

Прикладная биохимия и микробиология, 2020, T. 56, № 3, стр. 211-222
Механизмы иммуномодулирующей и мембранотропной активности трегалолипидных биосурфактантов (обзор)
М. С. Куюкина 1, 2, *, О. А. Кочина 1, С. В. Гейн 1, 2, И. Б. Ившина 1, 2, В. А. Черешнев 1, 2, 3
1 Институт экологии и генетики микроорганизмов,
Пермский федеральный исследовательский центр УрО РАН
614081 Пермь, Россия
2 Пермский государственный национальный исследовательский университет
614990 Пермь, Россия
3 Институт иммунологии и физиологии УрО РАН
620049 Екатеринбург, Россия
* E-mail: kuyukina@iegm.ru
Поступила в редакцию 04.10.2019
После доработки 20.12.2019
Принята к публикации 23.12.2019
Аннотация
Среди гликолипидых биосурфактантов, которые могут быть использованы в медицине, интенсивно исследуются трегалолипиды актинобактерий рода Rhodococcus, менее токсичные по сравнению с микобактериальными ди- и мономиколатами трегалозы. Иммуномодулирующая активность трегалолипидов обусловлена их взаимодействием с поверхностными рецепторами на мембране иммунных клеток и запуском сигнальной системы активации продукции цитокинов, активных форм кислорода, антителогенеза. Благодаря амфифильной природе трегалолипиды воздействуют на липидный бислой клеточных мембран, изменяя его проницаемость. Рассмотренные молекулярные механизмы взаимодействия трегалолипидов с клетками иммунной системы важны для разработки подходов к иммунотерапии.
Поверхностно активные вещества (ПАВ) – это соединения, которые снижают поверхностное натяжение двухфазных систем (газ/жидкость, жидкость/жидкость, твердая фаза/жидкость) [1]. ПАВ, сурфактанты, имеют широкое применение в различных областях промышленности. Однако большинство из них являются загрязнителями окружающей среды, а также дороги в производстве [2–4], поэтому в последние годы наблюдается повышенный интерес к биогенным сурфактантам. Биосурфактанты имеют ряд преимуществ по сравнению с синтетическими аналогами, а именно: высокую биодеградабельность, отсутствие токсичности, термо- и рН-стабильность и биологическую активность [6].
Благодаря уникальным физико-химическим свойствам биосурфактанты проявляют биологическую активность, не обнаруживаемую у синтетических аналогов, например, антимикробную, иммуномодулирующую и противоопухолевую [2, 5, 7].
Среди изученных биосурфактантов наиболее интересны в биомедицинском применении гликолипиды [7]. Гликолипидные биосурфактанты имеют общую структуру: гидрофобная часть представлена длинной цепочкой жирных кислот (они могут быть разветвленными, гидроксилированными, ненасыщенными, циклосодержащими), а гидрофильный углеводный компонент может быть представлен остатком трегалозы, глюкозы, галактозы, софорозы, рамнозы [8]. Различаются гликолипиды также по количеству углеродных атомов в составе своих цепей (С20–С90) [9].
Первый микробный гликолипид был описан в 1933 г. при изучении туберкулезной палочки Mycobacterium tuberculosis [10]. В 1956 г., была установлена химическая структура “корд-фактора” M. tuberculosis, представляющего собой смесь трегалозо-6,6-дикориномиколатов (ТДМ) общей формулы C186H366O17 ± 10CH2 [11]. Поскольку трегалоза также обладает биологической активностью, являясь частью трегалолипидов, она способствует ее проявлению [5]. Позднее “корд-фактор” и “лизо-корд-фактор”, представленный монокориномиколатом трегалозы, были обнаружены в клетках других патогенных микобактерий (включая, так называемых, “мягких” оппортунистических патогенов группы Mycobacterium avium – M. intracellulare, нокардий (Nocardia asteroides) и коринебактерий (Corynebacterium diphtheriae, C. matruchotii, C. xerosis) [12–15]. Однако явная или потенциальная патогенность штаммов-продуцентов, а также высокая токсичность синтезируемых гликолипидов существенно ограничивали их биомедицинское применение [16]. В связи с этим был проведен поиск новых продуцентов гликолипидных сурфактантов среди непатогенных представителей группы “миколат”, то есть актинобактерий, содержащих в составе клеточных липидов высокомолекулярные α-разветвленные β-гидроксилированные жирные (миколовые) кислоты [17], в частности представителей родов Rhodococcus, Gordonia, Dietzia, Tsukamurella, Skermania, Williamsia и др. [9]. Наиболее интенсивно в последние годы исследуются трегалолипидные биосурфактанты, синтезируемые родококками [9, 18, 19]. Следует отметить, что среди родов актинобактерий, входящих в семейства Corynebacteriaceae, Dietziaceae, Gordoniaceae, Mycobacteriaceae, Nocardiaceae и Tsukamurellaceae, объединенные в подпорядок Corynebacterineae [20], род Rhodococcus характеризуется минимальным числом патогенных и условно патогенных видов и, следовательно, представляют интерес в качестве промышленных продуцентов биосурфактантов [21].
Трегалолипиды родококков характеризуются чрезвычайно высоким структурным разнообразием, они часто представлены комплексами, состав которых варьирует в зависимости от физиологии и условий роста бактериальной культуры [18]. Кроме ТДМ, моно- и тримиколата трегалозы также описаны другие моно-, ди-, тетра-, гекса-, окта-ацилированные производные трегалозы, составляющие большую группу неионогенных трегалолипидов [22].
Исследуя связь между структурой молекулы и токсичностью гликолипидных биосурфактантов, Б. Мюнстерманн с соавт. показали с помощью биотестирования, что ТДМ и трегало-тетраэфиры из Rhodococcus erythropolis были менее токсичными для Photobacterium fischeri, чем рамнолипиды из Pseudomonas aeruginosa и синтетические ПАВ [23]. Позднее в работе [24] было показано, что неочищенный гликолипидный комплекс из Rhodococcus ruber был менее токсичен, чем все биосурфактанты, приведенные в предыдущей работе. Низкая токсичность гликолипидов из Rhodococcus spp. была выявлена и в отношении высших организмов. Так трегалолипиды, выделенные из штамма R. ruber ИЭГМ 231, не влияли на смертность, изменение массы тела или поведение беспородных белых мышей-самцов и не оказывали цитотоксического действия на лимфоциты человека [25, 26]. Трегалолипиды из R. erythropolis 51T7 были менее токсичны для мышиных фибробластов и кератиноцитов человека, и, следовательно, вызывали меньшее раздражение кожи, чем коммерческий синтетический сурфактант додецилсульфат натрия [27]. Сукциноил-трегалолипид из R. erythropolis CD-74 не оказывал токсичного воздействия на нормальные фетальные клетки легких человека, но заметно ингибировал рост лейкозных моноцитоидных клеток, что свидетельствует о его избирательной цитотоксичности в отношении раковых клеток [28]. В тестах на токсичность in vivo показано, что ТДМ из Rhodococcus sp. 4306 был в 40 раз менее токсичен для мышей, чем ТДМ из M. tuberculosis, предположительно, вследствие значительно меньшей длины углеродной цепи миколовых кислот (С34–С38 против С74–C86) [29]. Позднее группой авторов [30] было высказано предположение, что длина молекулы миколовых кислот в ТДМ и трегалозомономиколате (ТММ) из Rhodococcus sp. 4306 является ключевым фактором, определяющим более низкую токсичность и меньшую грануломатогенную активность по сравнению с ТДМ из микобактерий [30].
Влияние трегалолипидов на реакции врожденного и адаптивного иммунитета. Известно, что клетки врожденного иммунитета представляют собой основную мишень для бактериальных антигенов. Однако в литературе встречаются данные о влиянии микробных и синтетических трегалолипидов на клетки врожденного иммунитета в системах in vitro и in vivo, которые существенно различаются по своей направленности. Так в опытах in vitro гликолипидные биосурфактанты актинобактерий стимулировали продукцию цитокинов IFN-γ, IL-6, TNF-α, IL-12 и IL-10 мононуклеарами и моноцитами периферической крови [18, 25, 31]. Также показана способность биосурфактантов R. ruber модулировать цитокинпродуцирующую и поглотительную функции гранулоцитов [29, 32–34].
Под влиянием ТДМ из M. tuberculosis было зарегистрировано повышение микробицидной активности, а именно продукции NO в супернатантах мышиных клеток [31]. Аналогичный стимулирующий эффект на продукцию активных форм кислорода в спонтанных культурах лейкоцитов и гранулоцитов человека оказывал гликолипидный комплекс из R. ruber ИЭГМ 231 [34]. Результаты работы [35] также указывают на усиление продукции гранулоцитарного колониестимулирующего фактора и NO макрофагами в ответ на присутствие компонентов клеточной стенки коринебактерий. Трегалолипиды активировали цитокинпродуцирующую функцию дендритных клеток [36], тем самым индуцируя их противоопухолевую активность [28, 37, 38].
В то же время, в системе in vivo трегалолипид из R. ruber ИЭГМ 231 оказывал угнетающее влияние на динамику респираторного взрыва и продукцию цитокинов перитонеальными клетками мышей при внутрибрюшинном и внутримышечном способах введения [39].
Влияние гликолипидов на адаптивный иммунитет в большей степени является опосредованным, в первую очередь через активацию клеток врожденного иммунитета. Тем не менее, бактериальные гликолипиды модулируют антителогенез, выраженность реакции гиперчувствительности замедленного типа (ГЗТ), продукцию Th1/Th2-цитокинов (IL-2, IL-4, IFN-γ) [40–42].
Таким образом, представленные данные свидетельствуют об активирующем влиянии трегалолипидов на функциональную активность макрофагов в системе in vitro. Однако направленность их иммуномодулирующего действия в значительной степени различается в моделях in vitro и in vivo. Так, в системе in vivo гликолипиды могут проявить иммуносупрессорные свойства, что, по-видимому, зависит от их химической структуры и агрегатного состояния биосурфактантных мицелл, а также процессов межклеточной кооперации и особенностей взаимодействия с мембраной иммунокомпетентных клеток. По литературным данным, биосурфактанты, имеющие в составе остатки трегалозы и цепи миколовых кислот, контактируют с клетками врожденного иммунитета через ряд поверхностных рецепторов. Распознавание этих гликолипидов осуществляется рецепторами Mincl и MCL, экспрессирующимися на клеточных мембранах гранулоцитов. При контакте рецептора с микробным гликолипидом запускается сигнальная система, приводящая к активации клеток, продукции цитокинов, активных форм кислорода, усилению поглотительной активности гранулоцитов. Опосредованно может изменяться пролиферативная активность лимфоцитов и продукция ими цитокинов.
Рецепторы трегалолипидных биосурфактантов. Паттерн-распознающие рецепторы (PRRs) играют важную роль во врожденном иммунитете. Они способны к распознаванию специфических молекулярных структур, таких как: самоассоциированные молекулярные фрагменты (SAMPs), молекулярные фрагменты, ассоциированные с повреждениями (DAMPs), микробные молекулярные фрагменты (MAMPs) и патоген-ассоциированные фрагменты (PAMPs) [43]. К группе PRRs относятся Толл-подобные рецепторы (TLRs), внутриклеточные нуклеотид-связывающие NOD-подобные рецепторы (NLRs), RIG-I-подобные рецепторы и лектиновые рецепторы С-типа (CLRs) [44, 45]. Одновременный запуск нескольких PRRs вызывает внутриклеточный каскад реакций, приводящих к активации врожденного иммунитета, необходимого для формирования эффективного адаптивного иммунного ответа [46].
CLRs представляют большое семейство консервативных рецепторов позвоночных животных, которые связывают остатки углеводов [47]. Они распознают различные эндогенные и экзогенные лиганды патогенов, таких как грибы, бактерии и паразиты [48].
Макрофагальный индуцибельный лектин С-типа (Mincle или CLEC4e, Clecsf9) и макрофагальный лектин С-типа (MCL или CLEC4d, ClecSf8) являются рецепторами TДM [49]. Данные рецепторы относятся к отдельной группе лектинов С-типа, так как для осуществления связывания лиганда в распознающем углевод домене им необходимо присутствие ионов Ca2+ [47].
Mincle-рецептор. Впервые рецептор Mincle был идентифицирован в работе [50] как белок, который экспрессируется в ответ на стимуляцию липополисахаридом (ЛПС) и некоторыми провоспалительными цитокинами, такими как IFN-γ, IL-6, и TNF-α. Он относится ко II типу трансмембранных лектиновых рецепторов С-типа, которые экспрессируются у человека и грызунов на макрофагах, моноцитах, нейтрофилах, дендритных клетках и некоторых субпопуляциях В-лимфоцитов при их стимуляции [50–53]. У морских свинок Mincle также присутствует на клетках лимфоузлов, селезенки и перитонеальных макрофагах [54].
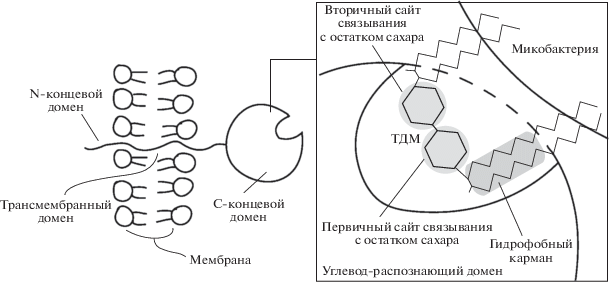
Структурно Mincle состоит из внеклеточной части, которая несет углеводраспознающий домен (CRD), отделенный от мембраны короткой ножкой, а также трансмембранный участок и короткий цитоплазматический хвост, не имеющий сигнальной последовательности (рис. 1) [52, 55]. В своем трансмембранном участке Mincle содержит положительно заряженный аргинин, который опосредует взаимодействие рецептора с иммунорецепторной тирозиновой активирующей последовательностью (ITAM) Fc рецептора γ-цепи (FcRγ), тем самым осуществляя передачу сигнала внутрь клетки [53]. Распознавание лиганда приводит к фосфорилированию ITAM и активации тирозинкиназы Syk, запуская каскад реакций через сигнальный комплекс Card9-Bcl10-MALT1 и высвобождая ядерный фактор каппа B (NFκB) [56].
Рис. 1.
Структура рецептора Mincle, состоящая из короткого N-концевого цитоплазматического домена, трансмембранного домена и С-терминального участка CRD, отделенного от мембраны короткой ножкой [55].

Детальные исследования лигандов Mincle показали, что он связывает гликолипиды, выделенные из M. tuberculosis, такие как трегалозодимиколат, трегалозомономиколат и глицеролмономиколат (ГММ) [57]. Однако ГММ приводит к меньшей активации Mincle и распознается избирательно только человеческим рецептором, но не мышиным. Кроме того, лигандом Mincle является и синтетический аналог ТДМ – трегалозодибегенат (ТДБ) [58]. Также Mincle способен распознавать целый ряд грибных патогенов, включая Candida albicans и нестандартные глицерогликолипиды гриба Malassezia [59–61].
Взаимодействие лигандов с человеческим или мышиным CRD-доменом Mincle осуществляется благодаря наличию консервативной для лектинов последовательности: глутаминовая кислота–пролин–аспарагин (EPN, остатки 169–171), которая является также необходимой для связывания с ТДМ [57].
В работе [55] проведены сравнительные исследования биохимических особенностей связывания ТДМ с мышиными, человеческими и бычьими углевод-распознающими доменами рецептора Mincle. Результаты показали сохранение способности к распознаванию гликолипида у Mincle различного происхождения, что свидетельствует о возможности исследования данного рецептора на животных и интерпретации полученных результатов на человека [55].
Понимание механизмов распознавания углеводов рецептором Mincle получено с помощью комплекса бычьего рецептора Mincle с ТДМ. В этом комплексе, один из двух остатков глюкозы, входящих в состав ТДМ, распознается первичным углеводсвязывающим сайтом. Он состоит из 5 аминокислотных остатков, связанных с ионом Ca2+, 4 из которых (Glu-176, Asn-192, Glu-168 и Asn-170) образуют водородные связи с 3 и 4 ОН-группами глюкозы, также взаимодействующими с ионом Ca2+. Второй остаток глюкозы связывается вторичным углеводсвязывающим сайтом, в котором Glu-135 и Arg-182 формируют водородные связи со 2 OH- группой остатка глюкозы [58]. Вблизи первичного углеводсвязывающего сайта располагается небольшой участок, состоящий из гидрофобных аминокислот, отсутствующий у большинства классических доменов С-типа. Он вмещает в себя ацильные цепи, присоединяющиеся к 6 ОН-группе остатка глюкозы, в первичном сайте связывания (рис. 2). Авторами показано, что достаточно одной ацильной цепи для связывания с Mincle, однако, обязательным условием является длина липидного фрагмента, которая должна быть не менее 10 углеродных остатков. Эти сайты могут включать разветвленные ацильные цепи миколовых кислот, содержащиеся в ТДМ и ТММ [58].
Рис. 2.
Модель взаимодействия Mincle с микобактерией. Mincle, располагающийся на поверхности макрофага, образует связь с ТДМ на поверхности микобактерии [58].
Аналогично некоторым другим представителям CLRs, Mincle способен распознавать DAMPs. Поврежденные и мертвые клетки высвобождают белок, ассоциированный со сплайсосомой 130 (SAP 130), который передает внутриклеточный сигнал, связываясь с внедоменной частью рецептора [53]. Также Mincle связывает кристаллы холестерина, формирующегося на атеросклеротических бляшках при гиперхолестеринемии [62].
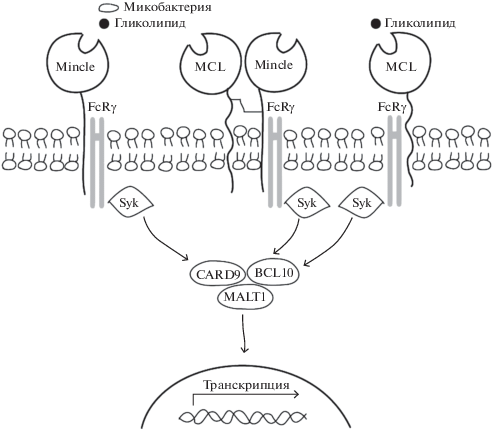
MCL-рецептор. Более 20 лет назад во время создания библиотеки мышиных макрофагальных генов был идентифицирован рецептор MCL, состоящий из 219 аминокислотных остатков [63]. MCL, как и Mincle, является С-лектиновым рецептором II типа, имеющим сходную структурную организацию: один распознающий домен на ножке и трансмембранный участок с коротким цитоплазматическим хвостом [64]. Данный рецептор также требует присутствия другого рецептора – FcRγ для осуществления передачи сигнала внутрь клетки через сигнальный путь Syk/CARD9 [65]. Однако природа взаимодействия с адаптерным белком не достаточно ясна, так как MCL не содержит необходимого остатка аргинина в трансмембранном участке для ассоциации с ITAM, тем самым ставя под вопрос непосредственную связь с FcRγ [66]. Предположительно, MCL и Mincle связываются ковалентно, образуя гетеродимер, в котором Mincle выступает в роли мостика между рецептором MCL и белком FcRγ (рис. 3) [66, 67]. Возможность такой связи доказывают данные о прямо-пропорциональной зависимости экспрессии этих двух рецепторов, полученные в спонтанных и ЛПС-стимулированных культурах перитонеальных макрофагов крысы [67].
Рис. 3.
Слева и справа: Связывание микробных гликолипидов с Mincle/MCL приводит к активации транскрипционного фактора NFκB, который вызывает продукцию цитокинов, активирующих наивные Т-клетки. По центру: MCL образует гетеродимер с Mincle, что позволяет передать сигнал через FcRγ [66].
MCL был первоначально охарактеризован как рецептор, конститутивно экспрессирующийся у мышей исключительно на макрофагах [63]. Однако дальнейшее изучение MCL у человека показало не только экспрессию на макрофагах (синовиальных, перитонеальных и пришедших из крови моноцитах), но и на клетках Лангерганса, нейтрофилах, моноцитах, а также зрелых и незрелых дендритных клетках [64]. У крыс MCL был обнаружен на макрофагах, нейтрофилах, дендритных клетках, B- и Т-лимфоцитах [58, 67].
В отличие от классических лектинов С-типа, MCL не имеет типичной последовательности EPN для распознавания углеводов. Она отличается остатком аминокислоты по последнему положению, например, у крыс аспарагин заменяется лизином, у человека аспартатом, а у мышей остается неизменным [43].
В целом, структура и локализация аминокислотных остатков и позиции ионов Ca2+ в углеводраспознающем домене у Mincle и MCL похожи, однако Mincle имеет два иона Ca2+ тогда как MCL – только один. Эти результаты указывают, что Mincle и MCL распознают углеводы через данные последовательности немного по-разному, но аналогичным образом [49].
М. Ямасаки и соавт. показали, что MCL, как и Mincle, является рецептором ТДМ и его синтетического аналога – ТДБ [61]. Однако прямое связывание ТДМ с MCL гораздо слабее, чем с Mincle, то есть MCL обладает меньшей аффинностью к ТДМ. Различия в аффинности эти двух CLR-рецепторов может быть объяснено разницей в размерах их гидрофобных зон. Mincle обладает большим гидрофобным участком (Val195, Thr196, Phe198, Leu199, Tyr201 и Phe202) по сравнению с MCL (Val197, Pro198 и Phe201) [49]. Однако оказалось затруднительным установить другие гликолипидные лиганды для MCL, так, в частности, не увенчался успехом поиск среди 326 углеводных лигандов и целого ряда моносахаридов [68]. По мнению Д.Г. Смита, такие результаты свидетельствовуют о том, что MCL не является углеводсвязывающим рецептором, а его взаимодействие с ТДМ осуществляется через липидный хвост последнего [43]. Следует отметить, что MCL также является рецептором, участвующим в эндоцитозе, что не отрицает возможности его участия в процессе переработки антигена [64].
По предположению Б. Мартина, γδТ-лимфоциты, экспрессирующие MCL и FcRγ, способны распознавать ТДМ и продуцировать в ответ цитокины независимо от Т-клеточного рецептора (TCR) [69].
В работе [70] показано взаимное влияние экспрессии рецепторов MCL и Mincle. Эксперименты in vivo и in vitro, проведенные в спонтанных и стимулированных ЛПС и БЦЖ (Бацилла Кальметта-Герена) культурах перитонеальных макрофагов мышей, показали, что экспрессия Mincle снижается в отсутствии MCL и наоборот [70]. Однако у клеток, лишенных Mincle, экспрессия генов MCL остается достоверно высокой на внутриклеточном уровне, а для локализации рецептора на поверхности требуется коэкспрессия рецептора Mincle. Таким образом, экспрессия обоих рецепторов носит взаимозависимый характер и формирует необходимый функциональный рецепторный комплекс на клеточной поверхности [64].
Сравнительно недавние исследования в области геномного секвенирования показали, что Mincle и MCL не только гомологичны друг к другу, но также высоко консервативны у разных организмов. Поиск местоположения генов данных CLRs у различных видов млекопитающих показал, что однопроходные и сумчатые обладают только рецепторами Mincle, тогда как плацентарные экспрессируют Mincle и MCL. У мышей и человека гены обоих рецепторов располагаются на 6 (6F3) и 12 (12p13) хромосоме соответственно [71].
CD1-рецептор. Молекулы CD1 представляют собой класс МНС-подобных молекул, которые развили способность к презентации липидов. У человека 5 генов CD1, которые впервые были клонированы в 1986 г. [72]. Эти гены расположены в первой хромосоме, и проявляют очень ограниченный полиморфизм. Возможно, недостаток функционального полиморфизма является результатом селективного давления в пользу способности связывать структурноконсервативные антигены и облегчать контакт с TCR (Т клеточный рецептор) [73]. Четыре изоформы CD1 участвуют в презентации липидов: CD1a, CD1b, CD1c и CD1d. Пятый, CD1е, являясь белком-переносчиком липидов, представляет собой единственный изотип, который напрямую не презентирует Т-клеткам антигены, однако играет важную роль в их процессинге и встраивании в CD1 [74, 75]. CD1a, CD1b и CD1c формируют первую группу молекул CD1, экспрессия генов которой индуцируется на миелоидных клетках, в то время как вторая группа, представленная CD1d, экспрессируется конститутивно. Каждая из изоформ имеет особенную молекулярную архитектуру, которая определяет специфичность молекулы CD1 к определенному спектру липидов (рис. 4) [76–78]. Так изоформы CD1 экспрессируются на различных типах клеток, таких как миелоидные дендритные клетки (имеют 5 изоформ белков CD1), клетки Лангерганса (CD1a, CD1e), и B-клетки (CD1c, CD1d) [73]. Гены CD1 также были обнаружены в тимусе, где они экспрессируются на тимоцитах [79].
Рис. 4.
Схематичное изображение структуры человеческой молекулы CD1 в 4х изоформах: CD1a, b, c, d [76].
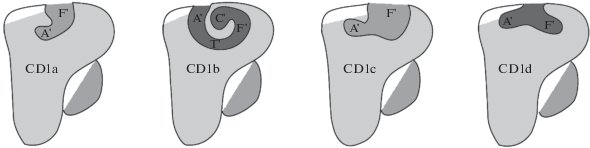
Молекулы CD1 – это гетеродимеры, состоящие из тяжелой цепи нековалентно связанной с β2-микроглобулином. Тяжелая цепь CD1состоит из трех доменов: α1, α2, и α3 и трансмембранного домена, который заякорен в мембране. В то время как домены α1 и α2 обладают большим разнообразием изотипов и образуют центральную связывающую полость, домен α3 наиболее консервативен и ассоциирован с β2-микроглобулином [80, 81]. У CD1 антигенсвязывающая область образована сетью гидрофобных карманов, отвечающих за связывание липидных молекул [73]. Объем таких областей варьирует среди изоформ с тенденцией: CD1a < CD1d < CD1c < CD1b. Размер и форма этих полостей коррелируют с размером гидрофобных цепей липидов, которые они могут презентировать [82]. Как правило, каждая молекула CD1 содержит, по меньшей мере, 2 гидрофобных кармана (так называемые А' и F'), которые связывают антиген [83]. В то время как карман А' имеет больший размер и более глубокое погружение, область F' является более открытой и доступной для захвата антигена [84].
Информация, находящаяся в молекуле CD1 считывается через распознавание Т-клетками [78, 85, 86]. В презентации миколовых липидов, включая миколовые кислоты и глюкозо- и глицерол-мономиколаты, выделенные из бактерий родов Mycobacterium, Corynebacterium, Rhodococcus и Nocardia, участвует изоформа CD1b [87, 88]. CD1b, являясь самым сложным членом семейства CD1, имеет самую большую связывающую полость среди всех изотипов. Впервые структура CD1b была изучена на примере комплекса с фосфотидилинозитолом или ганглиозидами, благодаря чему было установлено, что связывающая полость состоит из 4 соединенных карманов: A', C', F', и T' [76]. Такая уникальная архитектура CD1b позволяет связывать очень большие антигены длиной до 80 атомов углерода [82].
Таким образом, Mincle и MCL являются основными рецепторами клеточной мембраны иммунных клеток, связывающими трегалолипидные биосурфактанты. Распознавание гликолипидных молекул реализуется также посредством рецептора CD1b, который презентирует миколовые гликолипиды Т-лимфоцитам, обеспечивая иммунный ответ на липид-содержащие антигены.
Взаимодействие трегалолипидов с биологическими мембранами. Для объяснения разноплановых иммунорегуляторных эффектов трегалолипидных биосурфактантов важно понимание молекулярных механизмов, лежащих в основе их взаимодействия с поверхностными структурами иммунокомпетентных клеток. Выраженная амфифильная природа и биологическая активность трегалолипидов определяют направленность их воздействия на липидные молекулы биологических мембран. К основным структурным компонентам мембран эукариотных клеток относятся глицерофосфолипиды: фосфатидилхолин (ФХ), фосфатидилэтаноламин (ФЭА), фосфатидилсерин (ФС), фосфатидилинозитол (ФИ) и фосфатидовая кислота (ФК). Их гидрофобной частью является диацилглицерин, содержащие насыщенные или цис-ненасыщенные жирные кислоты с различной длиной цепи [89].
Группой исследователей [90, 91] были изучены физико-химические свойства трегалолипидного биосурфактанта из Rhodococcus sp. 51Т7, а также особенности его взаимодействия с мембранами, состоящими из цвиттерионных фосфолипидов (ФХ или ФЭА) с различными длинами ацильных цепей. Выявлено, что сукционил-трегалолипид встраивается между молекулами мембраны с образованием сурфактантных доменов. Эти биосурфактантные структуры хорошо визуализировались методом атомно-силовой микроскопии (АСМ), однако не изменяли макроскопическую двухслойную организацию мембраны [92].
Полученные в работе [93] АСМ-изображения трегалолипидного биосурфактанта из R. ruber ИЭГМ 231 выявили образование трех типов молекулярных структур в зависимости от концентрации (рис. 5). Так при низкой концентрации (100 мг/л) наблюдались мицеллоподобные структуры со средним диаметром 30–50 нм, которые либо располагались одиночно, либо были собраны в длинные тяжи. При более высокой концентрации (1000 мг/л) биосурфактант агрегировался в крупные везикулы неправильной формы диаметром 100–300 нм. Механизмы спонтанного перехода монодисперсных мицелл в агрегаты более высокого порядка при увеличении концентрации сурфактантов рассмотрены в работе [94].
Рис. 5.
АСМ-изображения трех типов молекулярных структур трегалолипидного биосурфактанта из R. ruber ИЭГМ 231 [93]. а – общий вид; б − мицеллы размером 30–50 нм; в − тяжи длиной 0.2–2 мкм; г − везикулы диаметром 100–300 нм.

По данным [92, 95], в присутствии трегалолипида из Rhodococcus sp. 51Т7 происходили следующие изменения мембранных структур: нарушалась конформация и кооперативность ФХ и ФЭА, увеличивалась текучесть их ацильных цепей, наблюдалась дегидратация межфазового пространства и повышалась толщина липидного бислоя.
Кроме незаряженных фосфолипидов в мембранах присутствуют в значительных количествах анионные фосфолипиды, которые являются ключевыми функциональными компонентами. В эукариотических мембранах основным анионным фосфолипидом является фосфатидилсерин [96]. Трегалолипид из Rhodococcus sp. 51Т7 оказывал аналогичное влияние на структурные свойства ФС, как и на нейтрально заряженные фосфолипиды, но, кроме того, понижал температуру фазового перехода из жидкокристаллической фазы в гель и наоборот [27, 97].
Этими же авторами изучена мембранотропная активность трегалолипидного биосурфактанта из Rhodococcus sp. 51Т7. Было установлено, что этот трегалолипид вызывает гемолиз человеческих эритроцитов через коллоидно-осмотический механизм [98]. С использованием искусственных мембран было выявлено, что трегалолипид образует пористые структуры при встраивании в мембранный бислой, тем самым индуцируя его пермеабилизацию. Возможно, трегалолипиды повышают проницаемость мембран эритроцитов по механизму формирования пермеабилизующих доменов. Однако липидный состав клеточной мембраны оказывает влияние на мембранотропную активность трегалолипидов. Так, наличие холестерола препятствовало повышению проницаемости мембраны под действием трегалолипида, что, по видимому, обуславливает его различную активность в отношении мембран прокариотных и эукариотных клеток [99]. С помощью сканирующей электронной микроскопии выявлены морфологические изменения красных кровяных телец при инкубации с трегалолипидом, в частности сфероцитоз (раздутые шарообразные) и эхиноцитоз (шиповатой формы) клеток (рис. 6) [98]. Следует отметить, что в литературе отсутствуют сведения о взаимодействии бактериальных гликолипидов, в частности трегалолипидов, с мембранами иммунных клеток.
Рис. 6.
Влияние трегалолипидов на морфологию эритроцитов человека: СЭМ контрольных эритроцитов в отсутствие (а) и в присутствии ПЭГ 3350 (в), эритроцитов, инкубированных с 60 мкм трегалолипида в отсутствие (б) и в присутствии ПЭГ 3350 (г). Масштабная линейка соответствует 5 мкм [98].

Таким образом, актинобактерии рода Rhodococcus, синтезирующие трегалолипидные биосурфактанты, могут рассматриваться как перспективные продуценты новых биоактивных молекул для возможного применения в биомедицине. Высокая иммуномодулирующая активность TДМ, TMM и других трегалолипидов родококков, их низкая токсичность по сравнению с микобактериальными производными определяют существенные преимущества их клинического применения. Недавними исследованиями на мембранах клеток врожденного иммунитета показано, что бактериальные трегалолипиды связываются с высокоаффинными рецепторами, к которым относятся Mincle и MСL-рецепторы лектинового семейства. Взаимодействие с данными рецепторами обеспечивает высокую иммунорегуляторную активность трегалолипидов за счет экспрессии транскрипционного фактора Nf-kb, который вызывает продукцию провоспалительных цитокинов. Основываясь на расшифровке молекулярных механизмов взаимодействия трегалолипидов с клетками иммунной системы, могут быть открыты новые подходы к иммунотерапии.
Работа выполнена в рамках госзаданий AAAAA19-119112290010-7 (ИЭГМ УрО РАН) и 6.3330.2017/4.6 (ПГНИУ) при частичной поддержке гранта Российского научного фонда 18-14-00140.
Список литературы
Banat I.M., De Rienzo M.A., Quinn G.A. // Appl. Microbiol. Biotechnol. 2014. V. 98. № 24. P. 9915–9929.
Gudiña E.J., Rodrigues A.I., Alves E., Domingues M.R., Teixeira J.A., Rodrigues L.R. // Bioresour. Technol. 2015. V. 177. № 2. P. 87–93.
Lechuga M., Fernández–Serrano M., Jurado E., Olea J.N. // Ecotoxicol. Environ. Saf. 2016. V. 125. № 3. P. 1–8.
Liu J., Chen Y., Xu R., Jia Y. // Indian J. Microbiol. 2013. V. 53. № 2. P. 168–174.
Paulino B.N., Pessôa M.G., Mano M.C. // Appl. Microbiol. Biotechnol. 2016. V. 100. № 24. P. 10265–10293.
Soberón-Chávez G., Maier R.M. Biosurfactants: from Genes to Applications. Ed. G. Soberón-Chávez. Berlin: Springer-Verlag, 2011. P. 1–11.
Cameotra S.S., Makkar R.S. // Curr. Opin. Microbiol. 2004. V. 7. № 3. P. 262–266.
Mukherjee A.K., Das K. // Advances in Experimental Medicine and Biology. 2010. V. 672. P. 54–64.
Franzetti A., Gandolfi I., Bestetti G., Smyth T.J. // Eur. J. Lipid Sci. Technol. 2010. V. 112. № 6. P. 617–627.
Anderson R.J., Newman M.S. // Biol. Chem. 1933. V. 101. P. 499–504.
Noll H., Bloch H, Asselineau J. // Biochim. Biophys. Acta. 1956. V. 20. № 2. P. 299–309.
Fujiwara N., Pan J., Enomoto K., Terano Y., Honda T., Yano I. // FEMS Immunol. Med. Microbiol. 1999. V. 24. № 2. P. 141–149.
Gerson D.F., Cooper D.G., Ramsay B., Zajic J.E. // Can. J. Microbiol. 1980. V. 26. № 12. P. 1498–500.
Margaritis A., Zajic J.E., Gerson D.F. // Biotechnol. Bioeng. 1979. V. 21. № 7. P. 1151–1162
Retzinger G.S., Meredith S.C., Takayama K., Hunter R.L., Kézdy F.J. // J. Biol. Chem. 1981. V. 256. № 15. P. 8208–8216.
Watanabe M., Kudoh S., Yamada Y., Iguchi K., Minnikin D.E. // Biochim. Biophys. Acta. 1992. V. 1165. № 1. P. 53–60.
Chun J., Kang S-O., Hah Y.C., Goodfellow M. // J. Industrial Microbiology. 1996. V. 17. № 3–4. P. 205–213.
Kuyukina M.S., Ivshina I.B. Application of Rhodococcus in bioremediation of contaminated environments / In: Microbiology Monographs. Ed. A. Steinbuchel. Dordrecht; London; N.Y.: Springer-Verlag, 2010. V. 16. P. 231–262.
White D.A., Hird L.C., Ali S.T. // J. Appl. Microbiol. 2013. V. 115. № 3. P. 744–755.
Stackebrandt E., Rainey F.A., Ward–Rainey N.L. // Int. J. Syst. Bacteriol. 1997. V. 47. № 2. P. 479–491.
Haburchak D.R., Jeffery B., Higbee J.W., Everett. E.D. // Am. J. Med. 1978. V. 65. № 2. P. 298–302.
Kügler J.H., Krasowska A., Czyżnikowska Ż., Łukaszewicz M. // Appl Microbiol Biotechnol. 2014. V. 98. № 21. P. 8905–8915.
Munstermann B., Poremba K., Lang S., Wagner F. Studies on Environmental Compatibility: Influence of (bio)surfactants on marine microbial and enzymatic systems / In Proceedings of the International Symposium on Soil Decontamination Using Biological Processes. Karlsruhe, Germany, 6–9 December 1992. P. 414–420.
Ivshina I.B., Kuyukina M.S., Philp J.C., Christofi N. // World J. Microbiol. Biotechnol. 1998. V. 14. № 5. P. 711–717.
Gein S.V., Kuyukina M.S., Ivshina I.B., Bayeva T.A., Chereshnev V.A. // Cytotechnology. 2011. V. 63. № 6. P. 559–566.
Kuyukina M.S. Ivshina I.B., Gein S.V., Baeva T.A., Chereshnev V.A. // Bull. Exp. Biol. Med. 2007. V. 144. № 3. P. 326–330.
Marques A.M., Pinazo A., Farfan M., Aranda F.J., Teruel J.A., Ortiz A. Manresa A., Espuny M.J. // Chem. Phys. Lipids. 2009. V. 158. № 2. P. 110–117.
Isoda H., Shinmoto H., Matsumura M., Nakahara T. // Cytotechnology. 1996. V. 19. № 1. P. 79–88.
Sakaguchi I., Ikeda N., Nakayama M., Kato Y., Yano I., Kaneda K. // Infect. Immunity. 2000. V. 68. № 4. P. 2043–2052.
Ueda S., Fujiwara N., Naka T., Sakaguchi I., Ozeki Y., Yano I., Kasama T., Kobayashi K. // Microb. Pathog. 2001. V. 30. № 2. P. 91–99.
Lima V.M., Bonato V.L., Lima K.M., Dos Santos S.A., Dos Santos R.R., Gonçalves E.D., Faccioli L.H., Bran-dão I.T., Rodrigues-Junior J.M., Silva C.L. // Infect. Immun. 2001. V. 69. № 9. P. 5305–5312.
Chereshnev V.A., Gein S.V., Baeva T.A., Galkina T.V., Kuyukina M.S., Ivshina I.B. // Bull. Exp. Biol. Med. 2010. V. 149. № 6. P. 734–738.
Yasuda K. Complement activation by mycoloyl glycolipids from Mycobacterium tuberculosis and Rhodococcus ruber // Osaka City Med. J. 1999. V. 45. P. 159–174.
Баева Т.А., Гейн С.В., Куюкина М.С., Ившина И.Б., Кочина О.А, Черешнев В.А. // Бюллетень экспериментальной биологии и медицины. 2014. Т. 157. № 2. С. 202–206.
Schick J., Etschel P., Bailo R., Ott L., Bhatt A., Lepenies B., Kirschning C., Burkovski A., Lang R. // Infect Immun. 2017. V. 85. № 7. https://doi.org/10.1128/IAI.00075-17
Tima H.G., Al Dulayymi J.R., Denis O., Lehebel P., Baols K.S., Mohammed M.O., L’Homme L., Sahb M.M., Potemberg G., Legrand S., Lang R., Beyaert R., Piette J., Baird M.S., Huygen K., Romano M. // J. Innate Immun. 2017. V. 9. № 2. P. 162–180.
Isoda H., Kitamoto D., Shinmoto H., Matsumura M., Nakahara T. // Biosci. Biotechnol. Biochem. 1997. V. 61. № 4. P. 609–614.
Sudo T., Zhao X., Wakamatsu Y., Shibahara M., Nomura N., Nakahara T., Suzuki A., Kobayashi Y., Jin C., Murata T., Yokoyama K.K. // Cytotechnology. 2000. V. 33. № 1–3. P. 259–264.
Кочина О.А., Куюкина М.С., Ившина И.Б. // Российский иммунологический журн. 2017. Т. 11. № 3. С. 396–398.
Bhunia D., Pallavi P.M., Bonam S.R., Reddy S.A., Verma Y., Halmuthur M.S. // Arch. Pharm. 2015. V. 348. № 10. P. 689–703.
Emoto M. Emoto Y., Buchwalow I.B., Kaufmann S.H. // Eur. J. Immunol. 1999. V. 29. № 2. P. 650–659.
Anderson B.L., Teyton L., Bendelac A., Savage P.B. // Molecules. 2013. V. 18. № 12. P. 15662–15688.
Smith D.G., Williams S.J. // Carbohydr. Res. 2016. V. 420. P. 32–45.
Akira S. TLR signaling // Curr. Top. Microbiol. Immunol. 2006. V. 311. P. 1–16.
Kawai T., Akira S. // Cell Death Differ. 2006. V. 13. № 5. P. 816–825.
Geijtenbeek T.B., Gringhuis S.I. // Nat. Rev. Immunol. 2009. V. 9. № 7. P. 465–479.
Zelensky A.N., Gready J.E. // FEBS J. 2005. V. 272. № 24. P. 6179–6217.
Kerscher B., Willment J.A., Brown G.D. // Int. Immunol. 2013. V. 25. № 5. P. 271–277.
Furukawa A., Kamishikiryo J., Mori D., Toyonaga K., Okabe Y., Toji A., Kanda R., Miyake Y., Ose T., Yamasaki S., Maenaka K. // Proc. Natl. Acad. Sci. USA. 2013. V. 110. № 43. P. 17438–43.
Kawata K., Illarionov P., Kenny T., Zhang W., Tsuda M., Ando Y., Leung P., Ansari A.A., Gershwin M.E. // J. Autoimmun. 2012. V. 39. № 4. P. 315–322.
Lee W.B., Kang J., Yan J., Lee M.S., Jeon B., Cho S., Kim Y.J. // PLoS Pathog. 2012. V. 8. № 4. P. 1–19.
Matsumoto M., Tanaka T., Kaisho T., Sanjo H., Copeland N.G., Gilbert D.J., Jenkins N.A., Akira S. // J. Immunol. 1999. V. 163. № 9. P. 5039–5048.
Yamasaki S., Ishikawa E., Sakuma M., Hara H., Ogata K., Saito T. // Nat. Immunol. 2008. V. 9. № 10. P. 1179–1188.
Toyonaga K., Miyake Y., Yamasaki S. // PLoS One. 2014. V. 9. № 2. P. 1–11.
Rambaruth N.D., Jégouzo S.A, Marlor H., Taylor M.E., Drickamer K. // Molecules. 2015. V. 20. № 4. P. 6670–6682.
Kingeter L.M., Lin X. // Cell Mol. Immunol. 2012. V. 9. № 2. P. 105–112.
Ishikawa E., Ishikawa T., Morita Y.S., Toyonaga K., Yamada H., Takeuchi O., Kinoshita T., Akira S., Yoshikai Y., Yamasaki S. // J. Exp. Med. 2009. V. 206. № 13. P. 2879–2888.
Feinberg H., Jégouzo S.A., Rowntree T.J., Guan Y., Brash M.A., Taylor M.E., Weis W.I., Drickamer K. // J. Biol. Chem. 2013. V. 288. № 40. P. 28457–28465.
Bugarcic A., Hitchens K., Beckhouse A.G., Wells C.A., Ashman R.B., Blanchard H. // Glycobiology. 2008. V. 18. № 9. P. 679–685.
Wells C.A., Salvage-Jones J.A., Li X., Hitchens K., Butcher S., Murray R.Z., Beckhouse A.G., Lo Y.L., Manzanero S., Cobbold C., Schroder K., Ma B., Orr S., Stewart L., Lebus D., Sobieszczuk P., Hume D.A., Stow J., Blanchard H., Ashman R.B. // J. Immunol. 2008. V. 180. № 11. P. 7404–7413.
Yamasaki S. Mincle, as a sensor for damaged cells // Tanpakushitsu Kakusan Koso. 2009. V. 54. № 8. P. 1125–1259.
Kiyotake R., Oh-Hora M., Ishikawa E2, Miyamoto T., Ishibashi T., Yamasaki S. // J. Biol. Chem. 2015. V. 290. № 42. P. 25322–25332.
Balch S.G., McKnight A.J., Seldin M.F., Gordon S. // J. Biol. Chem. 1998. V. 273. № 29. P. 18656–18664.
Arce I., Martínez–Muñoz L., Roda–Navarro P. // Eur. J. Immunol. 2004. V. 34. № 1. P. 210–220.
Miyake Y., Toyonaga K., Mori D., Kakuta S., Hoshino Y., Oyamada A., Yamada H., Ono K., Suyama M., Iwakura Y., Yoshikai Y., Yamasaki S. // Immunity. 2013. V. 38. № 5. P. 1050–1062.
Richardson M.B., Williams S.J. // Front. Immunol. 2014. V. 5. № 288. P. 1–9.
Lobato–Pascual A., Saether P.C., Fossum S., Dissen E., Daws M.R. // Eur. J. Immunol. 2013. V. 43. № 12. P. 3167–3174.
Graham L.M., Gupta V., Schafer G., Reid D.M., Kimberg M., Dennehy K.M., Hornsell W.G., Guler R., Campanero-Rhodes M.A., Palma A.S., Feizi T., Kim S.K., Sobieszczuk P., Willment J.A., Brown G.D. // J. Biol. Chem. 2012. V. 287. № 31. P. 25964–25974.
Martin B., Hirota K., Cua D.J. // Immunity. 2009. V. 31. № 2. P. 21–30.
Kerscher B., Wilson G. J., Reid D.M., Taylor J.A., Besra G.S., Yamasaki S., Willment J.A., Brown G.D. // Eur. J. Immunol. 2016. V. 46. № 2. P. 381–389.
Flornes L.M., Bryceson Y.T., Spurkland A., Lorentzen J.C., Dissen E., Fossum S. // Immunogenetics. 2004. V. 56. № 7. P. 506–517.
Calabi F., Milstein C. // Nature. 1986. V. 323. № 6088. P. 540–543.
Salio M., Silk J.D., Yvonne J.E., Cerundolo V. // Annu. Rev. Immunol. 2014. V. 32. P. 323–366.
Angenieux C., Fraisier V., Maître B., Racine V., van der Wel N., Fricker D., Proamer F., Sachse M., Cazenave J.P., Peters P., Goud B., Hanau D., Sibarita J.B., Salamero J., de la Salle H. // Traffic. 2005. V. 6. № 4. P. 286–302.
Facciotti F., Cavallari M., Angénieux C., Garcia-Alles L.F., Signorino-Gelo F., Angman L., Gilleron M., Prandi J., Puzo G., Panza L., Xia C., Wang P.G., Dellabona P., Casorati G., Porcelli S.A., de la Salle H., Mori L., De Libero G. // Proc. Natl. Acad. Sci. USA. 2011. V. 108. № 34. P. 14228–14233.
Adams E.J. Lipid presentation by human CD1 molecules and the diverse T cell populations that respond to them // Curr. Opin. Immunol. 2014. V. 26C. № 1. P. 1–6.
Barral D.C., Brenner M.B. // Nat. Rev. Immunol. 2007. V. 7. № 12. P. 929–941.
Sugita M., Barral D.C., Brenner M.B. // Curr. Top. Microb-iol. Immunol. 2007. V. 314. P. 143–164.
Van Rhijn I., van Berlo, Hilmenyuk T., Cheng T.Y., Wolf B.J., Tatituri R.V., Uldrich A.P., Napolitani G., Cerundolo V., Altman J.D., Willemsen P., Huang S., Rossjohn J., Besra G.S., Brenner M.B., Godfrey D.I., Moody D.B. // Proc. Natl. Acad. Sci. USA. 2016. V. 113. № 2. P. 380–385.
Moody D.B., Zajonc D.M., Wilson I.A. // Nat. Rev. Immunol. 2005. V. 5. № 5. P. 387–399.
Zajonc D.M., Flajnik M.F. // Immunogenetics. 2016. V. 68. № 8. P. 489–490.
Gadola S.D., Zaccai N.R, Harlos K., Shepherd D., Castro-Palomino J.C., Ritter G., Schmidt R.R., Jones E.Y., Cerundolo V. // Nat Immunol. 2002. V. 3. № 8. P. 721–726.
Rossjohn J., Gras S., Miles J.J., Turner S.J., Godfrey D.I., McCluskey J. // Annu. Rev. Immunol. 2015. V. 33. № 1. P. 169–200.
Li Y., Girardi E., Wang J., Yu E.D., Painter G.F., Kronenberg M., Zajonc D.M. // J. Exp. Med. 2010. V. 207. № 11. P. 2383–2393.
Bendelac A., Savage P.B., Teyton L. // Annu. Rev. Immunol. 2007. V. 25. № 1. P. 297–336.
Tilloy F., Treiner E., Park S.H., Garcia C., Lemonnier F., de la Salle H., Bendelac A., Bonneville M., Lantz O. // J. Exp. Med. 1999. V. 189. № 12. P. 1907–1921.
Beckman E.M., Porcelli S.A., Morita C.T. Behar S.M., Furlong S.T., Brenner M.B. // Nature. 1994. V. 372. № 6507. P. 691–594.
Layre E. Collmann A., Bastian M., Mariotti S., Czaplicki J., Prandi J., Mori L., Stenger S., De Libero G., Puzo G., Gilleron M. // Chem. Biol. 2009. V. 16. № 1. P. 82–92.
Ashrafuzzaman M., Tuszynski J. Membrane Biophysics / Berlin: Springer-Verlag, 2012. P. 31–48.
Ortiz A., Teruel J.A., Manresa Á., Espuny M.J., Arandaa A.J. // Biochimica et Biophysica Acta. 2011. V. 1808. № 8. P. 2067–2072.
Teruel J.A., Ortiz A., Aranda F.J. // Chemistry and Physics of Lipids. 2014. V. 181. № 7. P. 34–39.
Aranda F.J., Teruel J.A., Espuny M.J., Palacios–Lidón E., Ortiz A. // Biochimica et Biophysica Acta. 2007. V. 1768. № 10. P. 2596–2604.
Kuyukina M.S., Ivshina I.B., Korshunova I.O., Stukova G.I., Krivoruchko A.V. // AMB Express. 2016. V. 6. № 14. P. 1–12.
Svenson S. Controlling surfactant self-assembly // Curr Opin Coll Interface Sci. 2004. V. 9. № 3. P. 201–212.
Ortiz A., Teruel J.A., Espuny M.J., Marqués A., Manresa A., Aranda F.J. // Biochimica et Biophysica Acta. 2008. V. 1778. № 12. P. 2806–2813.
Buckland A.G., Wilton D.C. // Biochim. Biophys. Acta. 2000. V. 1483. № 2. P. 199–216.
Ortiz A., Teruel J.A., Espuny M.J., Marqués A., Manresa A., Aranda F.J. // Chemistry and Physics of Lipids. 2009. V. 158. № 1. P. 46–53.
Zaragoza A., Aranda F.J., Espuny M.J., Teruel J.A., Marqués A., Manresa A., Ortiz A. // Langmuir. 2010. V. 26. № 11. P. 8567–8572.
Zaragoza A.F., Aranda J., Espuny M.J., Teruel J.A. // Langmuir. 2009. V. 25. № 14. P. 7892–7898.
Дополнительные материалы отсутствуют.
Инструменты
Прикладная биохимия и микробиология