

Прикладная биохимия и микробиология, 2020, T. 56, № 1, стр. 96-104
Анализ взаимодействия фаговых антител с комплементарными антигенами клеток Herbaspirillum seropedicae Z78 электрооптическим датчиком
О. И. Гулий 1, 2, *, Н. С. Величко 1, Ю. П. Федоненко 1, В. Д. Бунин 3
1 Институт биохимии и физиологии растений и микроорганизмов РАН
410049 Саратов, Россия
2 Саратовский государственный аграрный университет им. Н.И. Вавилова
410012 Саратов, Россия
3 EloSystem GbR
13407 Берлин, Германия
* E-mail: guliy_olga@mail.ru
Поступила в редакцию 06.06.2019
После доработки 23.08.2019
Принята к публикации 30.08.2019
Аннотация
Электрооптическим методом с применением фаговых антител к основным антигенам клеток Herbaspirillum seropedicae Z78 (ЭПСI, КПСI и ЛПС) проведена оценка их комплементарного взаимодействия в системе антиген–антитело. Показано, что электрооптический анализатор позволяет разграничивать присутствие/отсутствие специфичного взаимодействия фаговых антител с основными эпитопами бактериальной поверхности. Выявленные закономерности изменения электрофизических параметров хорошо согласовывались с компонентным составом антигенов Herbaspirillum, их топографическим распределением, а также были подтверждены результатами электронной микроскопии и дот-анализа. Продемонстрирована возможность применения метода электрооптического анализа для детекции Herbaspirillum spp. с экспонированными на их поверхности антигенами. Полученные результаты могут служить основой при создании теста для быстрого определения данных микроорганизмов.
В результате снижения иммунорезистентности населения планеты все чаще возникают инфекции, вызываемые условно-патогенными бактериями. Основными оппортунистическими агентами становятся представители почвенной микрофлоры. Грамотрицательные β-Proteobacteria, включающие Burkholderia, Ralstonia, Herbaspirillum и другие почвенные микроорганизмы, широко представлены в биосфере и играют важную роль в различных областях народного хозяйства [1–3]. Herbaspirillum spp. широко населяет биосферу как в свободноживущем состоянии [1, 4], так и в тканях экономически и агрономически значимых сельскохозяйственных культур [5–7]. Увеличивается количество работ, отмечающих способность Herbaspirillum провоцировать и утяжелять течение септическиех процессов [8–13]. Первоначальная ошибочная идентификации гербаспирилл как представителей Burkholderia spp. была связана с их близким филогенетическим и фенотипическим сходством с Burkholderia cepacia [11, 13]. Диагностика гербаспирилл осложняется отсутствием для них коммерчески доступных тест-систем. В связи с этим улучшение методов детекции Herbaspirillum для создания экспресс-метода оценки бимолекулярной реакции антиген–антитело и их обнаружения представляет интерес с практической точки зрения.
Эффективным инструментом для решения проблем биотехнологии и прикладной микробиологии оказываются методы, основанные на анализе электрофизических параметров клеток в переменном электрическом поле [14–17]. Регистрация оптическим способом изменений электрических характеристик суспендированных клеток носит название электрооптического (ЭО) анализа. Ранее была показана возможность применения метода ЭО анализа для идентификации микробных клеток при их взаимодействии с поликлональными и моноклональными антителами [15, 18, 19]. При развитии ЭО метода дифференциальной диагностики микробных клеток важен подбор соответствующих антител (Ат).
Основными антигенами (Аг) поверхности бактерий являются экзо- и эндотоксины гликополимерной природы. Капсула бактерий содержит капсульные полисахариды (КПС), а микробная слизь – внеклеточные или экстраклеточные полисахариды (ЭПС). К гликополимерам, преобладающим на поверхности у грамотрицательных бактерий, наряду с КПС и ЭПС, относят липополисахариды (ЛПС), формирующие наружный слой внешней мембраны. Капсульные гликаны ряда бактерий представляют собой экстраклеточную форму ЛПС [20, 21], а некоторые бактерии при определенных условиях способны выделять ЛПС в окружающую среду [22]. Все они играют важную роль в иммунохимическом поведении микроорганизмов [23–27].
Традиционно для серологических и иммунохимических исследований используют поликлональные или моноклональные Ат, получение которых включает этап иммунизации животных, который может быть затруднителен при низкой иммуногенности или высокой токсичности Аг. Известно, что для эффективной диагностики достаточно одной антигенсвязывающей области (Fab- или Fv-фрагмента), то есть присутствие Fc-фрагмента Ат необязательно [28]. В качестве альтернативы классической иммунизации можно осуществлять эффективный отбор Ат в формате фагового дисплея [29, 30]. Селекция Аг-связывающих фрагментов Ат (cFv, Fab), представленных на поверхности нитевидного фага, проводится, как и селекция В-лимфоцитов, по способности взаимодействовать с Аг. Подобно плазматическим клеткам, нарабатывающим Ат, бактериальные клетки, инфицированные фагами, в геноме которых содержатся соответствующие гены, способны секретировать растворимые фрагменты Ат, успешно применяемые для идентификации микроорганизмов [14, 31, 32].
Получение фаговых антител (м-АтЭПСI, м-АтКПСI и м-АтЛПС) на основные антигены бактерий рода Herbaspirillum (ЭПСI, КПСI и ЛПС) и их дальнейшее применение для быстрого скрининга бактерий методом электрооптического анализа представляет значительный интерес.
Цель работы – получение м-АтЭПСI, м-АтКПСI и м-АтЛПС к основным антигенам Herbaspirillum seropedicae Z78 (ЭПСI, КПСI и ЛПС) и оценка возможности их применения для детекции Herbaspirillum spp. методом электрооптического анализа.
МЕТОДИКА
Микробные клетки и условия их культивирования. В работе использовали культуры Herbaspirillum seropedicae Z78 (IBPPM217), Azospirillum brasilense Sp245 (IBPPM 219) и Escherichia coli XL-1 (IBPPM 632), полученные из коллекции ризосферных микроорганизмов ИБФРМ РАН (http:// collection.ibppm.ru).
H. seropedicae Z78 выращивали в жидкой синтетической питательной среде с витаминами [33] в течение 24 ч при 30°С, что соответствовало окончанию экспоненциальной фазы роста бактерий. A. brasilense Sp245 и E. coli XL-1 выращивали на жидкой среде LB в течение 18–20 ч при 30°С, как описано [34].
Выделение и очистка ЛПС. Клетки осаждали центрифугированием (3000 g, 40 мин), осадок трижды ресуспендировали в 0.15 М NaCl, механически перемешивали и переосаждали. Экстракцию ЛПС из высушенной ацетоном бактериальной массы проводили горячим 45%-ным водным фенолом по модифицированной методике [35]. ЛПС очищали двукратным ультрацентрифугированием (105000 g, 4 ч). Препараты ЛПС лиофилизировали с использованием Benchtop 2K (“Virtis”, США).
Выделение внеклеточных гликополимеров. Гликополимеры выделяли как описано в работе [33]. Все фракции, содержащие углеводы, которые не поглощали между 240 и 260 нм, объединяли, концентрировали и лиофилизировали. В результате были получены и охарактеризованы экстраклеточный (ЭПСI) и капсульный (КПСI) гликополимеры [33].
Получение фаговых антител. Фаговые антитела к ЛПС, ЭПСI и КПСI получали с использованием овечьей фаговой библиотеки, любезно предоставленной профессором Харрисом (W.J. Harris, Griffin. 1, Великобритания; 1012 вирионов/мл) [36]. В качестве твердой фазы для закрепления Аг использовали полистирольный 96-луночный планшет для иммуноферментного анализа (ИФА). В лунку вносили 200 мкл 1.0 мг/мл соответствующего Аг и инкубировали в течение 12 ч при 4°С. ЛПС, ЭПСI и КПСI связывались с подложкой за счет электростатического взаимодействия. Неспецифичное связывание исключали блокированием в течение 1 ч пространства на стенках планшета, не занятого Аг, 2%-ным раствором БСА в фосфатно-солевом буфере. Затем в лунку вносили 200 мкл фаговой библиотеки в солевом буфере с твином (TBS-T) и после инкубации в течение 12 ч при 4°С планшет трехкратно отмывали этим буфером. Связавшиеся фаговые частицы элюировали 0.1 М глицин-HCl-буфером, pH 2.5, и использовали для инфицирования клеток E. coli штамма XL-1 Blue.
Аликвоты объемом 200 мкл с плотностью клеток E. coli штамм XL 1 Blue не менее 1012 кл./мл высевали на чашки Петри с TYE агаром, в который добавляли антибиотики: тетрациклин, ампицилин и канамицин для предотвращения заражения E. coli хелперными и библиотечными фагами. Клетки культивировали в течение 18 ч при 37°С. Рост E. coli отмечался только на среде с тетрациклином. Отбирали единичную колонию E. coli и высевали на питательную среду 2TY, после чего культивировали в течение ночи на термошейкере для достижения экспоненциальной фазы роста бактерий при 37°С. После этого культуру (из расчета 1 часть культуры на 5 частей среды) высевали на свежую питательную среду 2TY и культивировали в течение 4 ч при 37°С. Культуру помещали в термостат на 30 мин при 37°С без перемешивания для образования F-пилей. Далее E. coli заражали фаговой библиотекой и культивировали в течение 30 мин при 37°С. Затем высевали (из расчета 1 часть культуры на 5 частей среды) на свежую питательную среду 2TY, содержащую 1% глюкозы и 100 мкг/мл ампицилина, и культивировали в течение ночи при 37°С, после чего термостатировали 30 мин при 37°С. В бактериальную культуру, полученную на предыдущем этапе, вносили 200 мкл 1011 вирионов/мл фага М13К07 и инкубировали в течение 1.5 ч при 37°С. Клетки осаждали центрифугированием (1000 g, 10 мин), осадок ресуспензировали в 2TY среде, содержащей 100 мкг/мл ампицилина, 25 мкг/мл канамицина и 1.25 мкл/мл изопропил-β-D-тиогалактозида. Это позволило размножаться клеткам, содержащим как специфичный, так и вспомогательный (хелперный) фаг.
После инкубации клетки центрифугировали (1000 g, 10 мин), а к супернатанту, содержащему фаги, добавляли 1/5 объема ПЭГ/NaCl и инкубировали 1 ч при 4°С. Затем суспензию фаговых антител центрифугировали при 17 000 g 20 мин и 4°С. Полученный осадок ресуспензировали в 3 мл 0.01 M фосфатно-солевого буфера (PBS). Фаговую суспензию диализовали в течение 24 ч против 0.01 M PBS. Полученные фаговые частицы использовали для последующих раундов селекции, осуществляемых в аналогичных условиях, но с уменьшением вдвое концентрации антигенов, как описано в работе [37] для увеличения специфичности фаговых антител. Было проведено 4 раунда селекции бактериофагов, при этом с каждым раундом вдвое уменьшалась концентрация Аг для увеличения специфичности Ат. Начальная концентрация Аг составляла 1 мг/мл.
Для количественной характеристики полученных Ат использовали соотношение (A269–A320) × 5 × × 1014/15, где A320 – оптическая плотность суспензии при 320 нм и A269 – при 269 нм. Концентрацию фаговых частиц определяли из расчета A269‒A320 = 30 оптических единиц, что соответствовало 2 × 1014 вирионам/мл [38]. Концентрация фаговых антител составила 1.2 × 1013, 2.2 × × 1013 и 2.6 s× 1013 вирионов/мл для ЛПС, КПСI и ЭПСI соответственно.
Титр сыворотки определяли с помощью ИФА по общепринятой методике [39]. Измерение проводили на Specord BS-250 UV (“Analytic Jena”, Германия) при 490 нм. Титр полученных фаговых мини-Ат составил 1 : 4000.
Дот-иммуноанализ. Для дот-анализа использовали мембрану Western S (“Sigma-Aldrich”, США) и культуру клеток H. seropedicae Z78 с концентрацией 1.06 × 1011 кл./мл (по эталону мутности), как описано ранее [14].
Просвечивающая электронная микроскопия (ПЭМ). Подготовку препаратов для электронно-микроскопических снимков осуществляли при помощи просвечивающего электронного микроскопа Libra 120 (“CarlZeiss”, Германия) при ускоряющем напряжении 120 кВ, как описано в работе [34], в Центре коллективного пользования научным оборудованием, применяемым в области физико-химической биологии и нанобиотехнологии “Симбиоз” ИБФРМ РАН.
Проведение электрооптического анализа клеточных суспензий. Перед проведением ЭО осуществляли трехкратную отмывку клеток всех используемых штаммов от культуральной среды центрифугированием (2800 g, 5 мин). Для этого использовали дистиллированную воду с электропроводностью 1.6–1.8 μS/см. Электропроводность определяли с помощью кондуктометра HI 8733 (“HANNA”, США). Полученную суспензию центрифугировали (1000 g, 1 мин) для устранения конгломератов. Оптическую плотность при А670 полученного супернатанта доводили дистиллированной водой до значения ~0.42–0.45, что соответствовало 4.5 × 108 и 1.6 × 109 кл./мл для A. brasilense Sp245 и H. seropedicae Z78 соответственно. Ориентационные спектры клеток измеряли на электрооптическом анализаторе ELOAnalyser (“EloSystemGbR”, Германия). Параметры измерения: напряженность электрического поля (E) 93.1 В/см, длина волны света 670 нм (относительно вакуума), время (t) приложения электрического поля 3.0 с. Объем измерительной ячейки составлял 1 мл. В экспериментах оперировали дискретным набором частот (f) ориентирующего электрического поля: 740, 1000, 1450, 2000 и 2800 кГц.
Логарифмическая зависимость разности значений оптической плотности суспензии (δА), измеренной при распространении пучка неполяризованного света (I) вдоль и поперек направления электрического ориентирующего поля, была получена при последовательном изменении частоты радиоимпульсов. Эта разность была нормирована на значение оптической плотности при хаотической ориентации клеток [16].
Для оценки взаимодействия антигенных детерминант микробных клеток с фаговыми антителами в измерительную ячейку к 1 мл суспензии H. seropedicae Z78 (1.6 × 109 кл./мл) вносили 3 мкл м-Ат (1.2 × 1013, 2.2 × 1013 и 2.6 × 1013 вирионов/мл для ЛПС, КПСI и ЭПСI соответственно) и измеряли ЭО сигнал. При проведении исследований с клетками A. brasilense Sp245 условия экспериментов были аналогичными с H. seropedicae Z78, однако количество клеток A. brasilense Sp245 в измерительной ячейке составляло 4.5 × 108 кл./мл.
Для каждой серии экспериментов проводили не менее пяти измерений. Анализ и представление данных осуществляли при помощи программы Microsoft Excel 2010. Доверительные интервалы определены для надежности 95%.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Клеточная оболочка бактерий представляет собой сложную структуру с экспонированными на поверхности углеводными остатками, защищающую и в то же время осуществляющую взаимодействие клетки с окружающей внешней средой. Многие микроорганизмы образуют капсулы вокруг своих клеточных стенок и выделяют специфическую слизь, в которой происходят межклеточные взаимодействия. Бактериальные капсулы содержат КПС, тогда как микробная слизь состоит из ЭПС. Структурное разнообразие позволяет ЭПС и КПС функционировать в клеточных взаимодействиях в качестве сигнальных молекул [23, 40, 41]. Наряду с КПС и ЭПС, к основным поверхностным антигенам относятся ЛПС. Они участвуют в процессах адгезии, механизмах узнавания чужеродных объектов и индукции защитных реакций макропартнера. Однако данные, касающиеся состава экстраклеточных, капсульных и мембранных гликополимеров Herbaspirillum, фрагментарны [42, 43].
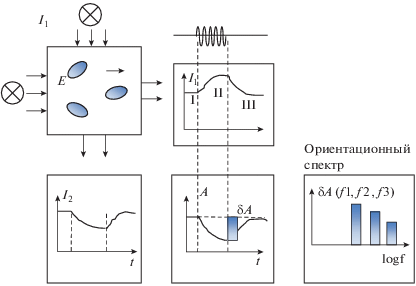
Для изучения различной экспонированности углеводных антигенных детерминант бактериальной поверхности с помощью метода ЭО анализа использовали штамм H. seropedicae Z78. Суть метода ЭО анализа микробных суспензий заключалась в измерении вариаций их оптических свойств при воздействии на суспендированные клетки переменного электрического поля. Электрическое поле на границах смежных клеточных структур вызывало появление в нем индуцированных зарядов. Их величина зависела от электрических свойств смежных структур, и, в частности, от параметров слоя поверхностного взаимодействия [15]. Регистрируемый ЭО сигнал можно охарактеризовать двумя параметрами: его стационарной величиной после завершения ориентационных процессов ΔА и формой релаксационной кривой (рис. 1).
Рис. 1.
Схема измерения оптической плотности микробной суспензии в процессе ЭО-анализа в моменты приложения электрического поля (I, хаотическая ориентация клеток), его выключения (II, клетки в ориентированном состоянии) и возвращения клеток в состояние с хаотической ориентацией (III).
Ранее было показано, что бактерии H. seropedicae Z78 выделяют два экстраклеточных гликополимера липополисахаридной природы. Один из них сохраняет связь с поверхностью клетки и обнаруживается в капсуле – КПСI, а второй, ЭПСI, выделяется бактериями в культуральную жидкость. При этом эти полимеры отличаются как составом моносахаридов, так составом и соотношением жирных кислот [33]. Детальная характеристика КПСI, ЭПСI и ЛПС H. seropedicae Z78 представлена в работах [33, 35].
В предыдущей работе было показано, что поликлональные кроличьи Ат к очищенному препарату ЛПС не проявляли специфичности ни к одному из изученных Аг, в том числе гомологичному [44]. В связи с трудностями, возникшими при получении поликлональных кроличьих Ат, была проведена селекция фаговых антител к очищенным индивидуальным препаратам ЭПСI, КПСI и ЛПС клеток H. seropedicae Z78 (м-АтЭПСI, м-АтКПСI и м-АтЛПС). Их подробные характеристики приведены в работе [44].
Увеличение специфичности фаговых антител оценивали с помощью ИФА. На одном уровне с гомологичным Аг взаимодействовали м-АтЛПС. Также отмечалось их взаимодействие с КПСI. ЭПСI отличались очень слабым (почти на уровне контроля) взаимодействием с м-АтЛПС. В то время как м-АтЭПСI взаимодействовал со всеми выделенными гликополимерами H. seropedicae Z78, при этом реакция с гомологичным Аг оказалась слабее, чем с КПСI, а с ЛПС – сильнее, чем КПСI.
Было показано, что м-АтКПСI взаимодействовали со всеми Аг H. seropedicae Z78, однако максимально с ЛПС. Результаты анализа полученных данных указывали на сложную природу полисахаридсодержащих Аг поверхности H. seropedicae Z78. Можно предположить, что ЛПС и КПСI обладают индивидуальными антенными детерминантами, которые узнаются м-АтЛПС, но отсутствуют в ЭПСI.
В предыдущей работе [15] была показана общая закономерность, заключающаяся в неизменности электрооптических параметров клеточной суспензии при отсутствии специфичного взаимодействия действующего агента с бактериальными клетками. В то же время связывание агента со строго определенной антигенной детерминантой клеток приводила к выраженному изменению величины электрооптического сигнала [15]. Тем не менее не было очевидным получение аналогичных закономерностей изменения ЭО-параметров при использовании фаговых антител, специфичных к различным антигенным детерминантам (ЭПСI, КПСI и ЛПС) микробных клеток.
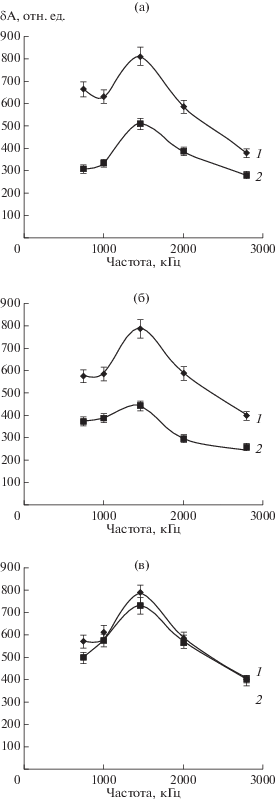
ЭО-датчиком оценивали взаимодействие антигенных детерминант клеток H. seropedicae Z78 с соответствующими фаговыми Ат. При анализе полученных данных решающей является разница величины ЭО-сигнала между контролем (клетки без добавления специфичных Ат) и экспериментом (клетки с добавлением Ат). Показано (рис. 2а), что при взаимодействии клеток H. seropedicae Z78 с м‑А-тЛПС величина ЭО-сигнала изменялась на 79% по сравнению с контролем (клетки без добавления Ат). Для удобства расчета полученных результатов использовали величины значений ЭО-сигнала суспензии клеток при частоте ориентирующего поля 1000 кГц.
Рис. 2.
Изменение величины ЭО-сигнала суспензии клеток H. seropedicae Z78 при взаимодействии с м‑АтЛПС (а), м-АтКПСI (б) и м-АтЭПСI (в): 1 (контроль) – суспензии клеток без добавления м-Ат ; 2 – после добавления м-Ат.
Помимо электрооптических данных об аффинности взаимодействия клеток H. seropedicae Z78 с м-АтЛПС методом электронной микроскопии визуализировали образование иммунных комплексов. Результаты, приведенные на рис. 3б, подтвердили высокий уровень взаимодействия м-АтЛПС с клетками данного штамма. Дополнительно на рис. 3а представлена фотография клеток H. seropedicae Z78 без Ат.
Рис. 3.
ПЭМ клеток H. seropedicae Z78 (×10 000) без Ат (а) и после инкубации с м-АтЛПС (б), м-АтКПСI (в) и м-АтЭПСI (г), меченными коллоидным золотом.
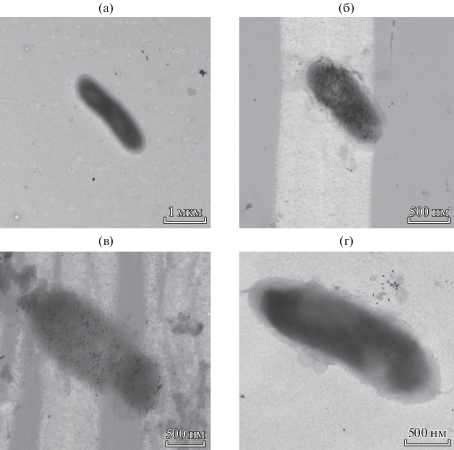
При изучении взаимодействия м-АтКПСI с клетками H. seropedicae Z78 (рис. 2б) было зафиксировано изменение величины ЭО-сигнала на 66% по сравнению с контролем (клетки без добавления Ат), то есть м-АтКПСI взаимодействовали с соответствующими Аг детерминантами клеточной поверхности. Электронно-микроскопический анализ также выявил высокую степень взаимодействия м-АтКПСI с клетками H. seropedicae Z78 (рис. 3в).
При оценке взаимодействия м-АтЭПСI с клетками H. seropedicae Z78 при использовании значений на частотах 740 и 1000 кГц были зафиксированы изменения ЭО-параметров в диапазоне 4%, а на остальных частотах их изменений практически не наблюдали (рис. 2в). Можно предположить, что это связано со структурными различиями мембранных и экстраклеточных липополисахаридов. На рис. 3г представлены результаты электронной микроскопии, демонстрирующие отсутствие взаимодействия клеток с м-АтЭПСI. Полисахаридные части ЛПС и КПСI состояли, помимо галактозы, глюкозы и глюкозамина, из остатков полиспиртов эритрита и глицерина. В то время как в составе ЭПСI помимо указанных сахаров присутствовали только остатки глицерина. Мажорным компонентом ЭПСI была глюкоза [33]. В отличие от ЛПС и КПСI, расположенных вблизи и на поверхности внешней мембраны клеточной стенки, ЭПСI секретируется бактериальной клеткой в окружающую среду, поэтому антигенные детерминанты, соответствующие этому полисахариду, могут полностью отсутствовать на клеточной поверхности.
На основании ранее проведенных измерений, регистрирующих взаимодействия Ат с микробными клетками, можно было ориентировочно определить критерий специфичного взаимодействия как изменение величины ЭО сигнала суспензии клеток на величину не менее ~5% при добавлении в суспензию клеток м-Ат. При анализе полученных данных решающей становится разница величин ЭО-сигнала между контролем (клетки без добавления специфичных м-Ат) и экспериментом (клетки с добавлением м-Ат). Данные, представленные на рис. 2, показали, что при взаимодействии м-АтЭПСI с клетками H. seropedicae Z78 изменения ЭО-параметров зафиксировались в диапазоне 4% при использовании значений на частотах 740 и 1000 кГц, а на остальных частотах они практически отсутствовали. Для исключения ложноположительных результатов на основании ранее проведенных многократных измерений с привлечением микробных клеток других штаммов, был выработан критерий специфического взаимодействия, как изменение величины ЭО-сигнала суспензии клеток на величину не менее ~5% при внесении в суспензию специфичных антител. Указанный порог разграничивал лишь конкретные иммунохимические реакции, изученные в этой работе. Данный критерий может быть применен для тех препаратов, в которых распознаваемые бактерии присутствовали в смеси с другими бактериями или иными примесями, влияющими на А670 и, соответственно, определяющими их разведение. Необходимо отметить, что для определения критерия следовало принимать во внимание значение сигнала не на одной частоте, а на всем используемом наборе частот.
На следующем этапе проверялась активность м-АтЛПС, м-АтКПСI и м-АтЭПСI по отношению к представителю других почвенных азотфиксирующих бактерий A. brasilense Sp245. Выбор этой культуры был обусловлен достаточно полным описанием ее серологических и иммунохимических свойств, что позволило корректно интерпретировать полученные результаты.
Специфичные изменения ЭО-параметров клеточных суспензий штамма Sp245 при воздействии м-АтЛПС, м-АтКПСI и м-АтЭПСI не были зафиксированы. Сравнение результатов ЭО- и дот-иммуноанализа показало совпадение данных, полученных двумя независимыми методами (табл. 1).
Таблица 1.
Выявление взаимодействия м-Ат с клетками методами дот-анализа и электрооптического анализа суспензий микробов
| м-Ат | Микроорганизмы | |||
|---|---|---|---|---|
| H. seropedicae Z78 | A. brasilenseSp245 | |||
| дот-анализ | ЭО-анализ | дот-анализ | ЭО-анализ | |
| м-АтЛПС | +* | + | – | – |
| м-АтКПСI | + | + | – | – |
| м-АтЭПСI | – | – | – | – |
Поскольку данные ЭО-анализа микробных суспензий были подтверждены стандартными методами дот-анализа и электронной микроскопией, можно было утверждать, что м-АтЛПС и м‑АтКПСI взаимодействовали с клетками H. seropedicae Z78, а м-АтЭПСI нет. При этом все изученные м-Ат не взаимодействовали с клетками A. brasilense Sp245.
В предыдущей работе [44] было показано, что в моносахаридном составе ЛПС и КПСI присутствуют рамноза, глюкозамин и галактозамин, которые могут входить и в состав АгД этих гликанов. Это объясняет взаимодействие м-АтЛПС, м‑АтКПСI не только с гомологичными Аг, но и перекрестные реакции с ЛПС и КПСI. Можно предположить, что из-за отсутствия в составе ЭПСI рамнозы и присутствия глюкозамина и галактозамина лишь в следовых количествах не наблюдалось их реакции с м-АтЛПС, а взаимодействие м-АтЭПСI с ЛПС и КПСI объяснялось присутствием в составе указанных Аг глюкозы и галактозы. На основании всего выше изложенного можно предположить, что м-АтЛПС должны проявлять большую специфичность к детерминантам, содержащим рамнозу, глюкозамин и галактозамин, м-АтЭПСI – к детерминатам, содержащим глюкозу и галактозу, а м-АтКПСI – к детерминантам, несущим в равной степени и те, и другие сахара.
В работе на примере фаговых антител (м-АтЭПСI, м-АтКПСI и м-АтЛПС) к различным антигенным детерминантам клеточной поверхности H. seropedicae Z78 показана возможность регистрации ЭО-датчиком взаимодействия Ат с комплементарными антигенами (ЭПСI, КПСI и ЛПС). Установлено, что специфичные изменения ЭО-параметров клеточных суспензий при их взаимодействии с фаговыми Ат происходили только у микробных клеток, обладающих соответствующими Аг-детерминантами. Таким образом, ЭО-анализатор позволил отличать ситуации, когда бактериальные клетки взаимодействовали со специфичными фаговыми Ат, и контрольные эксперименты, когда такое взаимодействие отсутствовало. Следует отметить, что с помощью предлагаемого анализа детектировались именно микробные клетки с экспонированными на их поверхности антигенами, а не антигены.
Принципиальными преимуществами предлагаемого подхода являются его оперативность (время анализа составляло около 5 мин), отсутствие необходимости дополнительной иммобилизации компонентов и возможность многократного использования датчика.
Адаптация электрооптического метода для анализа потенциально опасных для человека микроорганизмов открывает возможность скрининогового выявления идентичных антигенных детерминант в образцах, структуры которых еще не были изучены, и соотносить их с образцами полисахаридов известного строения. Результаты представляют интерес с практической точки зрения для создания экспресс-метода оценки бимолекулярной реакции Аг–Ат и могут служить основой при создании теста для быстрого обнаружения бактерий.
Список литературы
Baldani J.I., Baldani V.L.D., Seldin L., Dobereiner J. // Int. J. Syst. Bacteriol. 1986. V. 36. № 1. P. 86–93.
Falk E.C., Johnson J.L., Baldani V.L.D., Dobereiner J., Krieg N.R. // Int. J. Syst. Bacteriol. 1986. V. 36. № 1. P. 80–85.
Pedrosa F.O., Benelli E.M., Yates M.G., Wassem R., Monteiro R.A., Klassen G., Steffens M.B., Souza E.M., Chubatsu L.S., Rigo L.U. // J. Biotechnol. 2001. V. 91. № 1–2. P. 189–195.
Ding L., Yokota A. // Int. J. Syst. Evol. Microbiol. 2004. V. 54. № 6. P. 2223–2230.
James E.K., Olivares F.L. // Crit. Rev. Plant Sci. 1997. V. 17. № 1. P. 77–119
Boddey R.M., de Oliveira O.C., Urquiaga S., Reis V.M., Olivares F.L., Baldani V.L.D., Döbereiner J. // Plant Soil. 1995. V. 174. № 1–2. P. 195–209.
Rothballer M., Schmid M., Klein I., Gattinger A., Grudmann S., Hartmann A. // Int. J. Syst. Evol. Microbiol. 2006. V. 56. № 6. P. 1341–1348.
Baldani J.I., Pot B., Kirchhof G., Falsen E., Baldani V.L., Olivares F.L., Hoste B., Kersters K., Hartmann A., Gillis M., Döbereiner J. // Int. J. Syst. Bacteriol. 1996. V. 46. № 3. P. 802–810.
Tan M., Oehler R. // Infect. Dis. Clin. Pract. 2005. V. 13. № 5. P. 277–279.
Coenye T., Goris J., Spilker T., Vandamme P., LiPuma J.J. // J. Clin. Microbiol. 2002. V. 40. № 6. P. 2062–2069.
Spilker T., Uluer A.Z., Marty F.M., Yeh W.W., Levison J.H., Vandamme P., Lipuma J.J. // J. Clin. Microbiol. 2008. V. 46. № 8. P. 2774–2777.
Chen J., Su Z., Liu Y., Sandoghchian S., Zheng D., Wang S., Xu H. // Curr. Microbiol. 2011. V. 62. № 1. P. 331–333.
Ziga E.D., Druley T., Burnham C.A. // J. Clin. Microbiol. 2010. V. 48. № 11. P. 4320–4321.
Dykman L.A., Staroverov S.A., Guliy O.I., Ignatov O.V., Fomin A.S., Vidyasheva I.V., Karavaeva O.A., Bunin V.D., Burygin G.L. // J. Immunoassay. Immunochem. 2012. V. 33. № 2. P. 115–127.
Guliy O.I., Bunin V.D., Korzhenevich V.I., Ignatov O.V. // Curr. Immun. Rev. 2017. V. 13. № 2. P. 153–162.
Bunin V.D., Voloshin A.G. // J. Colloid. Interface Sci. 1996. V. 180. № 1. P. 122–126.
Мирошников А.B., Фомченков В.М., Иванов А.Ю. Электрофизический анализ и разделение клеток. М.: Наука, 1986. 185 с.
Bunin V.D., Ignatov O.V., Guliy O.I., Voloshin A.G., Dykman L.A., O’Neil D., Ivnitski D. // Biosens. Bioelectron. 2004. V. 19. № 2. P. 1759–1761.
Guliy O.I., Matora L.Yu., Burygin G.L., Dykman L.A., Ostudin N.A., Bunin V.D., Ignatov V.V., Ignatov O.V. // Anal. Biochem. 2007. V. 370. № 2. P. 201–205.
Konnova S.A., Makarov O.E., Skvortsov I.M., Ignatov V.V. // FEMS Microbiol. Lett. 1994. V. 118. № 2. P. 93–99.
Смолькина О.Н., Качала В.В., Федоненко Ю.П., Бурыгин Г.Л., Здоровенко Э.Л., Матора Л.Ю., Коннова С.А., Игнатов В.В. // Биохимия. 2010. Т. 75. № 5. С. 707–716.
Здоровенко Г.М. // Микробиол. журн. 1988. Т. 50. № 4. С. 98–107.
Оводов Ю.С. // Биохимия. 2006. Т. 71. № 9. С. 1155–1174.
Hols O., Ulme A.J., Brade H., Fla H-D., Rietsche E.T. // FEMS Immunol. Med. Microbiol. 1996. V. 16. № 2. P. 83–104.
Mora L., Newton W.E. New Horizons in Nitrogen fixation / Ed. B. Eckert. Dordrecht: Kluwer acad. pub., 2008. 705 p.
Saunder N.J., Pede J.F., Hoo D.W., Moxon E.R. // Mol. Microbiol. 1998. V. 27. № 6. P. 1091–1098.
Kannenberg E.L., Carlson R.W. // Mol. Microbial. 2001. V. 39. № 2. P. 379–391.
Глик Б., Пастернак Дж. Молекулярная биотехнология. Принципы и применение. М.: Мир, 2002. 589 с.
Smith G.P., Petrenko V.A. // Chem. Rev. 1997. V. 97. № 2. P. 391–410.
McCafferty J., Griffiths A.D., Winter G., Chiswell D.J. // Nature. 1990. V. 348. № 6301. P. 552–554.
Nanduri V., Sorokulova I.B., Samoylov A.M., Simonian A.L., Petrenko V.A., Vodyanoy V. // Biosens. Bioelectron. 2007. V. 22. № 6. P. 986–992.
Paoli G.C., Chen C.Y., Brewster J.D. // Immunol. Methods. 2004. V. 289. № 1–2. P. 147–155.
Смолькина О.Н., Шишонкова (Величко) Н.С., Юрасов Н.А., Игнатов В.В. // Микробиология. 2012. Т. 81. № 3. С. 345–352.
Guliy O.I., Zaitsev B.D., Borodina I.A., Shikhabudinov A.M., Teplykh A.A., Staroverov S.A., Fomin A.S. // Talanta. 2018. V. 178. P. 569–576.
Величко Н.С., Суркина А.К., Федоненко Ю.П., Здоровенко Э.Л., Коннова С.А. // Микробиология. 2018. Т. 87. № 5. С. 511–518.
Charlton K.A., Moyle S., Porter A.J., Harris W.J. // J. Immunol. 2000. V.164. № 9-12. P. 6221–6229.
Griep R.A., van Twisk C., van Beckhoven J.R., van der Wolf J.M., Schots A. // Phytopathology. 1998. V. 88. № 8. P.795–803.
Smith G.P., Scott J.K. // Meth. Enzymol. 1993. V. 217. P. 228–257.
Beatty J.D., Beatty B.G. and Vlahos W.G. // J. Immunol. Methods. 1987. V. 100. № 1–2. P. 173–179.
Jiang C.H., Fan Z.H., Xie P., Guo J.H. // Front. Microbiol. 2016. V. 7:664. https://doi.org/10.3389/fmicb.2016.00664
Maruzani R., Sutton G., Nocerino P., Marvasi M. // J. Microbiol. 2019 V. 57. № 1. P. 1–8.
Serrato R.V., Sassaki G.L., Cruz L.M., Carlson R.W., Muszyński A., Monteiro R.A., Pedrosa F.O., Souza E.M., Iacomini M. // Can. J. Microbiol. 2010. V. 56. № 4. P. 342–347.
Balsanelli E., Tuleski T.R., de Baura V.A., Yates M.G., Chubatsu L.S., Pedrosa Fde O., de Souza E.M., Monteiro R.A. // PLoS One. 2013. V. 8. № 10. P. e77001. https://doi.org/10.1371/journal.pone.0077001
Velichko N.S, Fedonenko Yu.P. // Ann. Microbiol. 2019. V. 69. № 7. https://doi.org/10.1007/s13213-019-01490-7
Дополнительные материалы отсутствуют.
Инструменты
Прикладная биохимия и микробиология