 3) превращение зародышей в кристаллиты, 4) сращивание индивидуальных кристаллитов и формирование сплошной пленки, 5) дальнейший рост сплошной пленки. Для гетерогенных подложек инкубационная стадия сводится преимущественно к образованию промежуточного слоя алмазоподобного углерода [58, 59] или графита [60, 61], либо, в случае металлов, карбидного слоя [62, 63].
В общем случае, в этот период происходит насыщение поверхности подложки углеродом и образование буферного слоя, препятствующего оттоку углерода внутрь. Далее происходит нуклеация центров роста на различного рода дефектах (микроостриях, царапинах и т.д.). В случае предварительного искусственного внесения центров зародышеобразования, как, например, методом механической обработки подложки алмазными частицами [64], ультразвукового засеивания поверхности частицами наноалмаза [65] или более сложной техники нуклеации ростовых центров посредством ионной бомбардировки в процессе роста [66, 67] (так называемая BEN-нуклеация (bias enhanced nucleation), инициированная электрическим смещением), инкубационный период минимален, и практически сразу начинается стадия превращения зародышей в кристаллиты.
3. ОБРАЗОВАНИЕ И РОСТ ПОЛИКРИСТАЛЛИЧЕСКИХ АЛМАЗНЫХ ПЛЕНОК
Из-за высокой поверхностной энергии алмаза его двумерный (монокристаллический) рост на неалмазных подложках крайне сложно реализовать. Кроме того, энергия связи атомов углерода в алмазе столь высока, что вероятность сращивания отдельных кристаллитов в монокристалл очень низка, и для этого требуется практически идеально правильная их взаимная ориентация.
Из всех известных материалов наиболее близким к алмазу по своим свойствам и кристаллическому строению является кубический нитрид бора (c-BN), представляющий собой двухэлементный аналог алмаза, который в кубической аллотропной модификации обладает сопоставимой плотностью (3.51 г/см3) и очень близким параметром решетки (3.616 Å против 3.567 Å у алмаза, т.е. несоответствие параметров не превышает 1.3%). Первые сообщения об успешной гетероэпитаксии алмаза на кристаллах c-BN относятся еще к началу 90-х годов XX века [68–70].
Так, на гранях (100) и (111) c-BN, без какой бы то ни было специальной подготовки, были синтезированы гетероэпитаксиальные монокристаллические алмазные слои высокого качества (без двойников, поликристаллизации и т.п.), причем ростовые условия ничем не отличались от условий роста поликристаллических пленок. Из описания работ следует, что рост алмаза на гранях (111) более восприимчив к изменениям ростового процента метана, температуры подложки и ростового давления, чем на гранях (100). Причиной, очевидно, является различие в энергиях связей C–C и C–N, B–C, B–H [71], ведущее к возникновению дефектов на границе слоев c-BN–алмаз.
Несмотря на то, что монокристалл c-BN на данный момент является наиболее подходящей гетерогенной подложкой для гетероэпитаксии алмаза, его применение бесперспективно по причине сложности его синтеза и обработки, а также потому, что максимально достижимый размер монокристалла c-BN не превышает такового для алмаза.
Поскольку гетероэпитаксия монокристаллического алмаза по современным данным возможна лишь на кубическом нитриде бора, рассмотрим подробно процесс формирования поликристаллической пленки (так как только она формируется на всех прочих подложках), ее текстурирования и перехода от поли- к монокристаллической структуре. Рост поликристаллического алмаза из газовой фазы обобщенно состоит из двух обязательных фаз: фазы нуклеации и фазы роста.
Рост сплошной пленки начинается с момента сращивания большинства отдельных алмазных кристаллитов (каждый из которых представляет собой отдельный монокристалл) друг с другом, т.е. с момента, когда свободный рост кристаллита во всех (кроме направления, перпендикулярного поверхности подложки) направлениях начинает ограничиваться соседними. С этого момента начинается соревновательный рост в направлении, перпендикулярном подложке, в результате чего растущая пленка имеет столбчатую (или колонную) микроструктуру, в которой толщина пленки фактически равна высоте отдельного алмазного кристаллита (см. рис. 4). У каждого растущего кристаллита с активированным газом контактирует только верхняя часть (рис. 4г), т.е. возможен рост только тех алмазных граней, которые контактируют напрямую с атомарным водородом и углеводородными радикалами.
Рис. 4.
Схема роста алмазной пленки из газовой фазы (a), микрофотография начальной стадии сращивания алмазных кристаллитов в сплошную пленку (б), микрофотография профиля синтезированной алмазной пленки (в) и топография поверхности растущей алмазной пленки (г).

Поскольку известно, что различные грани алмазного кристалла растут с различной скоростью в зависимости от условий синтеза [72, 73], то вполне очевидно, что через определенное время одни грани заблокируют рост других и произойдет текстурирование поверхности. Согласно подавляющему количеству работ [72–76] динамика роста алмазного кристаллита (т.е. системы граней) определяется преимущественно отношением скоростей роста граней (100) и (111).
Рост алмазного кристалла может быть охарактеризован параметром α = $\sqrt 3 $V100/V111, где V100 и V111 – скорости роста граней (100) и (111), соответственно. Влияние параметра α показано на рис. 5. Для α ≤ 1 идиоморфный кристалл растет в форме куба, в случае 1 < α < 3 – в форме кубооктаэдра, а при α ≥ 3 – в форме октаэдра. При изменении параметра α от 1 до 3 направление наиболее быстрого роста изменяется от 〈111〉 до 〈100〉, проходя через 〈110〉.
Рис. 5.
Идиоморфные кристаллические формы алмазного кристаллита, характерные для определенного значения параметра α. Стрелки указывают направление наиболее быстрого роста.
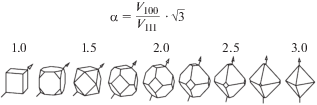
К факторам, определяющим значение отношения V100/V111, можно отнести два ключевых параметра: ростовую температуру и содержание углеводорода в газовой смеси. Данные зависимости, опубликованные в работе [72], наглядно видно на рис. 6. Как видно, наибольшей величины V100/V111 достигает при низких температурах синтеза (менее 700°С) и в этом диапазоне слабо зависит от процента метана. При этом в области высоких температур синтеза (а значит, и высоких скоростей роста алмаза) грань (100) растет тем быстрее, чем выше процент метана.
Рис. 6.
Влияние концентрации метана и температуры подложки на отношение скоростей роста граней алмаза V100/V111 (согласно [72]).
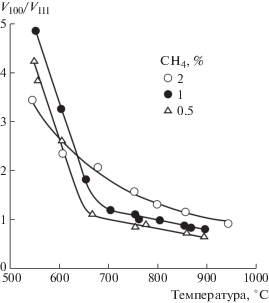
Следует отметить, что не стоит рассматривать температуру, процент метана и прочие параметры осаждения (например, расстояние подложки от активатора, давление в реакторе и др.), как совершенно независимые друг от друга. Каждый из них влияет на баланс атомарного водорода и углеводородных радикалов над подложкой, и изменение одного из параметров неизбежно влияет и на другие. Поэтому экспериментальная зависимость, отраженная на рис. 6, может весьма серьезно отличаться (и даже иметь совсем другой вид) при проведении аналогичных замеров скоростей роста в других условиях синтеза или на CVD-реакторов других типов. В связи с этим, во многих работах могут приводиться данные, не коррелирующие с приведенными, что отнюдь не указывает на их ошибочность.
4. ТЕКСТУРИРОВАНИЕ И ГЕТЕРОЭПИТАКСИАЛЬНЫЙ РОСТ
Открытие техник управления скоростью роста различных граней алмаза в поликристаллических пленках подтолкнуло исследователей использовать их для формирования текстурированных (т.е. состоящих из кристаллитов, которые ориентированы преимущественно в одном направлении) пленок. Текстурированная поликристаллическая пленка, выращенная на гетероподложке аналогичной ориентации, по сути и определению также является гетероэпитаксиальной, равно как и монокристаллическая. Более того, если плоскости текстуры и подложки точно совпадают, то поликристаллическая пленка по мере своего роста будет постепенно превращаться в монокристаллическую за счет снижения площади межзеренных границ.
Экспериментально было установлено, что при изменении ростовых условий действительно происходит изменение ориентации кристаллитов и формирование (или даже полная замена) текстуры в алмазных пленках [77–81]. Тем не менее, несмотря на создание условий преимущественного ориентированного роста алмазных кристаллитов, сформированные текстурированные пленки были далеки от гетероэпитаксии. Прежде всего, это обуславливалось существенными отклонениями ориентации текстуры синтезируемых пленок от плоскости подложки. В результате стало очевидно, что контроль ориентации кристаллитов нужно осуществлять еще на самых ранних стадиях роста пленки (нуклеация, формирование граней и сращивание в сплошную пленку), а также что для эффективного блокирования роста паразитных плоскостей плотность центров зародышеобразования должна быть как можно выше.
Кардинально изменить процесс синтеза текстурированных алмазных пленок помогла техника нуклеации зародышей посредством ионной бомбардировки (далее BEN-нуклеация). При помощи данной техники были получены высокотекстурированные алмазные пленки на самых разных монокристаллических гетероподложках [66, 72–74, 82–85].
Суть данной техники заключается в приложении к подложке прямо в процессе газофазного синтеза отрицательного электрического потенциала в диапазоне от одной до нескольких сотен вольт. При этом, из имеющейся либо создаваемой плазмы вытягиваются ионы, которые интенсивно бомбардируют поверхность подложки. Поскольку газовая среда в течение данного процесса насыщена углеродом, то происходит формирование тонкого слоя (в несколько нанометров) аморфной a-C:H углеродной пленки. Далее происходят параллельно идущие процессы вторичной нуклеации ростовых центров алмаза и их заращивание в слое аморфного углерода. Обычно эта стадия длится от десятков до сотен минут.
Далее отрицательный потенциал с подложки снимается и продолжается обычный процесс осаждения алмаза. Уже в первые секунды роста без воздействия бомбардирующих ионов весь аморфный углерод стравливается атомарным водородом, а оставшиеся ростовые центры алмаза начинают интенсивно расти, причем подавляющая часть растущих кристаллитов имеет латеральную ориентацию, совпадающую с ориентацией подложки. Дальнейший рост кристаллитов в свою очередь приводит к их сращиванию и образованию текстурированной сплошной пленки, степень текстурования которой возрастает по мере роста ее толщины.
Стоит отметить, что получаемый результат очень сильно зависит от вольт-амперных характеристик электрического смещения, длительности воздействия и непосредственно от техники осаждения алмаза. Упрощенно предполагаемая схема формирования высокоориентированных алмазных пленок с использованием BEN-нуклеации описана в работе [85] и представлена на рис. 7. Микрофотографии, отражающие процесс роста высокоориентированных пленок, представлены на рис. 8 (микрофотографии взяты из работы [72]).
Рис. 7.
Упрощенная схема, описывающая процесс формирования высокоориентированных алмазных пленок посредством BEN-нуклеации (согласно [85]).
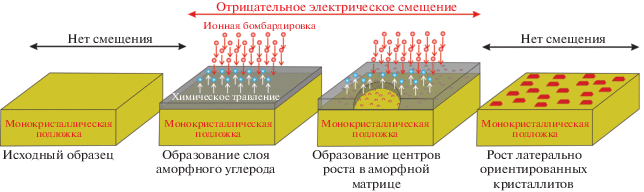
Рис. 8.
Микрофотографии высокоориентированных алмазных пленок на монокристаллическом кремнии после 2 ч (а) и 6 ч (б) осаждения (согласно [72]).

Как известно, гетероэпитаксиальный рост пленки на подложке возможен только при совпадении типов кристаллических решеток, а также при достаточно близком совпадении параметров решеток [86]. Известны случаи, когда гетероэпитаксия наблюдалась даже при несовпадении решеток в 20% (например, в случае осаждения пленки GaN на подложке GaAs (001) [87]) и даже в 30% (MnAs на подложке GaAs [88]). Тем не менее, столь высокие несоответствия решеток крайне затрудняют процесс синтеза, и экспериментально крайне затруднительно в таком случае осадить гетероэпитаксиальные пленки на сколько-нибудь существенной площади подложки. Наиболее приемлемым можно считать несовпадение решеток на уровне <20%. Классический пример это рост пленок GaN на сапфировых подложках при несовпадении решеток в 16% [89].
Одним из главных требований к подложке для гетероэпитаксии алмаза является ее кубическая гранецентрированная кристаллическая решетка. Параметр решетки при этом должен быть близок к 3.57 Å (алмаз). В табл. 2 приведен перечень материалов, являющихся наиболее подходящими кандидатами на роль гетероподложек для эпитаксии алмаза. Как видно, наиболее подходящими по параметру решетки материалами являются никель (3.524 Å) и медь (3.615 Å) (не считая c-BN), далее немного хуже подходят металлы платиновой группы: иридий (3.840 Å), палладий (3.890 Å) и платина (3.920 Å).
Таблица 2.
Материалы подложек с кубической гранецентрированной структурой решетки, наиболее подходящие для гетероэпитаксии алмаза
| Материал | Параметр решетки а, Å | Пространствен-ная группа | Несовпадение реше-тки с алмазом, % | Поверхностная энергия, Дж/м2 |
|---|---|---|---|---|
| Алмаз | 3.57 | Fd3m | 0 | 6.5 (110) [90] |
| Никель | 3.524 | Fm3m | –1.29 | 1.82 ± 0.2 [91]; 2.45 [92] |
| Медь | 3.615 | Fm3m | +1.26 | 1.72 ± 0.09 [91]; 1.83 [92] |
| Кубический нитрид бора | 3.615 | F43m | +1.26 | 5.20 (110) [90] |
| Иридий | 3.840 | Fm3m | +7.56 | 3.00 [92] |
| Палладий | 3.890 | Fm3m | +8.96 | 2.05 [92] |
| Платина | 3.920 | Fm3m | +9.8 | 2.34 ± 0.8 [91]; 2.48 [92] |
| Кремний | 5.4307 | Fd3m | +52.12 | 1.23 [93]; 1.24 [94] |
| Карбид кремния (β-SiC) | 4.3596 | F43m | +22.12 | 2.18 [95] |
При этом решетка кремния, как наиболее востребованного типа подложек для роста поликристаллического алмаза, имеет слишком большое несоответствие с алмазной (>52%). Доступность кремниевых подложек, близкий с алмазом коэффициент термического расширения, а также подобие кристаллических решеток кремния, карбида кремния (как неизбежно образуемого промежуточного слоя в результате насыщения кремния углеродом) и алмаза способствовали многочисленным [96, 97] попыткам гетероэпитаксии алмаза на данной подложке. При этом минимальный угол разориентации достигал 2.1° [96], но перехода от текстурированного поликристалла к монокристаллу, независимо от толщины пленки, не наблюдалось. По всей видимости, причиной является именно слишком большое различие в параметрах решеток.
Кроме того, контролируемость и повторяемость режимов проведения BEN-нуклеации на чистом или слабо легированном кремнии вызывает большие вопросы, так как технически крайне сложно обеспечить стабильный ионный ток на слабо проводящую подложку простой подачей отрицательного смещения на металлический подложкодержатель. Хорошо проводящий сильно легированный кремний, в свою очередь, также плохо подходит для гетеронуклеации из-за высокой доли легирующих примесей (B, As, P, Ga).
Никель, как уже писалось выше, по параметрам решетки наиболее близок к алмазу. Но первые же попытки [98] гетероэпитаксии на его монокристаллах алмазных пленок показали, что на его поверхности сначала образуется слой аморфного неалмазного углерода, нивелирующего все достоинства никеля. Впоследствии было установлено, что никель катализирует графитизацию алмаза [99] за счет обратной реакции разложения образуемого карбида никеля при температурах менее 700°С. Тем не менее, подбор оптимальных параметров нуклеации и роста позволил синтезировать высокоориентированные алмазные пленки достаточно высокого качества [100, 101].
Столь высокая зависимость от условий синтеза не позволяет с успехом использовать технику BEN-нуклеации на никеле (по крайней мере в ее современном виде). Получаемые на сегодняшний день образцы пленок на данном материале показывают скорее локальные области с гетероэпитаксиальным ростом, чем системную повторяемую от образца к образцу гетероэпитаксию. Очевидно, что более тонкий контроль стадии зародышеобразования и каталитической аморфизации на никеле может позволить сформировать гетероэпитаксиальные и даже монокристаллические алмазные пленки, но это, по-видимому, задача будущего.
Медь – второй по близости параметров решетки кандидат в качестве оптимальной подложки для роста гетероэпитаксиального алмаза. Теоретические расчеты показывают, что монокристалл меди с ориентациями (100) и (111) с точки зрения адгезионных и когезионных энергий, а также с его кристаллическим строением отлично подходит для наращивания эпитаксиальных слоев алмаза [102, 103]. Она не катализирует образование неалмазного углерода, как никель, и вообще химически с углеродом не взаимодействует. Множество работ указывают на успешный рост неориентированного алмаза на поликристаллической меди [104, 105].
Но попытки создания текстурированных алмазных пленок с плотностью ростовых центров, хотя бы отдаленно сопоставимой (не более 106 см–2 против 1011 см–2 на кремнии) с монокристаллическим кремнием, оказались безуспешными [106, 107]. Основной причиной предполагается отсутствие промежуточного карбидного слоя, что, впрочем, маловероятно, так как аналогичная невозможность образовывать карбиды совершенно не мешает гетероэпитаксии алмаза на иридии и платине. По мнению авторов данного обзора интерес к изучению гетероэпитаксии алмаза на меди и никеле вновь возникнет после более глубокого понимания процессов, происходящих при BEN-нуклеации и на границе раздела металл – углерод.
Платина в части химических свойств по отношению к углероду очень близка к меди. Она также инертна в плане карбидизации, но при этом имеет несовпадение решетки с алмазом на уровне +9.8%. Исследования особенностей BEN-нуклеации на платине показали, что максимально достижимая плотность зародышеобразования не превышает 108 см–2 [108], что также далеко от результатов, полученных на кремнии. При этом данные о синтезе высокоориентированных поликристаллических пленок алмаза на платине с помощью данной техники отсутствуют. Проведенные тем же коллективом эксперименты по росту алмаза на монокристалле платины (111) показали возможность получения высокоориентированных текстурированных пленок, но результаты рамановской спектроскопии указывают на существенную долю неалмазных углеродных примесей в них [109]. Высокая стоимость платиновых монокристаллов способствовала дальнейшему развитию работ в части перехода к тонким гетероэпитаксиальным слоям платины на сапфире [110]. В данной работе сообщается о синтезе (111) ориентированных алмазных пленок на платиновых слоях той же ориентации, причем по данным рентгеноструктурного анализа ширина алмазного пика на полувысоте составила около 1.1°.
Достаточно перспективным материалом для гетероэпитаксии алмаза можно назвать один из политипов карбида кремния – β(3С)-SiC, несовпадение решетки которого составляет около 22% по сравнению с алмазом. Данный материал известен как одна лучших подложек для роста поликристаллического алмаза, поскольку он обладает близким коэффициентом термического расширения и имеет аналогичную кристаллическую решетку. Применение BEN-нуклеации на данном материале позволяет достичь плотности зародышеобразования 1010 см–2 и ширины рентгеновской дифракционной линии алмаза на полувысоте менее чем 0.62° [111]. Гетероэпитаксия с подобной степенью структурного совершенства алмаза успешно проходила только на гранях (100), что скорее всего связано с достаточно высоким различием в параметрах кристаллических решеток.
5. ГЕТЕРОЭПИТАКСИЯ АЛМАЗА НА ИРИДИИ
Успешная гетероэпитаксия алмаза на иридии – один из феноменов современной науки. Несмотря на то, что данный чрезвычайно редкий и дорогой металл находится в ряду наиболее подходящих для гетероэпитаксии алмаза материалов, он по своим параметрам решетки более далек от алмаза, чем медь или никель (несовпадение решеток – 7.56%). Химически инертный, он никак не взаимодействует с углеродом, а коэффициент линейного термического расширения у него существенно выше, чем у алмаза (6.5 · 10–6 К–1 против 1 · 10–6 К–1 у алмаза при 20°С).
Единственно чем выделяется иридий – относительно высокой поверхностной энергией (3.00 Дж/м2 против 6.5 Дж/м2 у алмаза), но она лишь немного выше, чем у прочих близких к алмазу материалов. Тем не менее, среди подложек всех типов именно на иридии получены наиболее совершенные гетероэпитаксиальные пленки алмаза, которые многими исследователями признаются как монокристаллические.
Пионерские работы по гетероэпитаксии алмаза на тонких пленках иридия были проделаны японскими исследователями [112, 113]. Их результаты были столь многообещающими, что работы по данной тематике в кратчайшее время стали вестись по всему миру. В качестве первой подложки применялся монокристалл MgO в ориентации (100) с нанесенным эпитаксиальным слоем иридия толщиной около 500 нм. В данных работах также была апробирована BEN-нуклеация на данных подложках и была достигнута плотность зародышеобразования 108 см–2. В результате были получены гетероэпитаксиальные пленки алмаза толщиной до 1.5 мкм.
В более поздних работах MgO был заменен на SrTiO3 (100) для получения более совершенного по структуре 600-нм слоя иридия (угол разориентации <0.2°), напыляемого со скоростью 0.08 нм/с при температуре 950°С. Это позволило достичь плотности зародышеобразования 109 см–2 при BEN-нуклеации. Добавление в газовую смесь 30–50 ppm азота для преимущественного роста текстуры (100) позволило синтезировать гетероэпитаксиальные алмазные пленки толщиной до 8 мкм с азимутальным отклонением 0.65° [114]. Адгезия алмазных пленок в данном случае была низкой из-за существенного различия в коэффициентах термического расширения, в результате чего при охлаждении подложки после синтеза наблюдалось полное отделение пленки от подложки.
Ограничение в площади гетероэпитаксиального алмаза, обусловленное малыми размерами монокристаллических подложек MgO и SrTiO3, а также необходимость снижения внутренних напряжений, возникающих из-за различия в коэффициентах термического расширения, вынудили исследователей искать способ перехода на кремниевые подложки. Попытки непосредственного напыления иридия на монокристаллы кремния не увенчались успехом из-за их химического взаимодействия в условиях высокой температуры.
Для решения данной проблемы стали подбираться барьерные подслои, препятствующие диффузии иридия в кремний. Предпринимались попытки использования слоев CaF2 толщиной 200 нм [115], титаната стронция SrTiO3 толщиной 100 нм [116], а также диоксида циркония, стабилизированного иттрием (Yttria-stabilized zirconia – YSZ) [117]. В результате наибольшее распространение получили тонкие подслои на основе YSZ толщиной около 10–20 нм с последующим слоем иридия толщиной 100–150 нм. На рис. 9 показана микрофотография с атомарным разрешением, иллюстрирующая сечение такой системы c 20 нм иридия. Как видно, иридиевый слой растет эпитаксиально с самого начала его осаждения, а слой YSZ и сопутствующий слой SiO2 препятствуют диффузии иридия в кремний.
Рис. 9.
Микрофотография сечения системы Ir(20 нм)/YSZ/Si(001) [118], полученная на просвечивающем электронном микроскопе высокого разрешения.

Подобные результаты достигнуты для кремния с ориентациями как (100), так и (111). Слой YSZ выполняет также функцию постепенного выравнивания параметров решетки между кремнием и иридием, так как различие в параметрах решетки между ними составляет слишком большую для гетероэпитаксии величину – 29.3%. Слой YSZ снижает данное несовпадение на 5.3%. Несмотря на то, что YSZ при комнатной температуре является диэлектриком, в условиях роста алмаза (Tподложки > 700°С) он имеет достаточно высокую проводимость и не оказывает негативного влияния на процесс BEN-нуклеации.
Алмазные слои любой толщины, выращенные на системе Ir/YSZ/Si не отслаиваются с подложки при охлаждении, что позволяет использовать один и тот же образец в большом количестве последовательных циклов осаждения, наращивая таким образом (при необходимости) толщины более 1 мм. Несмотря на то, что начально превалирует островковый рост гетероэпитаксиальной пленки алмаза (см. рис. 10а), основная часть дефектов и межзеренных границ сосредоточена в прилегающем к подложке слое толщиной около 30 мкм, который впоследствии может быть удален механически. Остальная часть алмазного слоя практически не имеет дефектов и сопоставима с IIa-монокристаллом (плотность дислокаций около 107 см–2, при том, что IIa-монокристалл характеризуется их плотностью 108–109 см–2), имеющим примеси азота (так как обычно при наращивании толстых гетероэпитаксиальных пленок в газовую смесь добавляется несколько ppm азота).

Практика показывает, что ключом к получению эпитаксиальных алмазных слоев высокого качества является как можно более близкий к монокристаллическому слой иридия. Создание подобных слоев, в особенности на подложках большой площади, требует сверхвысоковакуумного напылительного оборудования (обычно это установки молекулярно-лучевой эпитаксии). Помимо этого, весь процесс синтеза (от очистки подложки до осаждения алмаза) должен проходить в помещении высокого класса чистоты для исключения пылезагрязнения подложек. Другим фактором является, безусловно, контроль параметров BEN-нуклеации, который также весьма затруднительно обеспечить на подложках площадью >50 см2. Типовая плотность тока на подложку составляет около 50 мА/см2 [85], что при напряжении смещения –300 В создает серьезные трудности с обеспечением равномерности температуры подложки и ионного тока на нее. Вкладываемая в подложку мощность при типовом токе составляет около 15 Вт/см2 (0.05 А, 300 В), что при площади подложки 50 см2 составляет уже 750 Вт, т.е. на стадии BEN-нуклеации подложка должна интенсивно охлаждаться.
6. ЗАКЛЮЧЕНИЕ
Массовое применение алмаза как конструкционного и функционального материала в современной технике станет возможным лишь в случае появления технологии его синтеза не в виде монокристаллов размером в несколько миллиметров, а в виде монокристаллических алмазных слоев и пластин площадью в десятки и сотни квадратных сантиметров. Единственной технологией, которая потенциально может позволить достичь этого, является метод осаждения из газовой фазы. Главным барьером является незавершенность и высокая трудоемкость технологии гетероэпитаксиального синтеза именно монокристаллического алмаза.
В настоящее время наилучшие успехи в этом направлении достигнуты с использованием многослойных подложек, основным функциональным элементом которых являются тонкие гетероэпитаксиальные слои иридия, одного из ближайших к алмазу материалов по структуре и параметрам кристаллической решетки. Несмотря на схожесть решетки иридия с алмазом, главные причины успехов синтеза алмаза на данном материале, равно как и неудач синтеза на более подходящих материалах (медь, никель), остаются, как минимум, не до конца понятными. Причины, по-видимому, кроются в сложности контроля процесса роста алмаза, недостаточной изученности процесса его нуклеации и начальной стадии роста, а также многочисленность факторов, кардинальным образом влияющих на процесс синтеза.
Очевидно, что более глубокое понимание процессов, происходящих при BEN-нуклеации, новые подходы к подготовке подложек, а также совершенствование техники осаждения гетероэпитаксиальных тонких пленок металлов могут позволить получить сопоставимые (а возможно, и превосходящие) с иридием результаты, но с использованием менее дорогих и экзотических материалов.
Список литературы
De Boer W., Bol J., Furgeri A., Müller S., Sander C., Berdermann E., Pomorski M., Huhtinen M. // Phys. Stat. Sol. (a). 2007. V. 204. № 9. P. 3004. https://doi.org/10.1002/pssa.200776327
Lagomarsino S., Bellini M., Corsi C., Cindro V., Kanxheri K., Morozzi A., Passeri D., Servoli L., Schmidt C.J., Sciortino S. // Appl. Phys. Lett. 2015. V. 106. P. 193509. https://doi.org/10.1063/1.4921116
Dore P., Nucara A., Cannavò D., de Marzi G., Calvani P., Marcelli A., Sussmann R.S., Whitehead A.J., Dodge C.N., Krehan A.J., Peters H.J. // Appl. Opt. 1998. V. 37. № 24. P. 5731. https://doi.org/10.1364/AO.37.005731
Thomas M.E., Tropf W.J. // Johns Hopkins APL Technical Digest. 1993. V. 14. № 1. P. 16.
Shvyd’ko Y., Blank V., Terentyev S. // MRS Bulletin. 2017. V. 42. P. 437. https://doi.org/10.1557/mrs.2017.119
Wort C.J.H., Balmer R.S. // Materialstoday. 2008. V. 11. № 1–2. P. 22. https://doi.org/10.1016/S1369-7021(07)70349-8
El-Hajj H., Denisenko A., Bergmaier A., Dollinger G., Kubovic M., Kohn E. // Diam. Relat. Mater. 2008. V. 17. № 4. P. 409. https://doi.org/10.1016/j.diamond.2007.12.030
Deneuville A. // Semiconductors and Semimetals. 2003. V. 76. P. 183. https://doi.org/10.1016/S0080-8784(03)80006-6
Braunstein G., Kalish R. // J. Appl. Phys. 1983. V. 54. № 4. P. 2106. https://doi.org/10.1063/1.332262
Pinault-Thaury M.-A., Temgoua S., Gillet R., Bensalah H., Stenger I., Jomard F., Issaoui R., Barjon J. // Appl. Phys. Lett. 2019. V. 114. P. 112106. https://doi.org/10.1063/1.5079924
Kato H., Makino T., Yamasaki S., Okushi H. // J. Phys. D: Appl. Phys. 2007. V. 40. P. 6189. https://doi.org/10.1088/0022-3727/40/20/S05
Koizumi S., Kamo M., Sato Y., Mita S., Sawabe A., Reznik A., Uzan-Saguy C., Kalish R. // Diam. Relat. Mater. 1998.V. 7. № 2–5. P. 540. https://doi.org/10.1016/S09259635(97)00250-1
Baranauskas V., Li B.B., Peterlevitz A., Tosin M.C., Durrant S.F. // J. Appl. Phys. 1999. V. 85. № 10. P. 7455. https://doi.org/10.1063/1.369378
Petherbridge J.R., May P.W., Fuge G.M., Robertson G.F., Rosser K.N., Ashfold M.N.R. // J. Appl. Phys. 2002. V. 91. № 6. P. 3605. https://doi.org/10.1063/1.1448679
Таблицы физических величин: Справочник / Под ред. акад. И.К. Кикоина. М.: Атомиздат, 1976.
Asmussen J., Reinhard D. Diamond films handbook. NY.: Marcel Dekker, 2002.
Liu H., Dandy D.S. Diamond chemical vapor Deposition. William Andrew, 1996. https://www.sciencedirect.com/book/9780815513803/diamond-chemical-vapor-deposition#book-description
Ivakin E.V., Sukhodolov A.V., Ralchenko V.G., Vlasov A.V., Khomich A.V. // Quantum electronics. 2002. V. 32. № 4. P. 367. https://doi.org/10.1070/QE2002v032n04ABEH002200
Klein C.A., Cardinale G.F. // Diam. Relat. Mater. 1993. V. 2. № 5–7. P. 918. https://doi.org/10.1016/0925-9635(93)90250-6
Novikov N.V., Dub S.N. // Diam. Relat. Mater. 1996. V. 5. № 9. P. 1026. https://doi.org/10.1016/0925-9635(95)00346-0
Лейпунский О.И. // Успехи химии. 1939. Т. 8. Вып. 10. С. 1519.
Варнин В.П. Вопросы истории естествознания и техники. 2015. Т. 36. Вып. 1. С. 124.
Deljanin B., Alessandri M., Peretti A., Åström M., Katrusha A. // Contributions to Gemology. 2015. № 15. P. 1.
Eaton-Magaña S., Shigley J.E., Breeding C.M. // Gems & Gemology. 2017. V. 53. № 3. P. 262. https://doi.org/10.5741/GEMS.53.3.262
Bray K., Kato H., Previdi R., Sandstrom R., Ganesan K., Oqura M., Makino T., Yamasaki S., Magyar P., Toth M., Aharonovich I. // Nanoscale. 2018. V. 10. P. 4028. https://doi.org/10.1039/C7NR09097H
Watanabe H., Nebel C.E., Shikata S. // Science. 2009. V. 324. № 5933. P. 1425. https://doi.org/10.1126/science.1172419
Berdermann E., Afanaciev K., Ciobanu M., Fischer M., Gsell S., Kiš M., Lagomarsino S., Lohmann W., Mayr M., Pomorski M., Rahman M.S., Schmidt C.J., Sciortino S., Schreck M., Stehl C., Träger M., The NoRHDia, CARAT, ADAMAS Collaborations // Diam. Relat. Mater. 2019. V. 97. P. 107420. https://doi.org/10.1016/j.diamond.2019.05.006
Bundy F.P. // J. Geophys. Res.: Solid Earth. 1980. V. 85. № B12. P. 6930. https://doi.org/10.1029/JB085iB12p06930
Bundy F.P., Bassett W.A., Weathers M.S., Hemley R.J., Mao H.U., Goncharov A.F. // Carbon. 1996. V. 34. № 2. P. 141. https://doi.org/10.1016/0008-6223(96)00170-4
Spitsyn B.V., Alexenko A.E. // Protection of Metals. 2007. V. 43. № 5. P. 415. https://doi.org/10.1134/S0033173207050025
Deryagin B.V., Fedoseev D.V. // Usp. Khim. 1970. V. 39. P. 1661. https://doi.org/10.1070/RC1970v039n09ABEH002022
Дерягин Б.В., Федосеев Д.В. М.: Наука, 1977. С. 116.
Schwander M., Partes K. // Diam. Relat. Mater. 2011. V. 20. № 9. P. 1287. https://doi.org/10.1016/j.diamond.2011.08.005
Gulbransen E.A., Andrew K.F., Brassart F.A. // J. Electrochem. Soc. 1965. V. 112. P. 49. https://doi.org/10.1149/1.2423463
Haubner R., Lux B. // Diam. Relat. Mater. 1993. V. 2. № 9. P. 1277. https://doi.org/10.1016/0925-9635(93)90008-P
Santos J.A., Neto V.F., Ruch D., Grácio J. // J. Nano Res. 2012. V. 18–19. P. 227. doi 10.4028/www.scientific.net/JNanoR.18-19.227
Liang Q., Chin C.Y., Lai J., Yan C.-S., Meng Y., Mao H.-K., Hemley R.J. // Appl. Phys. Lett. 2009. V. 94. P. 024103. https://doi.org/10.1063/1.3072352
Liu J., Hei L.F., Song J.H., Li C.M., Tang W.Z., Chen G.C., Liu F.X. // Diam. Relat. Mater. 2014. V. 46. P. 42. https://doi.org/10.1016/j.diamond.2014.04.008
Linnik S.A., Gaydaychuk A.V. // Diam. Relat. Mater. 2019.V. 94. P. 166. https://doi.org/10.1016/j.diamond.2019.03.011
Bolshakov A.P., Konov V.I., Prokhorov A.M., Uglov S.A., Dausinger F. // Diam. Relat. Mater. 2001. V. 10. № 9–10. P. 1559. https://doi.org/10.1016/S0925-9635(01)00408-3
Ashfold M.N.R., May P.W., Petherbridge J.R., Rosser K.N., Smith J.A., Mankelevich Y.A., Suetin N.V. // Phys. Chem. Chem. Phys. 2001. V. 3. P. 3471. https://doi.org/10.1039/B104265N
Prelas M.A., Popovici G., Bigelow L.K. Handbook of Industrial Diamonds and Diamond Films. N.Y.: Marcel Dekker, 1998. C. 1232.
Harris S.J. // Appl. Phys. Lett. 1990. V. 56. № 23. P. 2298. https://doi.org/10.1063/1.102946
Goodwin D.G. // Appl. Phys. Lett. 1991. V. 59. № 3. P. 277. https://doi.org/10.1063/1.105620
May P.W. // Phil., Trans., R., Soc., Lond. A. 2000. V. 358. P. 473. https://doi.org/10.1098/rsta.2000.0542
Skokov S., Weiner B., Frenklach M. // J. Phys. Chem. 1994. V. 98. P. 8. https://doi.org/10.1021/j100052a003
Pethrbridge J.R., May P.W., Pearce S.R., Rosser K.N., Ashfold M.N.R. // J. Appl. Phys. 2001. V. 89. № 2. P. 1484. https://doi.org/10.1063/1.1333031
Butler J.E., Mankelevich Y.A., Cheesman A., Ma J., Ashfold M.N.R. // J. Phys. Condens. Matter. 2009. V. 21. № 36. P. 364201. https://doi.org/10.1088/0953-8984/21/36/364201
Derjaguin B.V., Fedoseev D.V. // Scientific American. 1975. V. 233. № 5. P. 102.
Matsumoto S., Matsui Y. // J. Mater. Sci. 1983. V. 18. P. 1785. https://doi.org/10.1007/BF00542075
Mitura S. // J. Crys. Growth. 1987. V. 80. P. 417. https://doi.org/10.1016/0022-0248(87)90090-X
Buerki P.R., Leutwyler S. // J. Appl. Phys. 1991. V. 69. № 6. P. 3739. https://doi.org/10.1063/1.348468
Frenklach M., Kematick R., Huang D., Howard W., Spear K.E., Phelps A.W., Koba R. // J. Appl. Phys. 1989. V. 66. № 1. P. 395. https://doi.org/10.1063/1.343890
Frank F.C., Van der Merwe J.H. // Proc. Royal Soc. London Ser. A. 1949. V. 198. P. 205.
Volmer M., Weber A. // Z. Phys. Chem. 1926. V. 119. P. 277.
Baskaran A., Smereka P. // J. Appl. Phys. 2012. V. 111. P. 044321. https://doi.org/10.1063/1.3679068
Field J.E. The properties of diamond. London: Academic Press, 1979.
Barnes P.N., Wu R.L.C. // Appl. Phys. Lett. 1993. V. 62. № 1. P. 37. https://doi.org/10.1063/1.108811
Dubray J.J., Pantano C.G., Meloncelli M., Bertran E. // J. Vacuum Sci. & Technol. A. 1991. V. 9. № 6. P. 3012. https://doi.org/10.1116/1.577165
Belton D.N., Schmieg S.J. // Thin Solid Films. 1992. V. 212. № 1–2. P. 68. https://doi.org/10.1016/0040-6090(92)90502-3
Waite M.M., Shah S.I. // Appl. Phys. Lett. 1992. V. 60. № 19. P. 2344. https://doi.org/10.1063/1.107474
Kim D., Lee H., Lee J. // J. Mater. Sci. 1993. V. 28. № 24. P. 6704. https://doi.org/10.1007/BF00356418
Hirata G.A., Cota-Araiza L., Avalos-Borja M., Farfas M.H., Contreras O., Matsumoto Y. // J. Phys. Condens. Matter. 1993. V. 5. P. A305. https://doi.org/10.1088/0953-8984/5/33A/107
Arnault J.C., Demuynck L., Speisser C., Le Normand F. // Eur. Phys. J. B. 1999. V. 11. P. 327. https://doi.org/10.1007/s100510050943
Williams O.A., Douhéret O., Daenen M., Haenen K., Osawa E., Takahashi M. // Chem. Phys. Lett. 2007. V. 445. P. 255. https://doi.org/10.1016/j.cplett.2007.07.091
Wolter S.D., Glass J.T. // J. Appl. Phys. 1995. V. 77. № 10. P. 5119. https://doi.org/10.1063/1.359322
Yugo S., Kanai T., Kimura T., Muto T. // Appl. Phys. Lett. 1991. V. 58. P. 1036. https://doi.org/10.1063/1.104415
Koizumi S., Murakami T., Inuzuka T., Suzuki K. // Appl. Phys. Lett. 1990. V. 57. № 6. P. 563. https://doi.org/10.1063/1.103647
Inuzuka T., Koizumi S., Suzuki K. // Diam. Relat. Mater. 1992. V. 1. № 2–4. P. 175. https://doi.org/10.1016/0925-9635(92)90020-O
Argoitia A., Angus J.C., Ma J.S., Wang L., Pirouz P., Lambrecht W.R.L. // J. Mater. Res. 1994. V. 9. № 7. P. 1849. https://doi.org/10.1557/JMR.1994.1849
Koizumi S., Inuzuka T. // Jpn. J. Appl. Phys. 1993. V. 32. P. 3920. https://doi.org/10.1143/JJAP.32.3920
Maeda H., Ohtsubo K., Irie M., Ohya N., Kusakabe K., Morooka S. // J. Mater. Res. 1995. V. 10. № 12. P. 3115. https://doi.org/10.1557/JMR.1995.3115
Wild C., Kohl R., Herres N., Müller-Sebert W., Koidl P. // Diam. Relat. Mater. 1994. V. 3. P. 373. https://doi.org/10.1016/0925-9635(94)90188-0
Wild C., Koidl P., Müller-Sebert W., Walcher H., Kohl R., Herres N., Locher R. // Diam. Relat. Mater. 1993. V. 2. P. 158. https://doi.org/10.1016/0925-9635(93)90047-6
Smereka P., Li X., Russo G., Srolovitz D.J. // Acta Materialia. 2005. V. 53. P. 1191. https://doi.org/10.1016/j.actamat.2004.11.013
Delfaure C., Tranchant N., Mazellier J.-P., Ponard P., Saada S. // Diam. Relat. Mater. 2016. V. 69. P. 214. https://doi.org/10.1016/j.diamond.2016.08.013
Liu T., Raabe D. // Appl. Phys. Lett. 2009. V. 94. P. 021119. https://doi.org/10.1063/1.3072601
Cao G.Z., Schermer J.J., Van Enckevort W.J.P., Eist W.A.L.M., Gilling L.J. // J. Appl. Phys. 1996. V. 79. № 3. P. 1357. https://doi.org/10.1063/1.361033
Locher R., Wild C., Herres N., Behr D., Koidl P. // Appl. Phys. Lett. 1994. V. 65. № 1. P. 34. https://doi.org/10.1063/1.113064
Liu T., Raabe D., Mao W., Zaefferer S. // Adv. Funct. Mater. 2009. V. 19. P. 3880. https://doi.org/10.1002/adfm.200901231
Titus E., Mirsa D.S., Singh M.K., Tyagi P.K., Misra A., Le Normand F., Gracio J., Ali N. // J. Mater. Res. 2004. V. 19. № 11. P. 3206. https://doi.org/10.1557/JMR.2004.0433
Janischowsky K., Ebert W., Kohn E. // Diam. Relat. Mater. 2003. V. 12. P. 336. https://doi.org/10.1016/S0925-9635(02)00294-7
Yaita J., Suto T., Natal M.-R., Saddow S.E., Hatano M., Iwasaki T. // Diam. Relat. Mater. 2018. V. 88. P. 158. https://doi.org/10.1016/j.diamond.2018.07.011
Janischowsky K., Stammler M., Ley L. // Diam. Relat. Mater. 1999. V. 8 № 2–5. P. 179. https://doi.org/10.1016/S0925-9635(98)00259-3
Schreck M., Gsell S., Brescia R., Fischer M. // Scientific Reports. 2017. V. 7. P. 44462. https://doi.org/10.1038/srep44462
Rasic D., Narayan J. Epitaxial Growth of Thin Films // Crystal Growth (IntechOpen, 2019). https://doi.org/10.5772/intechopen.82745
Trampert A., Brandt O., Yang H., Ploog K.H. // Appl. Phys. Lett. 1997. V. 70. № 5. P. 583. https://doi.org/10.1063/1.118281
Trampert A., Ploog K.H. // Cryst. Res. Technol. 2000. V. 35. № 6–7. P. 793. https://doi.org/10.1002/1521-4079(200007)35:6/7<793::AID-CRAT793>3.0.CO;2-3
Kukushkin S.A., Osipov A.V., Bessolov V.N., Medvedev B.K., Nevolin V.K., Tcarik K.A. // Rev. Adv. Mater. Sci. 2008. V. 17. P. 1.
Prelas M.A., Gielisse P., Popovici G., Spitsyn B.V., Stacy T. Wide Band Gap Electronic Materials. Springer, 1994. P. 529. https://doi.org/10.1007/978-94-011-0173-8
Миссол В. Поверхностная энергия раздела фаз в металлах. М.: Металлургия, 1978. С. 176.
Skriver H.L., Rosengaard N.M. // Phys. Rev. B. 1992. V. 46. № 11. P. 7157. https://doi.org/10.1103/PhysRevB.46.7157
Jaccodine R.J. // J. Electrochem. Soc. 1963. V. 110. № 6. P. 524. https://doi.org/10.1149/1.2425806
Gilman J.J. // J. Appl. Phys. 1960. V. 31. № 12. P. 2208. https://doi.org/10.1063/1.1735524
Oshcherin B.N. // Phys. Status Solidi A. 1976. V. 34. P. K181. https://doi.org/10.1002/pssa.2210340266
Jiang X., Schiffmann K., Klages C.-P. // J. Appl. Phys. 1998. V. 83. № 5. P. 2511. https://doi.org/10.1063/1.367012
Trürer K.-H., Schreck M., Stritzker B. // Phys. Rev. B. 1998. V. 57. P. 15454. https://doi.org/10.1103/PhysRevB.57.15454
Belton D.N., Schmieg S.J. // J. Appl. Phys. 1989. V. 66. № 9. P. 4223. https://doi.org/10.1063/1.343962
Romanyuk O., Varga M., Tulic S., Izak T., Jiricek P., Kromka A., Shakalova V., Rezek B. // J. Phys. Chem. C. 2018. V. 122. P. 6629. https://doi.org/10.1021/acs.jpcc.7b12334
Yang P.C., Zhu W., Glass J.T. // J. Mater. Res. 1993. V. 8. № 8. P. 1773. https://doi.org/10.1557/JMR.1993.1773
Yang P.C., Zhu W., Glass J.T. // J. Mater. Res. 1994. V. 9. № 5. P. 1063. https://doi.org/10.1557/JMR.1994.1063
Zavodinsky V.G. // Comp. Mater. Sci. 2006. V. 36. P. 139. https://doi.org/10.1016/j.commatsci.2005.02.015
Zavodinsky V.G. // Diam. Relat. Mater. 2006. V. 15. № 9. P. 1201. https://doi.org/10.1016/j.diamond.2005.04.005
Pereira E., Fan Q.H. // J. Gracio, Mat. Res. Coc. Symp. Proc. 1997. V. 436. P. 323.
Hartsell M.L., Plano L.S. // J. Mater. Res. 1994. V. 9. № 4. P. 921. https://doi.org/10.1557/JMR.1994.0921
Wolter S.D., Stoner B.R., Glass J.T. // Diam. Relat. Mater. 1994. V. 3. P. 1188. https://doi.org/10.1016/0925-9635(94)90167-8
Chuang K.-L., Chang L., Lu C.-A. // Mater. Chem. Phys. 2001. V. 72. P. 176. https://doi.org/10.1016/S0254-0584(01)00431-X
Tachibana T., Yokota Y., Hayashi K., Miyata K., Kobashi K., Shintani Y. // Diam. Relat. Mater. 2000. V. 9. P. 251. https://doi.org/10.1016/S0925-9635(00)00207-7
Tachibana T., Yokota Y., Nishimura K., Miyata K., Kobashi K., Shintani Y. // Diam. Relat. Mater. 1996. V. 5. P. 197. https://doi.org/10.1016/0925-9635(95)00473-4
Tachibana T., Yokota Y., Hayashi K., Kobashi K. // Diam. Relat. Mater. 2001. V. 10. P. 1633. https://doi.org/10.1016/S0925-9635(01)00444-7
Kawarada H., Wild C., Herres N., Locher R., Koidl P. // J. Appl. Phys. 1997. V. 81. № 8. P. 3490. https://doi.org/10.1063/1.365047
Ohtsuka K., Suzuki K., Sawabe A., Inuzuka T. // Jpn. J. Appl. Phys. 1996. V. 35. № 8B. P. L1072. https://doi.org/10.1143/JJAP.35.L1072
Ohtsuka K., Fukuda H., Suzuki K., Sawabe A. // Jpn. J. Appl. Phys. 1997. V. 36. № 9A/B. P. L1214. https://doi.org/10.1143/JJAP.36.L1214
Schreck M., Roll H., Strizker B. // Appl. Phys. Lett. 1999. V. 74. № 5. P. 650. https://doi.org/10.1063/1.123029
Lee C.H., Qi J., Lee S.T., Hung L.S. // Diam. Relat. Mater. 2003. V. 12. P. 1335. https://doi.org/10.1016/S0925-9635(03)00083-9
Bauer T., Gsell S., Schreck M., Goldfuß J., Lettieri J., Schlom D.G., Stritzker B. // Diam. Relat. Mater. 2005. V. 14. P. 314. https://doi.org/10.1016/j.diamond.2004.10.028
Gsell S., Bauer T., Goldfuß J., Schreck M., Stritzker B. // Appl. Phys. Lett. 2004. V. 84. № 22. P. 4541. https://doi.org/10.1063/1.1758780
Gsell S., Fischer M., Brescia R., Schreck M., Huber P., Bayer F. // Appl. Phys. Lett. 2007. V. 91. P. 061501-1. https://doi.org/10.1063/1.2768003
Schreck M., Schury A., Hörmann F., Roll H., Stritzker B. // J. Appl. Phys. 2002. V. 91. № 2. P. 676. https://doi.org/10.1063/1.1424059
Дополнительные материалы отсутствуют.
Инструменты
Приборы и техника эксперимента