

Прикладная биохимия и микробиология, 2019, T. 55, № 4, стр. 315-337
Биотехнологические аспекты ферментативного получения биоактивных хитоолигосахаридов (обзор)
Г. Э. Актуганов 1, *, А. И. Мелентьев 1, В. П. Варламов 2
1 Уфимский Институт биологии Уфимского федерального исследовательского центра РАН
450054 Уфа, Россия
2 Институт биоинженерии, Федеральный исследовательский центр
“Фундаментальные основы биотехнологии” Российской академии наук
119071 Москва, Россия
* E-mail: gleakt@anrb.ru
Поступила в редакцию 19.09.2018
После доработки 14.11.2018
Принята к публикации 20.02.2019
Аннотация
В обзоре рассмотрены современные технологии ферментативного получения хитоолигосахаридов с помощью хитозаназ. Освещены также особенности микробного синтеза хитозаназ и подходы к реализации их биотехнологического производства. Отмечены тенденции в развитии методов и технологий направленного получения олигомеров хитозана с использованием биореакторов и иммобилизованных ферментов.
Водорастворимый олигомерный хитозан и хитоолигосахариды (ХОС) со степенью полимеризации n ≥ 5–6, образующиеся при частичной деструкции высокомолекулярного аналога, рассматриваются в силу их разнообразной биологической активности как потенциальные продукты с высокой добавленной стоимостью, перспективные для использования в области медицины, фармацевтики и сельского хозяйства [1–6]. Целевые характеристики хитоолигосахаридов и олигомерного хитозана во многом определяются методами их получения. Одним из наиболее перспективных способов получения этих соединений является ферментативная деполимеризация хитозана, которая может быть осуществлена с помощью различных групп гидролаз, в том числе коммерческих препаратов неспецифических ферментов – липаз, папаина, пепсина, целлюлаз (Целловиридин Г20х), лизоцима и др. [7–16]. Высокоспецифичные хитозаназы (КФ 3.2.1.132) способны осуществлять глубокую деструкцию исходного полимера при удельной активности, превышающей активность неспецифических ферментов в десятки и сотни раз [17].
Особенности деполимеризации хитозана различными группами ферментов для получения ХОС подробно были рассмотрены в предыдущем обзоре [18]. Настоящая работа посвящена изучению проблем применения микробных хитозаназ для получения биоактивных олигомеров хитозана.
Реализация технологии ферментативной деполимеризации хитозана и стоимость процесса неразрывно связана с этапами биотехнологического получения ферментного препарата, предварительной подготовки субстрата, проведения процесса деполимеризации, выделения смеси ХОС и, при необходимости, очистки и высушивания целевого олигомерного продукта (рис. 1). Серьезным ограничением для широкого применения специфических ферментов является их стоимость. Хитозаназы до настоящего времени остаются дорогостоящими, так как еще не созданы крупнотоннажные производства этих ферментов.
Рис. 1.
Общая технологическая схема ферментативного получения биологически активных ХОС с использованием микробных хитозаназ.
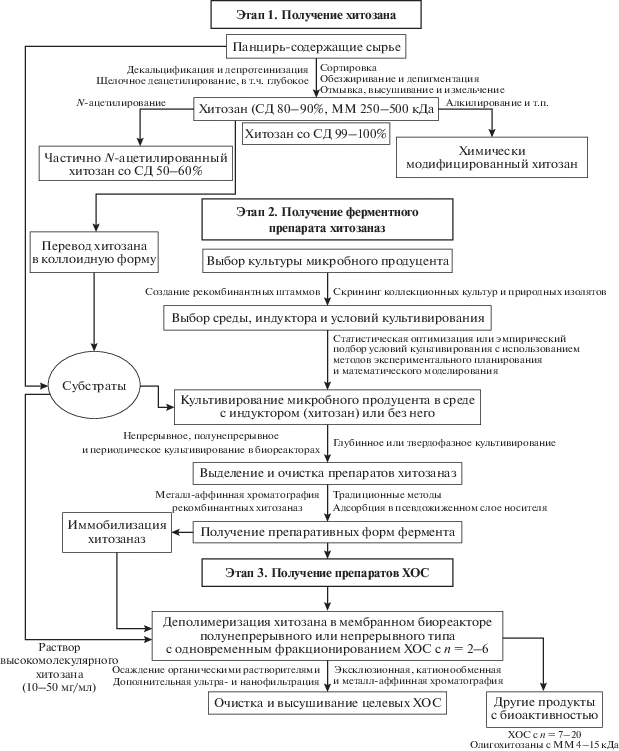
Хитозан, являющийся исходным сырьем для получения хитозаназ и ХОС, представляет собой относительно дорогостоящий субстрат, вносящий существенный вклад в издержки в процесс производства. Получение хитозана представляет собой многостадийный процесс, включающий этапы декальцификации, депротеинизации и обесцвечивания панцирь-содержащего сырья с последующим щелочным деацетилированием и др. [19]. Значительный вклад в стоимость процесса вносит предварительная подготовка хитозана, которая состоит из перевода его в растворимую или коллоидную форму, или применение специально подготовленного хитозана с нехарактерной степенью деацетилирования, например, 40 или 99%. Другая проблема связана с низкой стабильностью многих хитозаназ при использовании их в свободном состоянии при длительных периодических процессах гидролиза хитозана. Эффективность конверсии хитозана может быть увеличена за счет применения высококонцентрированных растворов субстрата, при этом поддержание его вязкости на функциональном уровне обеспечивается значительным повышением температуры процесса, что в свою очередь требует повышения термостабильности применяемых хитозаназ [20, 21]. При использовании свободной хитозаназы продукты гидролиза, как правило, загрязнены белками ферментного препарата и сопутствующими микробными метаболитами, поэтому для таких областей, как медицина и фармацевтика, требуется глубокая очистка целевых ХОС. В настоящее время разработаны высокоэффективные методы очистки и разделения ХОС, различающихся по степени полимеризации, количеству ацетильных групп и порядку их распределения. В основном, эти методы включают ультрафильтрацию и эксклюзионную, ионообменную и металл-хелатную аффинную хроматографию [2, 22]. Часто для получения гомогенных фракций ХОС необходимо комбинированное использование различных подходов. Современные носители для препаративной эксклюзионной хроматографии позволяют разделять смеси олигомеров со степенью полимеризации до 40 [2]. Это также может сильно влиять на повышение стоимости конечного продукта. Несмотря на удачные подходы, в целом получение фракций чистых ХОС остается кропотливой и дорогостоящей процедурой.
Необходимо отметить несколько подходов в реализации биотехнологического получения и применения хитозаназ, сложившихся к настоящему времени. Они включают: (1) повышение продуктивности природных изолятов бактерий и грибов и создание рекомбинантных штаммов – сверхпродуцентов хитозаназ, (2) поиск естественных штаммов и создание генноинженерных культур, способных к конститутивному синтезу хитозаназ в отсутствие хитозана, (3) поиск и молекулярное конструирование термостабильных хитозаназ, (4) совершенствование технологий периодического и непрерывно-проточного гидролиза хитозана ферментами для увеличения выхода целевых продуктов и повышения рентабельности процесса деполимеризации, (5) совершенствование методов и средств иммобилизации хитозаназ и (6) разработку новых подходов и методов выделения, очистки и анализа индивидуальных фракций ХОС.
БИОТЕХНОЛОГИЧЕСКОЕ ПОЛУЧЕНИЕ МИКРОБНЫХ ХИТОЗАНАЗ ДЛЯ ФЕРМЕНТАТИВНОЙ КОНВЕРСИИ ХИТОЗАНА
Особенности продукции хитозаназ микроорганизмами при периодическом культивировании. Микробные хитозаназы представляют собой, как правило, внеклеточные белки, выделяемые из культуральной среды продуцента без необходимости предварительного разрушения его клеток. Хитозаназы, в основном, являются индуцибельными ферментами. В качестве индуктора синтеза хитозаназ часто используется коллоидный хитозан, влияние которого на экспрессию генов хитозаназы хорошо изучено [23]. Примерами детально изученных ферментов, выделенных из супернатанта культуральной жидкости (КЖ) природных штаммов и требующих добавления хитозана в питательную среду, являются хитозаназы GH46 Bacillus circulans MH-K1 и GH8 Paenibacillus fukuinensis D2 [24, 25]. Микробные хитозаназы могут индуцироваться также в присутствии таких субстратов, полученных из отходов переработки морепродуктов, как хитин, измельченные до порошкообразного состояния панцири и головы креветок и гладиус кальмара [26, 27]. Синтез хитозаназ в присутствии этих субстратов был отмечен у штаммов бактерий Acinetobacter calcoaceticus TKU024, B. cereus TKU022, Pseudomonas sp. TKU017, Serratia sp. TKU020, Streptomyces roseolus DH и др. [22]. Рассматривались возможность и перспективы микробной биоконверсии вышеупомянутых хитин-содержащих отходов для получения биоактивных соединений и ферментов, в том числе, хитозаназ [28].
Продукция хитозаназы штаммами Paenibacillus ehimensis IB-739 (неопубликованные данные) и Penicillium sp. IB-37-2 наблюдалась в присутствии, как хитозана, так и коллоидного хитина из панциря краба и очищенного хитина из подмора пчел, при этом в последних случаях уровень синтеза фермента был выше [29]. У продуцента целлюлаз Trichoderma reesei продукция экзо-хитозаназы отмечалась только в присутствии аминосахаров, входящих в состав хитозана [30]. У гриба Penicillium sp. IB-37-2 в присутствии D-глюкозамина и N-ацетил-D-глюкозамина также наблюдался синтез экзо-хитозаназы, достигающий 20–25 и 10% соответственно от уровня активности, регистрируемого в среде с хитозаном [29]. В некоторых случаях природные штаммы продуцируют хитозаназы в отсутствие хитозана. Так, штамм Bacillus sp. MET1299, выделенный на среде LB с хитозаном, обнаруживал способность к конститутивной продукции хитозаназы семейства гликозил-гидролаз GH8 (молекулярная масса, ММ, – 52 кДа) [31]. Штамм Bacillus sp. KCTC 0377BP способен продуцировать хитозаназу семейства GH8 (ММ 45 кДа) при замещении хитозана растворимым крахмалом (0.5%) в качестве источника углерода, при этом продуктивность штамма возрастала с 1.2 до 4 ед/мл [32]. Известен мутантный штамм B. subtilis IMR-NK1, показывающий высокую хитозаназную активность при твердофазном культивировании без хитозана в присутствии пшеничных отрубей [33]. Можно предположить, что многие штаммы вида Bacillus thuringiensis, являющиеся патогенами насекомых, способны к конститутивной продукции хитозаназ [34].
В работе [23] рассмотрены трудности, связанные с необходимостью включения хитозана в состав среды культивирования. С одной стороны, хитозан является ценным источником углерода и азота, а с другой – он способен подавлять рост культуры вследствие своей антимикробной активности. Результатом этих двух противоречивых эффектов может быть продукция хитозаназы рекомбинантными штаммами по определенной, воспроизводимой схеме. В начале ферментации, когда высокомолекулярный хитозан присутствует в среде в основном в виде суспензии (коллоидный хитозан), его ингибирующий эффект практически не проявляется, поэтому наблюдается быстрый рост бактериальной биомассы, сопровождаемый постоянным повышением концентрации общего белка и хитозаназной активности. Накопление внеклеточной хитозаназы в среде сопровождается ферментативным гидролизом хитозана и образованием водорастворимых олигомеров, активно взаимодействующих с поверхностью бактериальных клеток, что приводит к увеличению их ингибирующего действия. Это, в свою очередь, индуцирует реакцию стресса у бактериальных клеток, повышая продукцию протеаз, приводя иногда к лизису клеток. На поздней стадии ферментации уровень хитозаназы резко снижается, поскольку фермент разрушается протеазами. Завершение цикла культивирования на этом этапе будет приводить к низкому выходу хитозаназы, загрязненной сложной смесью белков, образующихся при лизисе бактериальных клеток. В то же время, по мнению авторов работы [35], возможность экспрессии хитозаназы природными штаммами бактерий или микромицетов придает им большую устойчивость к антимикробной активности хитозана, поскольку внеклеточная хитозаназа может деградировать хитозан до небольших малоактивных фрагментов, не оказывающих существенного влияния на развитие микроорганизмов. Этот взгляд был подкреплен данными предшествующего изучения антигрибной и антибактериальной активности хитозана. Так, ранее было показано, что мономер и димер хитозана не ингибируют рост двух штаммов гриба Fusarium solani, в то время как антигрибное действие начинает обнаруживаться только у тримера и далее резко возрастает у гекса- и гептамера, а также более крупных олигомеров [36]. Отмечено существенное увеличение антигрибной активности олигомерного хитозана против различных видов фитопатогенных и плесневых микромицетов при увеличении его ММ с 1 до 10 кДа [1]. При действии хитозана на Escherichia coli было установлено, что минимальная ингибирующая концентрация олигомерных фракций полимера со средней ММ в пределах 9.3–10.5 кДа составляла 40 мкг/мл, в то время как олигохитозан с ММ 2.2–4.1 кДа не оказывал ингибирующего действия даже при 5000 мкг/мл [37]. Изучение природного штамма Streptomyces lividans TK24, являющегося продуцентом внеклеточной хитозаназы, показало его большую устойчивость по сравнению с мутантным штаммом этого вида, несущим делецию в области гена хитозаназы csnA [38]. Для рекомбинантных штаммов E. coli, экспрессирующих хитозаназу, не секретируемую в культуральную среду, а накапливаемую в периплазматическом пространстве, тоже была отмечена повышенная резистентность к хитозану [39]. Таким образом, использование хитозана в качестве субстрата-индуктора при культивировании микробных продуцентов хитозаназ требует сбалансированного выбора между концентрациями полимера, способствующими максимальному уровню синтеза фермента и вызывающими заметное подавление роста продуцента.
Получение препаратов хитозаназы с помощью рекомбинантных культур микроорганизмов. Использование рекомбинантных штаммов, помимо повышения продукции хитозаназы, дает преимущество, позволяя отказаться от применения хитозана в качестве специфического индуктора генной экспрессии и заменить его более дешевыми субстратами. Примеры экспрессии генов некоторых рекомбинантных хитозаназ бактериального и грибного происхождения представлены в табл. 1.
Таблица 1.
Примеры рекомбинантной экспрессии генов некоторых бактериальных и грибных хитозаназ.
| Хитозаназа, семейство/ продуцент |
Реципиен-тный штамм | Вектор | Используемые праймеры (последовательность 5' → 3') |
Выделение и очистка фермента | Ссылка |
|---|---|---|---|---|---|
| GH8/B. thuringiensis (9 штаммов) | E. coli JM109
или E. coli Top10 |
pACE | BTC-F, GGAATTCCATAT-GAATGGAAAAAGAAAT; BTC-R, AACTGCAGTTAATTATCGTATCCTTCAT |
− | [34] |
| GH8/B. subtilis | E. coli BL21 | pGEM-T Easy (клонирование) | P1, GCTGCTGCAAAGGAAATG AAA; P2, TTAATTATCGTATCCTTCATAAATT |
Разрушение клеток ультразвуком, металл-аффинная хроматография (Ni2+ колонка) | [40] |
| pET-(экспрессия) | P3,AAAAAAACATATGGCTGCTGCAAAGGAAATGAAA; P4, GGCCGCGAATTCACTAGTG |
||||
| GH46/B. subtilis 168 | E. coli Top10 | pMY202 | B.subCsnOmpAHindIIIFw,
CTGTGCAAGCTTCGGCGGGACTGAAT AAAGATCAAAAGC; B.subCsnBamHIRv, GCACAGGGATCCTTTGATTACAAAATTACCGTAC TCGTTTGAAC |
Очистка рекомбинантного белка из лизата клеток E. coli металл-аффинной хроматографией на Ni2+-NTA-агарозе | [41] |
| GH46/S. lividans TK244; S. lividans ΔcsnR |
E. coli DH5α™ | pCSN106-2 (клонирование) pHM8aBΔM, pFDES (интегративные векторы) |
FwcsnN106, CCGGAGACCCGCATGCCCCGGAC RvcsnN106, CGGTGCGCCAAGCTTGCGTTCGG Для клонирования области промотора хитозаназы: FwPr-WT, GTCTGCGCGGATCCTGACGGCCC RvPr-WT, GTCCGGGGCATGCGGGTCTCCGG |
− | [43] |
| B. subtilis V26 | E. coli Top10 | pBad | Cho5,
AAAGGGGCTAGCATGAAAATCAGTATGCAAAAAGC; Cho6,GTTTGAATTCCTCACTGCTTATTTGATT |
Очистка рекомбинантного белка из КЖ последовательной эксклюзионной ВЭЖХ на колонках Aquagel 40 и Bio-Sil SEC125 | [44] |
| GH75/Penicillium sp. D-1 | E. coli BL21 (DE3) | pET28a | csn–F, CGCCATATGTACGATATCCCTGACAAC csn–r, ATTCTCGAGTTACAGAGACGCAACAAG |
Очистка из КЖ металл-аффинной хроматографией на колонке Ni2+-NTA | [49] |
| GH75/A. fumigatus Y2K | P. pastoris GS115 | pHBM905A | p1, GTCATACAACCTACCCAACAATTTGA; p24, GGCCATTAAGCCTTCAAACCAGCAACCAACTTGTCA CCAATAGACTTTATAGAATC |
Выделение рекомбинантного белка фракционированием при 30-80% сульфата аммония | [50] |
| GH75/A. fumigatus Y2K | E. coli Rosetta blue (DE3) | pHBM-csn
(сконструирован на основе pET-26b (+)) |
CSN-BamH-b1,
CGCGGATCCGTACAACCTACCCAACAATTTGAAGCA CSN-Sal I I-b2, ACGCGTCGACTTAAGCCTTCAAACCAGCAACCAA CSN-BamH-n1, CGCGGATCCGCACCACCACCACACCAC CSN-Sal-n2, ACGCGTCGACTTAAGCCTTCAAACCAGCAACAA CSN-BamH-c1, CGCGGATCCGTACAACCTACCCAACAATTTGAAGCA CSN-Sal-c2, ACGCGTCGACAGCCTTCAAAACCAGCAACCAA |
– | [51] |
| Целлобиогидролаза-хитозаназа (GH7) T. reesei QM9414 | A. oryzae niaD300 | pNEN142 pUC119 (экспрессия) |
Trcbh1-f, TTGGTACCATGTATCGGAAGTTGGCCGTCA Trcbh1-r, TTTCTAGATTACAGGCACTGAGAGTAGTAA |
Осаждение при 80% насыщении сульфатом аммония из КЖ, ДЭАЭ-сефароза FF, ультрафильтрация | [124] |
Так, гетерологичная экспрессия гена, кодирующего хитозаназу семейства GH8 из Bacillus sp., была реализована в E. coli под контролем промотора бактериофага Т7 [40]. В этой системе был достигнут высокий выход хитозаназы, составляющий 500 мг на 1 л среды LB при удельной активности 270–290 ед./мг (около 140 ед./мл). Согласно приведенным данным 1.0 г рекомбинантной хитозаназы, полученной таким образом, способен конвертировать до 100 кг хитозана в функциональные ХОС с n = 2–6 [40].
При клонировании в E.coli гена хитозаназы csn семейства GH46 штамма Bacillus subtilis 168 был достигнут высокий уровень экспрессии фермента (900 ед./мг) с одновременным повышением его рН- и термостабильности [41]. Неочищенный препарат этой хитозаназы гидролизовал коммерческий хитозан с быстрым накоплением ХОС (n ≥ 2–6).
Слабым местом технологии использования рекомбинантных хитозаназ для получения ХОС на промышленном уровне является их довольно высокая стоимость, обусловленная необходимостью внесения антибиотика в культуральную среду для поддержания плазмидных векторов в клетках, разрушения клеток ультразвуком и включения изопропил-β-тиогалактозида (ИПТГ) в среду для индукции транскрипции клонированного гена [23]. К необходимому и дорогостоящему этапу в этом случае относится также очистка фермента с помощью металл-ион-аффинной хроматографии.
В качестве альтернативных продуцентов для получения рекомбинантной хитозаназы рассматривались штаммы Brevibacillus brevis и S. lividans TK24. Первый штамм был использован для экспрессии дикого типа и мутантных форм хитозаназы GH46 из B. circulans MH-K1, который в отличие от E. coli секретировал фермент в культуральную среду [42]. Для гетерологичного штамма S. lividans TK24, несущего гены хитозаназ csn семейства GH46 известных продуцентов рода Kitasatospora N106 и N174 в плазмиде pFD666, являющейся челночным вектором между E. coli и S. lividans, была установлена возможность синтеза хитозаназы в отсутствие субстрата-индуктора [43]. Обнаружено, что транскрипция гетерологичных генов этой хитозаназы, наряду с эндогенными генами S. lividans TK24, регулируется особым репрессором CsnR, блокирующим синтез фермента вследствие связывания с последовательностью оператора данных генов при отсутствии хитозана в среде. Введение генов в клетки S. lividans TK24, из генома которых был удален CsnR, позволяло культуре стабильно продуцировать хитозаназу в отсутствие хитозана, как на богатых средах неопределенного состава, так и на минимальных, точно регламентированных средах [43]. Отмечено, что подобный эффект следует ожидать при синтезе актинобактериальных хитозаназ, относящихся к семействам GH5 и GH75, гены которых обладают оператором CsnR-типа.
Высокий уровень экспрессии и секреции (6.2 г/л) в E. coli был достигнут для гена термостабильной и антигрибной хитозаназы CSNV26 из B. subtilis [44].
Были сконструированы две плазмиды на основе гена хитозаназы Paenibacillus sp. IK-5 и экспрессированы в B. brevis с использованием вектора pNY301. В результате удалось добиться 180-кратного (с 0.27 до 48.3 ед./мл, или 5.9 г/л) повышения продукции внеклеточной хитозаназы у одного из рекомбинантных штаммов [45].
В работе [46] сообщалось о 500-кратном увеличении синтеза хитозаназы штамма Janthinobacterium sp. 4239 при гетерологичной экспрессии ее гена в E. coli DH 10B.
Помимо бациллярных и актинобактериальных хитозаназ, разработаны системы экспрессии ферментов некоторыми штаммами псевдомонад и грибов [22, 47–49]. О конструировании гибридных систем на основе грибных хитозаназ имеется существенно меньше данных в связи со сложностью регуляции экспрессии генов у эукариот.
В работе [26] сообщалось об экспрессии хитозаназы из штамма F. solani 0114 в промышленном штамме Saccharomyces cerevisiae для крупномасштабного производства данного фермента. Другим примером удачного конструирования подобных систем является синтез рекомбинантной термостабильной эндохитозаназы штаммом дрожжей Pichia pastoris GS115, несущим соответствующий ген из Aspergillus fumigatus [50].
Известны также примеры сверхэкспрессии генов грибных хитозаназ в клетках E. coli. Так, удалось осуществить экспрессию в E. coli термостабильной хитозаназы GH75 из A. fumigatus (MМ 25 кДа), эффективно гидролизующей хитозан с высокой степенью деацетилирования до тримера, тетрамера и пентамера [51, 52]. В отличие от упомянутых выше примеров в этом случае была достигнута секреция фермента из клеток E. coli в культуральную среду за счет использования в составе экспрессионного вектора pET-26b последовательности N-концевого сигнального пептида PelB, обеспечивающего эффективную секрецию хитозаназы из клеток. Для облегчения очистки целевого белка была внесена гистидиновая метка в состав N- или C-концевой последовательности фермента. Было отмечено, что рекомбинантный штамм E. coli, несущий ген хитозаназы pET26b-csn без гистидиновой вставки, при индукции 0.5 мМ ИПТГ демонстрировал максимальный уровень секреции фермента (2890 ед./мл), в то время как штамм pET26b-csn-N с N-концевой гистидиновой меткой обнаруживал существенно меньшую секреторную активность (1700 ед./мл). Штамм E. coli pET26b-csn-С с C-концевой гистидиновой меткой оказался неактивным, поскольку секретировал хитозаназу в составе нерастворимых телец включения. Были дополнительно оптимизированы параметры продукции рекомбинантной хитозаназы A. fumigatus в колбах при индукции фермента лактозой (1%) в среде TB, дополненной глицином (1%), при 25°С, что позволило добиться уровня продукции 6015 ед./мл. При культивировании (48 ч) в 5 л ферментере с периодическим добавлением оптимальных количеств лактозы и глицина выход хитозаназы достигал 14 000 ед./мл (табл. 2).
Таблица 2.
Продукция хитозаназ некоторыми природными и рекомбинантными штаммами микроорганизмов
| Фермент/oрганизм-источник/штамм-продуцент | Уровень максимальной продукции | Биосинтез/ индукция | Способ получения | Удельная продукция, ед./мл/сут | Ссылка |
|---|---|---|---|---|---|
| Экзо-хитозаназа A. fumigatus | 6 ед./мл | 1% хитозана или 1% D-глюкозамина | Глубинное культивирование (от 3 сут до 7 сут), 180 об./мин | 2.0 | [22] |
| Экзо-хитозаназа Penicillium sp. IB-37-2 | 3.5 ед./мл | 1.5–2% коллоидный крабовый хитин | Глубинное культивирование
в колбах Эрленмейера (6 сут, 220 об./мин) |
0.58 | [29] |
| Хитозаназа Bacillus sp. KCTC 0377BP | 100 ед./мл | Растворимый крахмал – 2%, полипептон – 2.5%, силиконовое масло – 0.005% | Глубинное культивирование
в 500-л ферментере (100 об/мин, аэрации 1 об./об./мин |
− | [32] |
| Рекомбинантная хитозаназа B. subtilis, экспрессируемая в E. coli BL21 | 140 ед./мл (500 мг/л) |
Среда LB с ампициллином (100 мг/мл) и изопропил-β-D-тиогалактозидом (ИПТГ, 1 мМ) | Глубинное культивирование в колбах (2 этапа, всего 25 ч, 250–150 об./мин), 5 л (по 0.5 л в колбе) | ~134 | [40] |
| Рекомбинантная хитозаназа B. subtilis V26, экспрессируемая в E. coli (изоформы 27 и 31 кДа) | (6.2 г/л) | Индуктор – арабиноза (125 мкг/мл)в среде LB с ампициллином (60 мкг/мл) | Глубинное культивирование в колбах Эрленмейера (50 мл, 200 об./мин) |
− | [44] |
| Рекомбинантная хитозаназа A. fumigatus, экспрессируемая в P. pastoris | 25 000 ед./мл | Индуктор – метанол | Глубинное культивирование в 14 л ферментере (5 сут) | 3600 | [50] |
| Рекомбинантная хитозаназа A. fumigatus, экспрессируемая в E. coli | 6015 ед./мл | 1% лактозы и 1% глицина В среде TB |
Глубинное культивирование в колбах | − | [51] |
| 14 000 ед./мл | Глубинное культивирование в 5 л ферментере (2 сут) | 7000 | |||
| Конститутивная хитозаназа Bacillus sp. RKY3 | 63.53 ед/мл | ~3% мальтозы и 1.5% говяжьего экстракта | Глубинное культивирование в колбах Эрленмейера (50 ч, 200 об./мин) | ~30.5 | [53] |
| Хитозаназа Microbacterium sp. OU01 | 118 ед/мл | 1% коллоидный хитозан | Глубинное культивирование в колбах (96 ч, 150 об./мин) | 29.5 | [54] |
| Хитозаназа T. koningii | 4.84 ед/г сухого субстрата | Среда с 60% пшеничных отрубей и 30% хитозана | Твердофазное стационарное культивирование (48 ч) | 0.54 (или 2.42 ед/г/день) | [55] |
| Иммобилизованные на пенополиуретане и альгинате кальция клетки Gongronella sp. JG | 242.9 ед./мл | 0.1% глюкозы и 0.1% глутаминовой кислоты | Глубинное культивирование в колбах по 100 мл (84 ч, 150 об./мин) | 69.4 | [98] |
| Рекомбинантная хитозаназа B. subtilis, экспрессируемая в L. plantarum с использованием векторов pSIP | 79 ед./мл | MRS среда с 2% глюкозы, индукция пептидным феромоном IP-673 (12.5 нг/мл) | Глубинное культивирование в биореакторе (3 л, 20 ч), 100 об./мин | 94.8 | [100] |
| Хитозаназа P. ehimensis NRRL B-23118 | 0.50 ед./мл | Индуктор – растворимый хитозан, 0.2% | Глубинное культивирование (36 ч, 120 об./мин) | ~0.32–0.33 | [125] |
| Хитозаназа S. zaomyceticus | 22 ед./мл | 2% хитозана или 2% глюкозы | Глубинное культивирование в биореакторе (максимум 72–84 ч) |
6.3–7.3 | [126] |
| Хитозаназа C. globosum KM651986 | 1758.75 ед./г сухого субстрата | 20% пшеничных отрубей и 1.5% хитозана | Твердофазное культивирование (9 сут) |
195.4 | [127] |
| Хитозаназа B. cereus TKU034 | ~0.70 ед./мл | Минеральная среда с 1% измельченного порошка гладиуса кальмара | Глубинное культивирование в колбах Эрленмейера (4–5 сут, 150 об./мин) | 0.175 | [128] |
Применение подходов статистической и эмпирической оптимизации для повышения продукции хитозаназы микроорганизмами и очистки целевого фермента. Оптимизация параметров культивирования и подбор состава питательной среды для выращивания продуцентов хитозаназ вносят существенный вклад в достижение повышенной продукции этих ферментов в коммерческих целях. Использование подходов экспериментального планирования, в том числе методологии поверхности отклика, позволяет добиться 3–4-кратного повышения продукции хитозаназы. Например, оптимизация накопления биомассы и продукции конститутивной хитозаназы штаммом Bacillus sp. RKY3 по параметрам культивирования (время ферментации) и компонентам питательной среды (мальтоза, сульфат магния и говяжий экстракт), предварительно отобранных в качестве наиболее значимых переменных методом Плаккетта–Бермана, приводила к 11-кратному повышению уровня продукции хитозаназы штаммом Bacillus sp. RKY3 от 5.63 до 63.53 ед./мл [53]. Подбор оптимальных значений выбранных переменных и их взаимного эффекта осуществлялся с помощью метода крутого восхождения и экспериментальным планированием по Боксу-Бенкену, при этом ожидаемый отклик (62.3 ед./мл) был практически идентичен значениям, полученным в реальном эксперименте [53].
Методом планирования по Плаккетту–Берману было установлено значительное влияние таких параметров, как концентрация сульфатов аммония и магния и начальное значение рН среды, на продукцию хитозаназы штаммом Microbacterium sp. OU01 [54]. Наилучшим индуктором для синтеза фермента оказался коллоидный хитозан. При оптимальном соотношении параметров продукция хитозаназы Microbacterium sp. OU01 возрастала с 3.6 до 118 ед./мл [54].
Путем последовательного подбора концентраций источников углерода (2% крахмала) и азота (2.5% полипептона) удалось добиться 38‑кратного повышения продукции хитозаназы (до 45.8 ед/мл) при выращивании штамма Bacillus sp. KCTC 0377BP в колбах (табл. 2) [32]. Дальнейшая оптимизация при масштабировании условий культивирования в ферментере объемом 500 л способствовала увеличению продукции хитозаназы примерно до 100 ед./мл, показывая в итоге 83-кратное повышение первоначального уровня активности штамма [32].
Проведена оптимизация продукции хитозаназы энтомопатогенным грибом Trichoderma koningii при твердофазном культивировании [55]. В качестве основных переменных изучали содержание хитозана, пшеничных отрубей и степени увлажнения субстрата. При подборе оптимальных условий экстракции фермента оценивали совместное влияние температуры, степени перемешивания субстрата и времени экстракции. С использованием анализа поверхности отклика были подобраны условия, способствующие 2.3-кратному увеличению выхода хитозаназы по сравнению с максимальными значениями, наблюдаемыми до оптимизации [55].
Эффективность статистической оптимизации, основанной на аналогичных методах планирования эксперимента, продемонстрирована для продукции хитозаназы грибом Aspergillus sp. QD-2 при глубинном культивировании [56]. Продукция хитозаназы некоторыми природными и рекомбинантными штаммами продуцентов и особенности их культивирования представлены в табл. 2.
Сходные подходы экспериментального планирования и математического моделирования используются для оптимизации процессов выделения микробных хитозаназ, в особенности, при разработке и изучении новых способов их очистки. Традиционные процедуры очистки хитозаназ широко известны и основаны на комбинированном применении методов солевого фракционирования, ультрафильтрации, хроматографии и т.п. [26]. Однако в последнее десятилетие активно изучаются новые подходы, позволяющие осуществлять прямое выделение хитозаназ из культуральной среды продуцентов. Например, с помощью экспериментального планирования по Боксу-Бенкену был оптимизирован способ очистки хитозаназ Bacillus cereus NTU-FC-4, основанный на обращенной мицеллярной экстракции неочищенного фермента, предварительно осажденного из культуральной жидкости 70%-ным ацетоном [57]. Было изучено влияние шести независимых переменных на выход хитозаназы в процессе экстракции, включая концентрацию белка, NaCl, рН водной фазы и концентрацию анионного сурфактанта (1,2-бис(-2-этилгексил)сульфосукцинат натрия) в органической фазе, а также температуру и длительность экстракции. Оптимизация условий экстракции позволила добиться 70%-ного выхода хитозаназы при 30-кратной степени очистки [57].
В качестве перспективного способа хроматографической очистки хитозаназ рассматривается адсорбция в псевдожиженном слое носителя, позволяющая осуществлять прямое выделение ферментов из вязкой и дисперсной культуральной среды без предварительных стадий осветления, удаления бактериальной биомассы и нерастворимых компонентов центрифугированием, фильтрацией и т.п. [58, 59]. Особенность данного метода состоит в обратном токе элюента и его подаче на колонку снизу, когда происходит стабилизация расширяющегося слоя адсорбента при достижении равновесия между скоростью осаждения частиц носителя и потоком элюента снизу. Принципы адсорбции по этому методу могут быть такими же, как и в различных вариантах классической хроматографии (ионообменной, гидрофобной, аффинной и т.п.). Эффективность метода адсорбции в расширяющемся слое носителя изучалась при одноэтапной очистке хитозаназ B. cereus, P. ehimensis и Metarhisium anisopliae [60–62]. Процедуру выделения этих ферментов оптимизировали с использованием различных методов экспериментального планирования и математического моделирования, однако, в целом, степень их очистки оставалась не очень высокой при выходе от 31 до 45%.
Получение и поиск хитозаназ со специальными характеристиками. Необходимым условием эффективной деполимеризации хитозана в промышленных условиях является высокая стабильность используемых хитозаназ при повышенных значениях температуры и при относительно низких значениях рН, ниже константы ионизации хитозана (pK ~ 6.2–6.3), обеспечивающих хорошую растворимость исходного субстрата.
Оптимум рН микробных хитозаназ варьирует в интервале рН 4–8, грибные и растительные ферменты характеризуются, как правило, более низкими значениями рН-оптимума [26]. Относительно редко встречаются хитозаназы с оптимумом в щелочной области. Следует ожидать синтез таких ферментов алкалофильными и алкалотолерантными микроорганизмами [63].
Оптимальная температура для действия хитозаназы микроорганизмов-продуцентов, как правило, взаимосвязана с температурным диапазоном их роста, и составляет обычно 30–60°С [17, 26]. Известна хитозаназа Janthinobacterium sp. 4239, активная при рН 10 и 30°С [46], однако большинство изученных хитозаназ являются термолабильными белками [26]. Особый интерес представляют термостабильные хитозаназы, имеющие преимущества для промышленного использования, поскольку повышенная температура может ускорять процесс деполимеризации и уменьшает вязкость раствора хитозана, что позволяет увеличивать растворимость исходного субстрата и снизить риск микробной контаминации. Известны природные штаммы микроорганизмов, продуцирующие термостабильные хитозаназы, например, штамм Paenibacillus sp. 1794, выделенный из торфяного компоста, содержащего панцири креветок, продуцирующий хитозаназу из семейства GH8 с ММ 40 кДа, оптимумом рН 4.8 и температуры 80–85°С [21]. Время полуинактивации этого фермента при 70°С в присутствии хитозана составляло ~20 ч, а время протекания гидролиза субстрата с постоянной скоростью при такой температуре – не менее 6 ч. Термостабильная хитозаназа гриба A. fumigatus, экспрессируемая в Pichia pastoris, характеризовалась временем полуинактивации 2.5 ч при 80°С и 1 ч при 90°С [49].
Конструирование термостабильных хитозаназ осуществляется также с помощью направленного мутагенеза и генетических манипуляций [64]. Так, при введении дополнительных дисульфидных связей в молекулу хитозаназы EAG1 B. ehimensis (сейчас P. ehimensis), пространственная модель которой была определена на основе структурного анализа, были получены мутантные формы фермента, отличающиеся значительно более длительным временем полужизни при 50°С и повышенной в 3 раза каталитической активностью [65]. Оказалось, что влияние дисульфидных связей на термостабильность хитозаназы зависит в основном от их локализации в молекуле белка. Введение дисульфидных связей в гибкие области обеспечивает жесткую поддержку и создает защищенное микроокружение, что приводит к улучшению термостабильности фермента и его каталитической активности [65].
Поиск и отбор хитозаназ с необходимыми свойствами может осуществляться на стадии целевого скрининга микроорганизмов-продуцентов, выделенных из соответствующих местообитаний. Так, термостабильные хитозаназы могут с наибольшей вероятностью обнаруживаться у термофильных микроорганизмов, а психрофильные – у психрофильных и психротолерантных микроорганизмов. Рациональность такого подхода подтверждается многочисленными данными об обнаружении термостабильных хитиназ и хитозаназ у бактерий, выделенных из областей с повышенной температурой – компостов, горячих источников, пустынь и т.д., включая представителей гипертермофильных архей [21, 66–70]. Для отбора активных продуцентов разрабатывались различные системы быстрого скрининга, основанные на использовании в качестве высокочувствительного субстрата окрашенных производных хитозана, например, коллоидного хитозана в комплексе с красителем ремазолом ярко-голубым R [71]. Удобство использования подобных субстратов заключается в возможности проведения экспресс-мониторинга активности большого количества активных изолятов на микропланшетном спектрофотометре. Известны усовершенствованные варианты целевого скрининга хитозаназ с активностью эндотипа, использующие водорастворимый хитозан, связанный с ремазолом ярко-голубым R, получаемый в результате частичного ферментативного гидролиза [72].
Описан высокопродуктивный метод быстрого скрининга ацидофильных мутантных штаммов гриба Aspergillus fumigatus, показывающих высокую продукцию хитозаназы при рН 4.0 [73]. Первичный отбор наиболее активных культур осуществляли по образованию вокруг грибных колоний значительных зон просветления в агаре с 1%-ным коллоидным хитозаном, окрашенным в парах металлического йода. Отобранные мутанты анализировали по уровню продукции хитозаназы в условиях глубинного культивирования, а наиболее активные из них далее сравнивали по образованию олигомерных продуктов при ферментолизе хитозана, используя экспресс-метод оценки в агаре после обработки 0.005%-ным раствором анилинового голубого. Диффундирующие в агар смеси олигомеров хитозана с n = 2–5 в зависимости от их концентрации, образовывали более крупные и интенсивно окрашенные по сравнению D-глюкозамином и хитобиозой концентрические зоны вокруг лунок [73].
Для поиска новых хитозаназ с определенными характеристиками перспективно использовать методы функциональной метагеномики, которая используется при изучении хитиназ [74]. Один из известных подходов основывается на метагеномном анализе микробных сообществ (в т.ч. некультивируемых форм) супрессивных почв. Так, с помощью фосмидной библиотеки метагенома супрессивной почвы была идентифицирована новая хитиназа Chi18H8, проявляющая антигрибную активность против возбудителей болезней сельскохозяйственных культур – Colletotrichum gloesporioides, Fusarium graminearum и F. oxysporum [75, 76]. Аналогичный подход может быть перспективен при поиске антигрибных хитозаназ, учитывая то, что такие ферменты были описаны у некоторых бактерий и микромицетов [77–81].
Таким образом, современные тенденции в оценке потенциала физико-химических и каталитических свойств хитозаназ связаны с направленной инженерией новых ферментов на основе обширной базы имеющихся сведений о генах, кодирующих хитозаназы различных семейств гликозил-гидролаз, и пространственной структуре их каталитических и субстрат-связывающих доменов. При этом следует особо подчеркнуть возможность создания моделей реакционной специфичности хитозаназ и конструирования на их основе гибридных и искусственных ферментов для специализированного получения целевых ХОС [2].
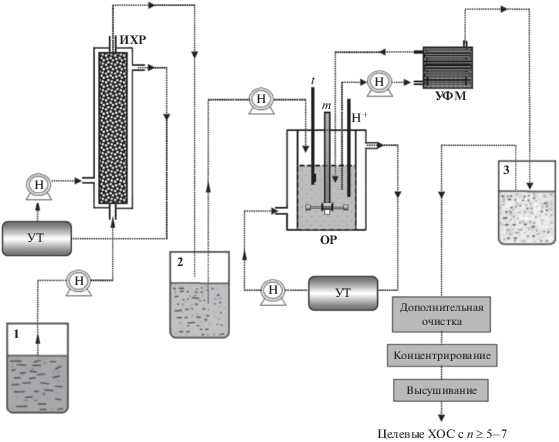
Технологии ферментативного получения олигомеров хитозана с использованием биореакторов. С 1980–1990 гг. ферментативная деполимеризация хитозана изучалась с применением реакторов периодического типа. При такой технологии наблюдался низкий выход продуктов и кроме того, необходимо было использовать большие количества дорогостоящей хитозаназы [82]. В начале нынешнего столетия изучали возможность использования ферментативных реакторов с ультрафильтрационной ячейкой для преодоления недостатков периодического метода. Данную схему можно охарактеризовать как реакторную систему полунепрерывного получения ХОС [83]. Такая технология рассматривалась как инструмент многократного использования фермента с возможностью его отделения от конечных продуктов. Метод позволял конвертировать до 11 объемов субстрата тем же количеством хитозаназы, которое использовалось для одного процесса в реакторе периодического типа. С помощью данной технологии была показана возможность получения ХОС c n = 3–6, обладающих антибактериальной активностью [83]. Наиболее важным параметром в системе является скорость мембранной фильтрации. Большое значение для образования ХОС имеет также время пребывания субстрата в реакторе. Однако данный метод не позволяет осуществлять непрерывный или длительный процесс получения ХОС из-за роста трансмембранного давления [83]. Лимитирующим фактором данной технологии было быстрое загрязнение и забивание мембраны вследствие высокой вязкости растворов хитозана. Оно проявлялось в необратимом падении скорости фильтрации при сохранении основных рабочих параметров, в том числе трансмембранного давления, скорости потока и т.д. Для преодоления этого недостатка была разработана усовершенствованная двойная реакторная система, включающая ферментный реактор, подсоединенный к ультрафильтрационному модулю, и колоночный реактор с хитозаназой, иммобилизованной на хитиновой матрице [84]. Первичный реактор, на который подается раствор субстрата, может также включать носитель с иммобилизованными клетками продуцента хитозаназы [17]. Процесс получения ХОС осуществлялся в два этапа, на первом этапе (1) в колоночном реакторе получали частично гидролизованный хитозан, и на втором (2) (в основном реакторе) – конечные ХОС из промежуточного продукта (рис. 2). За счет подбора скорости внешнего потока и использования мембран с различным размером пор удалось стабилизировать процесс при разделении целевых продуктов на три фракции: олигомеры с ММ 7–24 и 1.5–6 кДа и ХОС с n = 5–7 [84]. Хотя этот метод имеет определенные преимущества перед традиционными реакторами периодического типа, однако ограниченность доступа иммобилизованного фермента к хитозану по сравнению со свободным ферментом существенно снижала его эффективность.
Рис. 2.
Схема двойной реакторной системы для полунепрерывного получения ХОС, состоящей из колоночного термостатируемого реактора с иммобилизованной на твердом носителе хитозаназой (ИХР) и основного реактора (ОР) (адаптировано из [84]). 1 – расходный резервуар с подачей субстрата (раствор хитозана); 2 – резервуар для сбора раствора частично гидролизованного хитозана; 3 – резервуар для сбора конечного продукта (смесь целевых ХОС). Обозначения: н – насос; УФМ – мембранный модуль для ультрафильтрации; УТ – циркуляционный термостат; t – температурный датчик; m – смеситель; H+ – рН-метр.
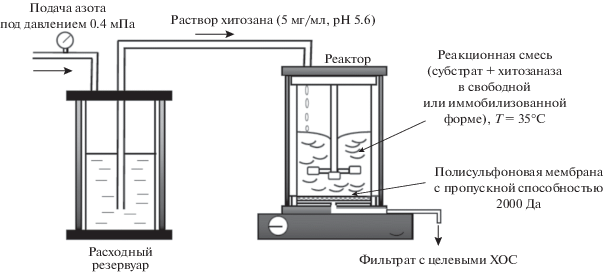
Для улучшения выхода олигомеров хитозана при селективном получении некоторых ХОС и стабилизации процесса были разработаны методы непрерывного гидролиза хитозана в мембранном биореакторе. Был сконструирован усовершенствованный реактор непрерывно-проточного типа с механическим перемешиванием, оснащенный ультрафильтрационной мембраной с пределом отсечения ММ 2000 Да (рис. 3). Развитие мембранных технологий привело к созданию нового типа биореактора, в котором непрерывная реакция происходит одновременно с выделением продукта из реакционной смеси [85]. В данном процессе субстрат непрерывно поступает в реакционную смесь, а продукт непрерывно извлекается из реактора [86]. Размер образуемых ХОС в фильтрате контролируется за счет использования мембран с подходящим размером пор. Тем не менее, в таких условиях остается проблема небольшой длительности процесса гидролиза, не превышающей 48 ч [86]. Это связывают с низкой стабильностью хитозаназ, обычно применяемых в свободной форме. Для стабилизации ферментативного процесса было предложено использование хитозаназы, иммобилизованной методом многоточечной сшивки на поверхности 1 мм кубиков агарового геля. В оптимизированных условиях использование иммобилизованной хитозаназы позволяло непрерывно получать 2.3 г/л (выход 46%) целевых пента- и гексаолигомеров хитозана в течение 1 мес. При этом время 50%-ного снижения продуктивности реактора составляло 50 сут [86]. Таким образом, использование мембранных биореакторных систем дает преимущества, как в плане селективности, так и стабильности самого процесса, и может быть направлено на решение различных задач, в том числе получение различных продуктов при гидролизе хитозана. Накопление и суммирование подобных технологических схем будет способствовать созданию полного технологического регламента энзиматической биотрансформации хитозана.
В рамках совершенствования технологии ферментативного получения ХОС был предложен электрохимический метод с использованием биполярной электродиализной мембраны, которой может быть интегрирован с системой мембранного биореактора [87]. Принцип этой технологии основан на диссоциации молекул воды на границе биполярных мембран при воздействии электрического поля в системе электродиализа. Использование этого метода позволит сначала после циркуляции в подкисляющем отсеке перевести хитозан в растворимое состояние in situ, а хитозаназа может быть инактивирована после циркуляции в подщелачивающем отсеке. Таким образом, основа метода состоит в отказе от традиционного кислотного растворения хитозана на подготовительном этапе деполимеризации и нагревательного оборудования для термоденатурации фермента на заключительной стадии. Это делает процесс ферментолиза хитозана безопаснее с экологической точки зрения.
Использование иммобилизованных препаратов хитозаназы для получения биоактивных ХОС. Применение иммобилизованных ферментов позволяет контролировать процесс деполимеризации хитозана, а также повысить выход целевых продуктов. Иммобилизация хитозаназ на различных носителях улучшает их стабильность, увеличивает сроки хранения, обеспечивая возможность регенерации фермента и его повторного использования, облегчает непрерывность процесса деполимеризации и делает более технологичным применение хитозаназ в условиях биореактора [22]. Ограничение применения иммобилизованных препаратов хитозаназы на промышленном и полупромышленном уровне обусловлено их относительно низкой каталитической активностью.
Одна из первых попыток проведения процесса непрерывного гидролиза хитозана осуществлялась хитозаназами Enterobacter sp. G-1, иммобилизованными на анионообменных смолах, гранулах хитозана и альгинате кальция [88]. Среди этих носителей наиболее высокая удельная активность отмечалась у хитозаназы, иммобилизованной путем ионного связывания с сильно щелочным анионообменником FE-3901, в то время как наибольшей стабильностью обладал фермент, иммобилизованный на гранулах хитозана, который сохранял более половины первоначальной активности после 17 сут хранения [88]. Однако деполимеризация хитозана оценивалась только по снижению его кинематической вязкости и общей концентрации образуемых продуктов гидролиза без детальной оценки конкретных групп олигомеров.
В конце 1990 гг. были получены смеси от тетра- до гексамеров хитозана с использованием хитозаназы, физически иммобилизованной на хитине [89]. Недостатком метода являлся низкий выход продуктов вследствие слабой аффинности и пониженной скорости реакции по сравнению со свободным ферментом. Ковалентная иммобилизация на хитине путем поперечной сшивки глутаральдегидом предпринималась ранее для хитозаназы из Penicillium sp. ZDZ1 [90]. В этом случае было отмечено существенное повышение стабильности иммобилизованной хитозаназы (сохранение 80% активности после 20 сут) по сравнению со свободным ферментом без оценки ее эффективности для получения биоактивных ХОС.
Высокостабильный препарат хитозаназы B. pumilus BN-262 был получен ковалентной иммобилизацией фермента на агаровом геле методом многоточечной сшивки [91]. В качестве сшивающего агента и активатора использовали глицидол (эпигидрин) и периодат натрия, в качестве восстановителя – борогидрид натрия. Показано расширение рН-оптимума (до 4–6), увеличение температурного оптимума (с 60 до 80°С) и термостабильности иммобилизованной хитозаназы по сравнению со свободным ферментом. Фермент сохранял 95% своей первоначальной активности после 225 ч инкубации при 50°С. Наибольшая эффективность иммобилизации достигалась при обработке 0.7 М глицидолом, когда активность хитозаназы составляла 11.9 ед./г носителя. Иммобилизованная на агаре хитозаназа была испытана в процессе непрерывного получения ХОС в условиях лаборатории при непрерывной подаче 0.5%-ного раствора хитозана (0.086 мл/мин, рН 5.6) в биореактор (t = 35°С) с колонкой (12.2 × × 39 мм), упакованной кубиками агара (1 мм3) с иммобилизованной хитозаназой. Степень деполимеризации хитозана составляла 44% и оставалась стабильной на протяжении 28 сут реакции, а общий выход пентамера и гексамера достигал 30% [91]. Таким образом, с начала 2000 гг. опыт применения бактериальной хитозаназы, иммобилизованной методом многоточечного ковалентного связывания на частицах агарового или агарозного геля, показал ее эффективность при непрерывном гидролизе хитозана в течение 1 мес. в биореакторе со слоем носителя (рис. 2). В дальнейшем были оптимизированы условия процесса, в результате которых при начальном содержании хитозана 20 кг/м3 достигался более высокий выход пента- и гексамеров, составляющий 7 кг/м3 [92]. Показано, что выход целевых ХОС в значительной степени определяется такими факторами, как поверхностная плотность иммобилизованного фермента и скорость внешнего массопереноса. В качестве важных параметров отмечают также размер частиц носителя, температуру и начальную концентрацию субстрата [22].
Считается, что продукция высоких концентраций целевых ХОС сохраняет время, снижает стоимость концентрирования продуктов и повышает производительность процесса в целом. Для улучшения массообмена и получения высоких концентраций олигомеров хитозана была разработана особая технология, основанная на использовании биореактора со специальной многолопастной мешалкой, дисковые лопасти которой покрывали агаровым гелем с иммобилизованной хитозаназой [20]. При этом иммобилизованная хитозаназа оказалась высоко стабильной и позволяла получать 9 кг/м3 ХОС с n = 5–6 из раствора хитозана 20 кг/м3, а также из высококонцентрированных (до 50 кг/м3) его растворов. С использованием одной и той же иммобилизованной хитозаназы периодические циклы гидролиза раствора хитозана (50 кг/м3) проводили 3 раза без какого-либо снижения уровня активности при повторных реакциях. Максимальная концентрация пента- и гекса-олигосахаридов составляла 20 кг/м3 (выход) в каждом цикле [20].
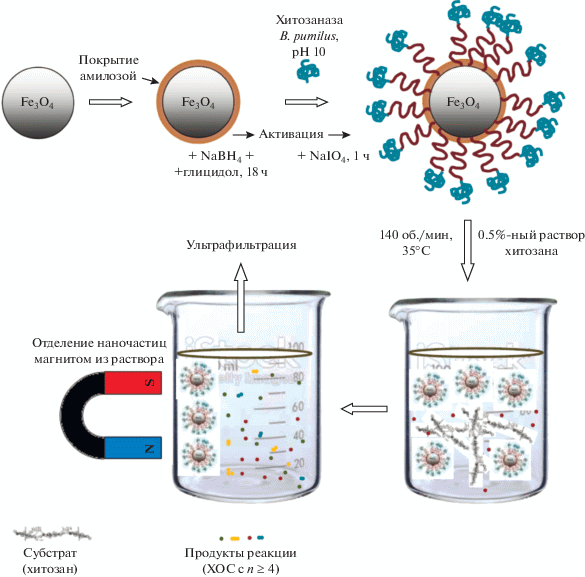
Вследствие высокой вязкости растворов хитозана степень его гидролиза существенно зависела от массопереноса вблизи иммобилизованного фермента, при этом выход целевых олигомеров может быть весьма низким, в том случае, если реакционная система находится в ограниченном режиме массообмена. Это предполагает, что для получения необходимых ХОС традиционные пористые подложки миллиметрового или субмиллиметрового размера не удобны для иммобилизации фермента, поскольку внутри частиц подложки существует резкая резистентность к массопереносу. Для преодоления этого предлагается использовать в качестве носителя магнитные наночастицы и нановолокна [93, 94]. Основные преимущества наночастиц в качестве носителя состоят в том, что они обладают хорошими массообменными характеристиками, позволяют легко восстанавливать и повторно использовать иммобилизованный фермент и имеют большую удельную площадь поверхности. Известны примеры использования магнитных наночастиц для иммобилизации липаз, протеаз и других ферментов [95]. Была проведена оценка эффективности получения ХОС с использованием хитозаназы, иммобилизованной на магнитных наночастицах из магнетита (Fe3O4) [93]. Гидроксильные группы амилозы, покрывающей поверхность наночастиц, химически активировали, а затем использовали для иммобилизации хитозаназы через многоточечные ковалентные связи (рис. 4). Данный метод позволял иммобилизовать в 1.4–2 раза больше хитозаназы по сравнению с традиционным методом физической адсорбции. За один цикл реакции выход целевых олигомеров достигал 40%. При этом отмечалась стабильность фермента, а относительная активность хитозаназы, иммобилизованной таким образом, составляла около 60 и 50% после второго и третьего циклов. Одно из преимуществ метода состоит в том, что магнитные наночастицы могут быть легко и быстро отделены от вязкого раствора хитозана при контакте сосуда с магнитом [93].
Рис. 4.
Схема получения ХОС с использованием хитозаназы, иммобилизованной методом многоточечной сшивки на магнитных наночастицах, покрытых амилозой (после завершения ферментативной реакции суспендированные в реакционной смеси наночастицы регенерируют и собирают с помощью контакта магнита с внешней стенкой реакционного сосуда [93]).
В качестве одного из новых материалов для иммобилизации хитозаназ рассматривались нановолокнистые мембраны из полиакрилонитрила [94]. Для иммобилизации использовали грибную хитозаназу. Фермент сохранял более 70% активности после 10 циклов и мог храниться до 60 сут при 4°С с минимальными потерями активности.
Вариантом применения хитозаназы для получения ХОС было создание ферментного комплекса, иммобилизованного на поверхности липосом при их прямом взаимодействии с цитоплазматической мембраной Streptomyces griseus − известного продуцента хитозаназы [96]. Установлено, что в составе полученного комплекса хитозаназа проявляет намного более высокую активность по сравнению со свободным ферментом. На активность хитозаназы значительное влияние оказывала гидратация поверхности липосом, при этом стабильность фермента значительно увеличивалась при различных значениях рН и температуры. Вместе с тем достоверных данных об эффективности липосомных комплексов для получения ХОС пока не получено.
Известен опыт применения иммобилизованных клеток Trichoderma reesei в колоночном реакторе пузырькового типа для одновременного получения фермента и деполимеризации хитозана преимущественно (73%) до D-глюкозамина [97]. Стабильность такой системы сохранялась в течение 10 циклов. В работе [98] было показано увеличение на 60% продукции хитозаназы грибом Gongronella sp. JG после иммобилизации его клеток в геле альгината кальция. Иммобилизованные клетки сохраняли 85% активности после 7 циклов периодического культивирования, в связи с этим предполагается возможным их применение в процессах полунепрерывного получения хитозаназы.
Одним из интересных примеров “клеточной” иммобилизации можно назвать экспрессию хитозаназы GH8 в Paenibacillus fukuinensis D2 на поверхности дрожжевых клеток Saccharomyces cerevisiae для получения ХОС из хитозана [99]. Для этой цели был сконструирован гибридный ген на основе секреционной сигнальной последовательности гена глюкоамилазы Rhizopus oryzae, основной последовательности гена хитозаназы GH8 и фрагмента гена, кодирующего С-концевой участок α-агглютинина. Полученный фрагмент встраивали в плазмиду pCAS-chi, трансформируемую в клетки дрожжей. Хитозаназа экспрессировалась на поверхности дрожжевых клеток, оставаясь связанной с клеточной стенкой через С-концевой фрагмент α-агглютинина. Подобный метод позволяет использовать дрожжи в качестве стабильного и высокоактивного клеточного катализатора, который может содержать от 10 до 100 тыс. молекул хитозаназы на своей поверхности и эффективно гидролизовать хитозан до целевых олигомеров. Сходный подход был реализован в одной из недавних работ [100]. В ней была продемонстрирована экспрессия хитозаназы из B. subtilis ATCC 3857 на поверхности клеток Lactobacillus plantarum WCFS1 в результате закрепления фермента с помощью C-концевой якорной последовательности (cwa2) липопротеина клеточной стенки Lp_2578 L. plantarum. Наиболее высокая активность хитозаназы составляла 1360 ед./г сухой клеточной биомассы (или 1.7 мг/мг по белку). Поверхностная локализация хитозаназы, как и в работе [99], подтверждалась с помощью иммунофлуоресцентной микроскопии, а также методом проточной цитометрии. По данным высокоэффективной анионообменной хроматографии и ЭСИ (электроспрей ионизации)-масс-спектрометрии клетки L. plantarum, содержащие на поверхности хитозаназу, эффективно конвертировали хитозан до ХОС с n = 2–6. Однако использование клеток L. plantarum для деполимеризации хитозана рассматривалось в данной работе в аспекте потенциального применения лактобацилл в составе пробиотических продуктов с учетом лечебно-профилактического и, возможно, пребиотического действия образуемых ими хитоолигомеров.
В целом, следует отметить, что проявление ферментативной активности гетерологичных хитозаназ на клеточной поверхности бактерий или дрожжей рассматривается как перспективный подход при развитии технологии клеточных биокатализаторов для промышленного получения биоактивных ХОС. Наиболее привлекательной особенностью данного подхода является то, что молекулы фермента одновременно синтезируются и сами “иммобилизуются” на поверхности клеток, а “биокатализатор” может быть легко получен и “размножен” обычным культивированием [101, 102]. При этом непосредственное использование микробных клеток для получения ХОС может осуществляться как в процессе их выращивания, так и после него. Помимо известных преимуществ иммобилизованных ферментов, эта стратегия избавляет от необходимости очистки хитозаназ, упрощает процесс деполимеризации и снижает общие издержки при получении ХОС.
Выделение и очистка индивидуальных фракций ХОС. Препаративное разделение хитоолигомеров, как правило, основывается на различии их ММ и степени деацетилирования, что определяет основные методологические подходы в получении очищенных фракций ХОС. Одним из методов, наиболее часто упоминаемых в литературе, является фракционирование ХОС в реакторах с ультрафильтрационной мембраной. К основным факторам, определяющим особенности мембранного разделения ХОС, относятся типы мембран, рН, температура и концентрация исходных растворов. Как показано в работе [103], сильное влияние на ММ олигохитозанов, фракционируемых методом ультрафильтрации, может оказывать природа соли в растворах, используемых в качестве растворителя. В некоторых случаях было отмечено, что фракции олигомеров могут иметь более высокую ММ, чем предел пропускания мембраны, используемой для их ультрафильтрации [104]. Обычно это связывают с преимущественной калибровкой УФ-мембран по глобулярным белкам [103].
Получение ХОС при непрерывном гидролизе хитозана с разделением продуктов на УФ-мембране обладает определенными преимуществами по сравнению с периодическим процессом вследствие возможности продукции более высокомолекулярных ХОС, предотвращения глубокого гидролиза субстрата и ингибирования образования нежелательных побочных продуктов [22]. Фракционирование ХОС ультрафильтрацией может осуществляться как самостоятельно, так и в комбинации с другими методами. Собственно ультрафильтрация позволяет отделять только определенную фракцию продуктов гидролиза хитозана в пределах пропускающей способности используемой мембраны. Фракционирование ХОС в более узком диапазоне ММ требует ее проведения на двух и более мембранах с довольно близкими параметрами пропускающей способности. Так, был оптимизирован метод разделения ХОС ультрафильтрацией на половолоконных мембранах с диаметром пор, соответствующим пропускной способности 2 и 3 кДа [105]. Гидролизаты частично деацетилированных хитозанов разделяли в системе мембранного реактора с использованием УФ-мембран с пропускающей способностью 10, 5 и 1 кДа [104]. Комбинированная система УФ-мембран с пропускной способностью 3 и 5 кДа была использована при очистке гетеро-хитоолигосахаридов с различной степенью деацетилирования [106]. Российскими исследователями показана возможность эффективного фракционирования низкомолекулярного хитозана ультрафильтрацией на мембранах Гидросарт “Sartorius” (Германия) с пропускной способностью 30, 10 и 5 кДа [103]. Для разделения биоактивных пента- и гексамеров от более низкомолекулярных ХОС (n = 2–4), продуктов ферментативного гидролиза хитозана, применялись также нанофильтрационные (НФ) мембраны (предел фракционирования ≥2 нм), функционирующие при более высоком давлении, чем УФ-мембраны [107]. После оптимизации таких основных параметров процесса непрерывной диафильтрации, как тип НФ-мембраны, рабочее давление и скорость разбавления ретентата, выход целевых ХОС (n = 5–6) в олигомерной смеси возрастал с 30 до 77% [107].
Для разделения ХОС предложен комбинированный метод ультрафильтрации и нанофильтрации [105]. На первом этапе взвешенные частицы и нерастворимые агрегаты молекул удаляли пропусканием через мембрану 0.22 мкм с последующей ультрафильтрацией на УФ-мембране с пределом фракционирования 1 кДа. Получаемый фильтрат разделяли нанофильтрацией, затем ХОС из фильтратов (с рН, доведенном до 8.3) осаждали 3-кратным объемом этанола и лиофилизировали. Лиофильно высушенные ХОС дополнительно очищали хроматографией на КМ-сефадексе С-25. Для селективного и высокоспецифичного разделения ХОС с n = 2–4 использован метод, сочетающий ультрафильтрацию и электродиализ при различных значениях рН [108].
Помимо ультрафильтрации для очистки ХОС широко применяется фракционирование органическими растворителями. Так, осаждение ХОС водными растворами метанола с различным объемным соотношением растворителя позволяет выделять фракции олигомеров, различающиеся по ММ [22]. Данный метод нельзя назвать высокоселективным, поскольку разделяемые фракции, как правило, включают несколько групп олигомеров, например, с n = 3–5 и n = 6–11 [109]. Использование органических растворителей в некоторых случаях может ограничивать применение целевых ХОС в областях, требующих высокой степени чистоты (медицина и фармацевтика).
Наиболее эффективно разделение ХОС может быть осуществлено с использованием методов жидкостной хроматографии. На ранних этапах для разделения смеси мономера и олигомеров (n = 2–4) хитозана использовалась хроматография на уголь-целитных колонках [22, 110]. Это метод позволял быстро разделять смеси ХОС на индивидуальные компоненты при элюировании водными растворами этанола в различных объемных соотношениях. Для фракционирования ХОС, имеющих n = = 2–12 и n = 2–6, проводилась хроматография на Toyopearl HW-40S, КМ-сефадексе С-25 и анионообменной смоле Дауэкс (H+) [111, 112]. С первой половины 2000 гг. вошли в практику современные методы эксклюзионной хроматографии, основанные на применении последовательно соединенных колонок с носителем СупердексTM30 (“GE Healthcare”, США) [2]. Описаны примеры использования трех спаренных колонок с этим сорбентом, общим размером 2.6 × 180 см, в которых в качестве элюента использовали аммоний-ацетатный буфер [113]. Олигомеры обычно детектируются с помощью рефрактометрического детектора в режиме реального времени. Для быстрого количественного анализа D-глюкозамина, образующегося при совместном гидролизе хитозана под действием эндо- и экзохитозаназы, был предложен метод высокоэффективной анионообменной хроматографии с пульсирующим амперометрическим детектором, позволяющим обнаруживать концентрации образуемого продукта на уровне 50 пМ, что на три порядка превышало уровень чувствительности методов, основанных на оценке индекса преломления [114].
Фракции индивидуальных ХОС могут быть идентифицированы с помощью MALDI-TOF-масс-спектрометрии [113]. Такой вариант детекции в эксклюзионной хроматографии позволяет вне зависимости от порядка расположения ацетильных остатков и степени деацетилирования разделять индивидуальные олигомеры со сходными значениями степени полимеризации в интервале от 2 до 20 остатков (а для смесей ХОС до n ≤ 40) [115].
С помощью катионообменной хроматографии олигомеры с одинаковой степенью полимеризации могут быть разделены в зависимости от относительного содержания у них деацетилированных звеньев. Дальнейшее частичное разделение изобарических ХОС (имеющих одинаковую степень ацетилирования, но различный порядок расположения ацетильных и деацетилированных групп) может быть достигнуто с помощью сильных катионообменников [116]. В качестве альтернативного подхода для разделения более коротких ХОС была успешно использована металлохелатная аффинная хроматография. Поскольку ХОС обладают сильным сродством к ионам меди (Cu2+), то их применение в качестве хелатирующего агента может обеспечивать 90% разделения полностью деацетилированных олигомеров с n ≤ 4 (95% – для димера и тримера) при выходе 60–95%, однако это не было показано для смесей N-ацетилированных ХОС [117]. Функционализация носителя полигидроксильной природы для металлохелатной хроматографии обеспечивается с помощью иминодиацетата, карбоксиметил аспартата и трис(карбоксиметил)этилендиамина [117].
Для характеристики ХОС по показателям деацетилирования, степени полимеризации и распределения ацетильных и деацетилированных групп применяются преимущественно ЯМР- и масс-спектрометрия (МС). В зависимости от сложности олигомерной смеси С13-ЯМР-спектрометрия позволяет определить степень деацетилирования и частично идентифицировать порядок расположения ацетилированных звеньев у коротких ХОС [118]. Современные методы МС обладают мощным инструментарием для анализа степени полимеризации и деацетилирования олигомеров [119, 120]. Так, МС с электроспрей ионизацией в режиме реального времени имеет большой потенциал для высокопроизводительного количественного мониторинга продуктов ферментативного расщепления олигомеров хитозана с n = 2–6 [121]. Разработан метод оценки последовательности ацетилированных и неацетилированных остатков в ХОС с n ≤ 12, основанный на маркировке редуцирующего конца олигомеров 2-аминоакридоном, использующимся в качестве флуоресцентной метки для анализа гликанов и олигосахаридов [120, 122]. С помощью этого флуорофора возможно определять пикомолярные концентрации олигомеров. Для идентификации последовательности ацетилированных остатков в смеси ХОС, гомогенных по степени полимеризации, может быть использована MALDI-TOF спектрометрия, однако отмечаются некоторые ограничения данного метода при анализе смесей олигомеров. Относительная концентрация и структурная идентичность смесей маркированных ХОС, стабильных в широком интервале рН, может оцениваться также с помощью мицеллярной электрокинетической капиллярной хроматографии, нормальнофазовой и обращеннофазовой хроматографии с последующей МС (в том числе МС с электроспрей ионизацией). Описан также метод восстановительного аминирования ХОС с использованием в качестве флуоресцентной метки 3-(ацетиламино)-6-аминоакридина с последующей идентификацией “изобарических” ХОС MALDI-TOF-спектрометрией с ионной ловушкой [116]. Этот метод позволяет одновременно проводить количественную оценку и анализ последовательности ацетильных групп в смесях ХОС с идентичной степенью полимеризации, но различными значениями степени деацетилирования и порядком расположения функциональных мономерных звеньев. Недавно сообщалось также о ферментативном секвенировании частично N-ацетилированных олигомеров хитозана [123], основанном на последовательном отщеплении остатков N-ацетил-D-глюкозамина и D-глюкозамина с нередуцирующего конца ХОС экзо-β-N-ацетилгексозаминидазой и экзо-β-D-глюкозаминидазой соответственно. Однако этот метод рассматривается как частный при анализе механизма действия специфичных хитиндеацетилаз на ХОС и в большей степени как дополнение к методам МС.
В настоящее время стала вполне реальной, в особенности для низших хитоолигомеров (n = 5–7), возможность осуществления на промышленном уровне эффективной и управляемой деполимеризации хитозана для получения препаратов ХОС, обогащенных определенными типами биологически активных олигомеров. Этому способствуют накопленные сведения о механизмах, кинетике и специфичности хитозаназ, хитиназ и неспецифических гидролаз, а также уровень развития современных технологий и материалов, связанных с новыми возможностями очистки, фракционирования и анализа ХОС, а также иммобилизации ферментов. В то же время промышленное получение высокочистых и гомогенных ХОС с определенной степенью деацетилирования и известным порядком расположения ацетилированных и деацетилированных остатков остается проблемой. В ближайшем будущем их изучение, по-видимому, будет связано с получением высокочистых ХОС (включая нативные ХОС и их производные) для медицины, фармацевтики, пищевой промышленности и т.д. Для коммерциализации применения ХОС дальнейшие работы будут включать оптимизацию биотехнологического получения хитозаназ, в том числе за счет использования рекомбинатных штаммов-сверхпродуцентов, улучшение выхода целевых продуктов деполимеризации хитозана, совершенствование методологии их очистки и анализа при снижении ценовых издержек. Получение целевых ХОС может обеспечиваться выбором подходящего фермента и хитозана (в основном по степени ацетилирования), а также контроля глубины и степени деградации субстрата. Потребность в определенных типах ХОС может быть удовлетворена использованием инженерных ферментов с особым типом реакционной специфичности или деацетилирования полученных продуктов с помощью хитиндеацетилаз. Важнейшими вопросами в технологии получения биоактивных ХОС останутся разработка новых типов реакторов для деполимеризации хитозана и применение иммобилизованных хитозаназ, в том числе в составе клеточных катализаторов.
В настоящее время считается [2], что получение смесей ХОС, обладающих разнообразной биологической активностью, с экономической точки зрения более выгодно, чем выделение высокоочищенных индивидуальных олигомеров, в особенности для тех областей промышленности (рис. 5), в которых эти характеристики не являются критичными для создания эффективного продукта (растениеводство, животноводство и т.п.).
Рис. 5.
Области применения хитозана и ХОС в зависимости от их качества и степени очистки. ХОС наивысшего качества с высокой добавленной стоимостью используются в области медицины и фармакологии [129].
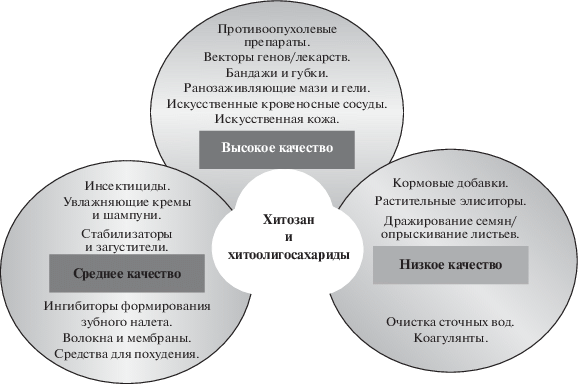
Современные возможности очистки и анализа ХОС и их дальнейшее развитие помогут выяснить зависимость степени проявления той или иной биологической активности у индивидуальных олигомеров от ММ, СД и последовательности расположения ацетилированных остатков. Создание баз данных индивидуальных ХОС на основе достоверной оценки их биологической активности, наряду с исследованиями фундаментального характера, позволяющими установить молекулярные и клеточные механизмы активности конкретного хитоолигомера, поможет коммерциализации этих соединений для применения в области медицины и фармакологии.
Работа выполнена при частичной финансовой поддержке гранта Российского научного фонда (проект № 16-14-00046).
Список литературы
Park Y., Kim M.H., Park S.C., Cheong H., Jang M.K., Nah J.W., Hahm K.S. // J. Microbiol. Biotechnol. 2008. V. 18. № 10. P. 1729–1734.
Aam B.B., Heggset E.B., Norberg A.L., Sørlie M., Vårum K.M., Eijsink V.G.H. // Mar. Drugs. 2010. V. 8. № 5. P. 1482–1517.
Yin H., Zhao X., Du Y. // Carbohydr. Pol. 2010. V. 82. № 1. P. 1–8.
Badawy M.E.I., Ravea E.I. // Int. J. Carbohydr. Chem. 2011. https://doi.org/10.1155/2011/460381
Xia W., Liu P., Zhang J., Chen J. // Food Hydrocolloids. 2011. V. 25. № 2. P. 170–179.
Kerch G. // Mar. Drugs. 2015. V. 13. № 4. P. 2158–2182.
Muzzarelli R.A.A., Tomasetti M., Ilari P. // Enzyme Microb. Technol. 1994. V. 16. № 2. P. 110–114.
Lee D.X., Xia W.S., Zhang J.L. // Food Chem. 2008. V. 111. № 2. P. 291–295.
Stokke B.T., Vårum K.M., Holme H.K., Hjerde R.J.N., Smidsrød O. // Can. J. Chem. 1995. V. 73. № 11. P. 1972–1981.
Ильина А.В., Ткачева Ю.В., Варламов В.П. // Прикл. биохимия и микробиология. 2002. Т. 38. № 2. С. 132–135.
Roncal T., Oviedo A., Lopez de Armentia I., Fernandez L., Villaran M.C. // Carbohydrate Res. 2007. V. 342. № 18. P. 2750–2756.
Xia W., Liu P., Liu J. // Bioresource Technol. 2008. V. 99. № 15. P. 6751–6762.
Pan A.D., Zeng H.Y., Foua G.B., Alain C., Li Y.Q. // Carbohydr. Polym. 2016. V. 135. № 1. P. 199–206.
Foua G.B., Zeng H.Y., Pan A.D. // Bioengineering. 2016. V. 3. № 3. P. 1-17. https://doi.org/10.3390/bioengineering3030017
Fernandez-Lucas J., Castaneda D., Hormigo D. // Trends Food Sci. Technol. 2017. V. 68. № 10. P. 91–101.
Santos-Moriano P., Fernandez-Arrojo L., Mengibar M., Belmonte-Reche E., Peñalver P., Acosta F.N., Ballesteros A.O., Morales J.C., Kidibule P., Fernandez-Lobato M., Plou F.J. // Biocatal. Biotransformation. 2018. V. 36. № 1. https://doi.org/10.1080/10242422.2017.1295231
Jung W.-J., Park R.-D. // Mar. Drugs. 2014. V. 12. № 7. P. 5328–5356.
Актуганов Г.Э., Мелентьев А.И. // Прикл. биохимия и микробиология. 2017. Т. 53. № 6. С. 551–567.
Cheung R.C.F., Ng T.B., Wong J.H., Chan W.Y. // Mar. Drugs. 2015. V. 13. № 7. P. 5156-5186.
Ming M., Kuroiwa T., Ichikawa S., Sato S., Mukataka S. // Food Sci. Technol. Res. 2006. V. 12. № 2. P. 85–90.
Zitouni M., Fortin M., Scheerle R.K., Letzel T., Matteau D., Rodrigue S., Brzezinski R. // Appl. Microbiol. Biotechnol. 2013. V. 97. № 13. P. 5801–5813.
Sinha S., Chand S., Tripathi P. // Appl. Biochem. Biotechnol. 2016. V. 180. № 5. P. 883–889.
Brzezinski R. // Bioeng. Bugs. 2011. V. 2. № 4. P. 226–229.
Yabuki M., Uchiyama A., Suzuki K., Ando A., Fujii T. // J. Gen Appl. Microbiol. 1998. V. 34. № 3. P. 255–270.
Kimoto H., Kusaoke H., Yamamoto I., Fujii T., Onodera T., Taketo A. // J. Biol. Chem. 2002. V. 277. № 17. P. 14695–14702.
Thadathil N., Velappan S.P. // Food Chem. 2014. V. 150. P. 392–399. https://doi.org/10.1016/j.foodchem.2013.10.083
Nguyen A.D., Huang C.-C., Liang T.-W., Nguyen V.B., Pan P-.S., Wang S.-L. // Carbohydr. Polym. 2014. V. 108. № 1. P. 331–337.
Wang S.-L., Liang T.-W., Yen Y.-H. // Carbohydr. Polym. 2011. V. 84. № 2. P. 732–742.
Актуганов Г.Э., Галимзянова Н.Ф., Терегулова Г.А., Мелентьев А.И. // Прикл. биохимия и микробиология. 2016. Т. 52. № 5. С. 520–526.
Nogawa M., Takahashi H., Kashiwagi A., Ohshima K., Okada H., Morikawa Y. // Appl. Environ. Microbiol. 1998. V. 64. № 3. P. 890–895.
Kim P., Kang T.H., Chung K.J., Kim I.S., Chung K.C. // FEMS Microbiol. Lett. 2004. V. 240. № 1. P. 31–39.
Choi Y.J., Kim E.J., Piao Z., Yun Y.C., Shin Y.C. // A-ppl. Environ. Microbiol. 2004. V. 70. № 8. P. 4522–4531.
Chiang C.-L., Chang C.-T., Sung H.-Y. // Enzyme Microb. Technol. 2003. V. 32. № 2. P. 260–267.
Lee H.-S., Jung J.S., Choi S.-K., Lee D.-W., Kim E.-J., Jung H.-C., Pan J.-G. // FEMS Microbiol. Lett. 2007. V. 277. № 2. P. 133–141.
Viens P., Lacombe-Harvey M.-E., Brzezinski R. // Mar. Drugs. 2015. V. 13. № 11. P. 6566–6587.
Kendra D.F., Hadwiger L.A. // Exp. Mycol. 1984. V. 8. № 3. P. 276–281.
Tokura S., Ueno K., Miyazaki S., Nishi N. // Macromol. Symp. 1997. V. 120. № 1. P. 1–9.
Ghinet M.G., Roy S., Poulin-Laprade D., Lacombe-Harvey M.-È., Morosoli M., Brzezinski R. // Biochem. Cell Biol. 2010. V. 88. № 6. P. 907–916.
Lacombe-Harvey M.-È., Fukamizo T., Gagnon J., Gh-inet M.G., Dennhart N., Letzel T., Brzezinski R. // FEBS J. 2009. V. 276. № 3. P. 857–869.
Liu Y.-L., Jiang S., Ke Z.-M., Wu H.-S., Chi C.-W., Guo Z.-Y. // Carbohydr. Res. 2009. V. 344. № 6. P. 815–819.
Pechsrichuang P., Yoohat K., Yamabhai M. // Bioresource Technol. 2013. V. 127. № 1. P. 407-414. https://doi.org/10.1016/j.biortech.2012.09.130
Fukamizo T., Amano S., Yamaguchi K., Yoshikawa T., Katsumi T., Saito J., Suzuki M., Miki K., Nagata Y., Ando A. // J. Biochem. 2005. V. 135. № 5. P. 563–569.
Dubeau M.-P., Guay I., Brzezinski R. // Microb. Cell Fact. 2011. V. 10. P. 7. https://doi.org/10.1186/1475-2859-10-7
Kilani-Feki O., Frikha F., Zouari I., Jaoua S. // Bioprocess Biosyst. Eng. 2013. V. 36. № 7. P. 985–992.
Kusaoke H., Shinya S., Fukamizo T., Kimoto H. // Int. J. Biol. Macromol. 2017. V. 104. Pt. B. P. 1633–1640.
Johnsen M.G., Hansen O.C., Stougaard P. // Microb. Cell. Fact. 2010. V. 9. P. 5. https://doi.org/10.1186/1475-2859-9-5
Liu G.L., Li Y., Zhou H.X., Chi Z.M., Madzak C. // J. Mol. Catal. B Enzym. 2012. V. 83. № 1. P. 100–107. https://doi.org/10.1016/j.molcatb.2012.07.012
Li S., Chen L., Wang C., Xia W. // Carbohydr. Res. 2008. V. 343. № 17. P. 3001–3004.
Zhu X-F., Tan H-Q., Zhu C., Liao L., Zhang X-Q., Wu M. // AMB Express. 2012. V. 2. № 1. P. 1–8.
Chen X., Zhai C., Kang L., Li C., Yan H., Zhou Y., Yu X., Ma L. // Biotechnol. Lett. 2012. V. 34. № 4. P. 689–694.
Huang L., Wang Q., Jiang S., Zhou Y., Zhang G., Ma Y. // Biprocess Biosyst. Bioeng. 2016. V. 39. № 11. P. 1679–1687.
Cheng C.Y., Li Y.-K. // Biotechnol. Appl. Biochem. 2000. V. 32. Pt. 3. P. 197–203.
Wee Y.-J., Reddy L.V.A., Chung K.-C., Ryu H.-W. // J. Chem. Technol. Biotechnol. 2009. V. 84. № 9. P. 1356–1363.
Sun Y., Han B., Liu W., Zhang J., Gao X. // Bioresuor. Technol. 2007. V. 98. № 8. P. 1548-1553.
Da Silva L.C., Honorato T.L., Franco T.T., Rodrigues S. // Food Bioprocess Technol. 2012. V. 5. № 5. P. 1564–1572.
Zhang H., Sang Q., Zhang W. // Ann. Microbiol. 2012. V. 62. № 1. P. 193–201.
Chen Y.-L., Su C.-K., Chiang B.-H. // Process Biochem. 2006. V. 41. № 4. P. 752–758.
Hjorth R. // Trends Biotechnol. 1997. V. 16. № 6. P. 230–235.
De Aurajo N.K., Pagnocelli M.G.B., Pimentel V.C., Xavier M.L.O., Padilha C.E.A., de Macedo G.R., dos Santos E.S. // Int. K. Biol. Macromol. 2016. V. 82. P. 291–298. https://doi.org/10.1016/j.ijbiomac.2015.09.063
Santana S.C., Filho R.C.S., Oliveira J.A., Macedo G.R., Padilha F.F., Santos E.S. // Biocatal. Agric. Biotechnol. 2015. V. 4. № 4. P. 727–736.
De Aurajo N.K., Pimentel V.C., da Silva N.M.P., De Aurajo Padilha C.E., dos Santos E.S. // J. Sep. Sci. 2016. V. 39. № 4. P. 709–716.
De Aurajo Padilha C.E., Fortunato Dantas P.V., de Sousa F.C. Junior, de Santana Souza D.F., de Oliveira J.A., de Macedo G.R., dos Santos E.S. // J. Chromatogr. B Analyt. Technol. Biomed Life Sci. 2016. V. 1039. P. 44–50. https://doi.org/10.1016/j.jchromb.2016.10.027
Goo B.G., Park J.K. // J. Biosci. Bioeng. 2014. V. 117. № 6. P. 684–689.
Yoon H.-G., Kim H.-Y., Lim Y.-H., Kim H.-K., Shin D.-H., Hong B.-S., Cho H.-Y. // Appl. Microbiol. Biotechnol. 2001. V. 56. № 1–2. P. 173–180.
Sheng J., Ji X., Zheng Y., Wang Z., San M. // Biotechnol. Lett. 2016. V. 38. № 10. P. 1809–1815.
Yoon H.-G., Kim H.-Y., Lim Y.-H., Kim H.-K., Shin D.-H., Hong B.-S., Cho H.-Y. // Appl. Environ. Microbiol. 2000. V. 66. № 9. P. 3727–3734.
Tanaka T., Fukui T., Atomi H., Imanaka T. // J. Bacteriol. 2003. V. 185. № 17. P. 5175–5181.
Sakai K., Yokota A., Kurokawa H., Wakayama M., Moriguchi M. // Appl. Environ. Microbiol. 1998. V. 64. № 9. P. 3397–3402.
Bhushan B. // J. Appl. Microbiol. 2000. V. 88. № 5. P. 800–808.
Toharisman A., Suhartono M.T., Spindler-Barth M., Hwang J.-K., Pyun Y.-R. // World J. Microbiol. Biotechnol. 2005. V. 21. № 5. P. 733–738.
Fen L.L., Illias R. Md., Kamaruddin K., Maskat M.Y., Hassan O. // Enzyme Microb. Technol. 2006. V. 38. № 1–2. P. 215–219.
Zitouni M., Fortin M., Thibeault J.-S., Brzezinski R. // Carbohydr. Polym. 2010. V. 80. № 2. P. 521–524.
Ding S., Chen G.-G., Liang Z.-Q., Zeng W., Cao M.-M., Chen G.-P., Xie S.-Y., Li W. //World J. Microbiol. Biotechnol. 2016. V. 32. № 11. P. 174. https://doi.org/10.1007/s11274-016-2134-0
Cretoiu M.S., Kielak A.M., Al-Soud W.A., Sørensen S.J., van Elsas J.D. // Appl. Microbiol. Biotechnol. 2012. V. 94. № 5. P. 1347–1358.
Hjort K., Presti I., Elväng A., Marinelli F., Sjöling S. // Appl. Microbiol. Biotechnol. 2014. V. 98. № 6. P. 2819–2828.
Berini F., Presti I., Beltrametti F., Pedroli M., Vårum K.M., Pollegioni L., Sjöling S., Marinelli F. // Microb. Cell. Fact. 2017. V. 16. № 1. P. 16. https://doi.org/10.1186/s12934-017-0634-8
Matsuda Y., Iida Y., Shinogi T., Kakutani K., Nonomura T., Toyoda T. // J. Gen. Plant Pathol. 2001. V. 67. № 4. P. 318–324.
Gao X.-A., Ju W.-T., Jung W.-J., Park R.-D. // Carbohydr. Polym. 2008. V. 72. № 3. P. 513–520.
Saito A., Ooya T., Miyatsuchi D., Fuchigami H., Terakado K., Nakayama S.-Y., Watanabe T., Nagata Y., Ando A. // FEMS Microbiol. Lett. 2009. V. 293. № 1. P. 79–84.
Tomita M., Kikuchi A., Kobayashi M., Yamaguchi M., Ifuku S., Yamashoji S., Ando A., Saito A. //Antonie Leeuwenhoek. 2013. V. 104. № 5. P. 737–748.
Rodriguez-Martin A., Acosta R., Liddell S., Nuñes F., Benito M. J., Asensio M.A. // Appl. Microbiol. Biotechnol. 2010. V. 88. № 2. P. 519–528.
Izume M., Ohtakara A. // Agric. Biol. Chem. 1987. V. 51. № 4. P. 1189–1191.
Jeon Y.-J., Kim S.-K. // Carbohydr. Polym. 2000. V. 41. № 2. P. 133–141.
Jeon Y.-J., Kim S.-K. // Process Biochem. 2000. V. 35. № 6. P. 623–632.
Giorno L., Drioli E. // Trends Biotechnol. 2000. V. 18. № 8. P. 339–349.
Kuroiwa T., Izuta H., Nabetani H., Nakajima M., Sato S., Mukataka S., Ichikawa S. // Process Biochem. 2009. V. 44. № 3. P. 283–287.
Shee L.F.T., Arul J., Brunet S., Basinet L. // J. Biotechnol. 2008. V. 134. № 3–4. P. 305–311.
Yamasaki Y., Fukumoto I., Kumagai N., Ohta Y., Nakagawa T., Kawamukai M., Matsuda H. // Biosci. Biotech. Biochem. 1992. V. 56. № 10. P. 1546–1551.
Jeon Y.J., Park P.J., Byun H.G., Song B.K., Kim S.K. // Korean J. Biotechnol. Bioeng. 1998. V. 13. № 2. P. 147–154.
Zeng J., Zheng L.-Y. // Process Biochem. 2002. V. 38. № 4. P. 531–535.
Ichikawa S., Takano K., Kuroiwa T., Hiruta O., Sato S., Mukataka S. // J. Biosci. Bioeng. 2002. V. 93. № 2. P. 201–206.
Kuroiwa T., Ichikawa S., Sato S., Mukataka S. // Biotechnol Bioeng. 2003. V. 84. № 1. P. 121–127.
Kuroiwa T., Noguchi Y., Nakajima M., Sato S., Mukataka S., Ichikawa S. // Process Biochem. 2008. V. 43. № 1. P. 62–69.
Sihna S., Dhakate S.R., Kumar P., Mathur R.B., Tripathi P., Chand S. // Bioresour. Technol. 2012. V. 115. P. 152–157. https://doi.org/10.1016/j.biortech.2011.11.101
Koneracka M., Kop P., Timko M., Ramchand C.N., de Sequeira A., Trevan M. // J. Mol. Catal. B: Enzym. 2002. V. 18. № 1–3. P. 13–18.
Ngo K.X., Umakoshi H., Shimanouchi T., Sugaya H., Kuboi R. // J. Biotechnol. 2010. V. 146. № 3. P. 105–113.
Wu M.B., Xia L.M., Cen P.L. // Gong Cheng Xue Bao. (Chinense J. Biotechnol., article in Chinese). 2000. V. 16. № 3. P. 368–372.
Zhang P., Zhou W., Wang P., Wang L., Tang M. // Braz. J. Microbiol. 2013. V. 44. № 1. P. 189–195.
Fukuda T., Isogawa D., Takagi M., Kato-Murai M., Kimoto H., Kusaoke H., Ueda M., Suye S.-I. // Biosci. Biotechnol. Biochem. 2007. V. 71. № 11. P. 2845–2847.
Nguyen H.-M., Mathiesen G., Stelzer E.M., Pham M.L., Kuczkowska K., Mackenzie A., Agger J.W., Eijsink V.G.H., Yamabhai M., Peterbauer C.K., Haltrich D., Nguyen T.-H. // Microb. Cell. Fact. 2016. V. 15. P. 169. https://doi.org/10.1186/s12934-016-0570-z
Lee S.Y., Choi J.H., Xu Z. // Trends Biotechnol. 2003. V. 21. № 1. P. 45–52.
Kuroda K., Ueda M. // Biomolecules. 2013. V. 3. № 3. P. 632–650.
Лопатин С.А., Дербенева М.С., Куликов С.Н., Варламов В.П., Шпигун О.А. // Журн. аналит. химии. 2009. Т. 64. № 6. С. 666–670.
Park P.-J., Lee H.-K., Kim S.-K. // J. Microbiol. Biotechnol. 2004. V. 14. № 1. P. 41–47.
Lin Y.W., Hsiao Y.C., Chiang B. H. // Food Res. Int. 2009. V. 42. № 9. P. 1355–1361.
Byun H.-G., Kim Y.-T., Park P.-J., Lin X., Kim S.-K. // Carbohydr. Polym. 2005. V. 61. № 2. P. 198–202.
Kuroiwa T., Izuta H., Nabetani H., Nakajima M., Sato S., Mukataka S., Ichikawa S. // Membrane. 2009. V. 34. № 6. P. 336–341. https://doi.org/10.5360/membrane.34.336
Aider M., Brunet S., Bazinet L. // Sep. Purif. Technol. 2008. V. 63. № 3. P. 612–619.
Lee M-Y., Var F., Shin-ya Y., Kajiuchi T., Jung J.-W. // Process Biochem. 1999. V. 34. № 5. P. 493–500.
Singh S., Packwood J., Samuel C.J., Critchley P., Crout D.H. // Carbohydr. Res. 1995. V. 279. P. 293–305.
Li K., Xing R., Liu S., Li R., Qin Y., Meng X., Li P. // Carbohydr. Polym. 2012. V. 88. № 3. P. 896–903.
Gao X.A., Zhang Y.F., Park R.D., Huang X., Zhao X.Y., Xie J., Jin R.D. // J. Appl. Biol. Chem. 2012.V. 55. № 1. P. 13–17.
Wu H., Aam B.B., Wang W., Norberg A.L., Sorlie M., Eijsink V.G.H., Du Y. // Carbohydr. Polym. 2012. V. 89. № 2. P. 511–518.
Mekasha S., Toupalová H., Linggadjaja E., Tolani H.A., Anděra L., Arntzen M.Ø., Vaaje-Kolstad G., Eijsink V.G.H., Agger J.W. // Carbohydr. Res. 2016. V. 433. P. 18-24. https://doi.org/10.1016/j.carres.2016.07.003
Sørbotten A., Horn S.J., Eijsink V.G., Vårum K.M. // FEBS J. 2005 V. 272. № 2. P. 538–549.
Haebel S., Bahrke S., Peter M.G. // Anal. Chem. 2007. V. 79. № 15. P. 5557–5566.
Le Dévédec F., Bazinet L., Furtos A., Venne K., Brunet S., Mateescu M.A. // J. Chromatogr. A. 2008. V. 1194. № 2. P. 165–171.
Vårum K.M., Holme H.K., Izume M., Stokke B.T., Smidsrød O. // Biochim. Biophys. Acta. 1996. V. 1291. № 1. P. 5–15.
Cederkvist F., Zamfir A.D., Bahrke S., Eijsink V.G.H., Sørlie M., Peter-Katalinic J., Peter M.G. // Angew. Chem. Int. Ed. Engl. 2006. V. 45. № 15. P. 2429–2434.
Bahrke S., Einarsson J.M., Gislason J., Haebel S., Letzel M.C., Peter-Katalinic J., Peter M.G. // Biomacromolecules. 2002. V. 3. № 4. P. 696–704.
Dennhart N., Fukamizo T., Brzezinski R., Lacombe-Harvey M.-E., Letzel T. // J. Biotechnol. 2008. V. 134. № 3–4. P. 253–260.
Okafo G., Langridge J., North S., Organ A., West A., Morris M., Camilleri P. // Anal. Chem. 1997. V. 69. № 24. P. 4985–4993.
Hamer S.N., Moerschbacher B.M., Kolkenbrock S. // Carbohydr. Res. 2014. V. 392. P. 16-20. https://doi.org/10.1016/ j.carres.2014.04.006
Ike M., Ko Y., Yokoyama K., Sumitani J.-I., Kawaguchi T., Ogasawara W., Okada H., Morikawa Y. // J. Mol. Cat. B: Enz. 2007. V. 47. № 3–4. P. 159–163.
Pagnocelli M.G.B., De Araujo N.K., Da Silva N.M.P., De Assis C.F., Rodrigues S., De Macedo G.R. // Braz. Arch. Biol. Technol. 2010. V. 53. № 6. P. 1461–1468.
Sinha S., Chand S., Tripathi P. // Biocatal. Biotransformation. 2014. V. 32. № 4. P. 208–214.
Shehata A.N., Abd El-Aty A.A. // J. Chem. Pharm. Res. 2015. V. 7. № 1. P. 727–740.
Liang T.-W., Lo B.-C., Wang S.-L. // Mar. Drugs. 2015. V. 13. № 8. P. 4576–4593.
Weinhold M.X., Sauvageau J.C.M., Keddig N., Matzke M., Tartsch B., Grunwald I., Kübel C., Jastorff B., Thö-ming J. // Green Chem. 2009. V. 11. № 4. P. 498–509.
Дополнительные материалы отсутствуют.
Инструменты
Прикладная биохимия и микробиология