

Прикладная биохимия и микробиология, 2021, T. 57, № 5, стр. 477-484
Ферментативная деструкция целлюлозы: особенности кинетического взаимодействия литических полисахаридмонооксигеназ и индивидуальных целлюлаз
М. В. Семёнова 1, А. В. Гусаков 2, *, В. Д. Телицин 2, А. П. Синицын 1, 2
1 Институт биохимии им. А.Н. Баха, Федеральный исследовательский центр
“Фундаментальные основы биотехнологии” Российской академии наук
119071 Москва, Россия
2 Московский государственный университет им. М.В. Ломоносова, химический факультет
119991 Москва, Россия
* E-mail: avgusakov@enzyme.chem.msu.ru
Поступила в редакцию 17.03.2021
После доработки 15.04.2021
Принята к публикации 23.04.2021
Аннотация
Исследованы закономерности кинетического взаимодействия литических полисахаридмонооксигеназ (ЛПМО) из Thielavia terrestris (TtLPMO), Penicillium verruculosum (PvLPMO) и Trichoderma reesei (TrLPMO) с очищенными целлобиогидролазами (ЦБГ I и ЦБГ II), а также эндоглюканазой II (ЭГ II) P. verruculosum при ферментативной деструкции микрокристаллической (МКЦ) и аморфной целлюлозы. TtLPMO относится к ЛПМО C1-типа, тогда как PvLPMO и TrLPMO являются ЛПМО смешанного C1/C4-типа, согласно общепринятой классификации данного класса оксидаз. При действии любой из трех ЛПМО совместно с ЦБГ II или ЭГ II на МКЦ или аморфную целлюлозу ферменты проявляли синергизм, результатом которого было увеличение выхода восстанавливающих сахаров (ВС), выраженное в наибольшей степени в начальный период реакции. При использовании смесей PvLPMO и TrLPMO с ЦБГ I и МКЦ в качестве субстрата также наблюдался синергизм, тогда как TtLPMO, напротив, ингибировала образование ВС из целлюлозы с ЦБГ I. Наблюдаемый синергизм и антагонизм между ЛПМО и ЦБГ II/ЦБГ I можно объяснить в рамках простой кинетической схемы, когда первый фермент образует дополнительные концы молекул полисахарида, служащие субстратом либо ингибитором для второго.
Закономерности биодеградации природных полисахаридов и, в частности, целлюлозы, как наиболее прочного и устойчивого к действию грибных и бактериальных ферментов компонента клеточной стенки растений, остаются темой интенсивных исследований в лабораториях разных стран и до сих пор преподносят сюрпризы для ученых, работающих в этой области. С момента открытия и начала широкого изучения ферментов – целлюлаз в середине прошлого века и вплоть до 2010–2011 гг. в научной литературе существовала общепринятая точка зрения, что ферментативную деструкцию целлюлозы катализируют целлюлазы, относящиеся к классу гидролаз, а именно эндо-1,4-β-глюканазы (ЭГ, КФ 3.2.1.4), экзо-целлобиогидролазы (ЦБГ, КФ 3.2.1.91 и 3.2.1.176) и β-глюкозидазы (БГ, КФ 3.2.1.21) [1, 2], причем последние, строго говоря, не относятся к собственно целлюлазам, поскольку не способны расщеплять полимерный субстрат, а осуществляют гидролиз целлобиозы и других олигосахаридов, образующихся под действием ЦБГ и ЭГ.
В результате открытия около 10 лет назад литических полисахаридмонооксигеназ (ЛПМО), катализирующих окислительную деструкцию хитина, целлюлозы и других полисахаридов [3–5], а также благодаря накопленным знаниям в области исследования ферментов, принимающих участие в биодеградации лигнина – другого важного компонента растений [6], стало ясно, что круг ферментов, благодаря которым в природе осуществляется эффективная биодеградация этих биополимеров, далеко не ограничивается гидролазами. Поэтому в 2013 г. в наиболее известную в мире базу данных CAZy (Carbohydrate-Active enZymes) по ферментам, активным по отношению к углеводам, были введены семейства так называемых вспомогательных активностей (Auxiliary Activities, AA) [7]. Большинство ЛПМО, для которых основным субстратом является целлюлоза, относятся к семейству AA9, тогда как ЛПМО “хитиназного” типа преимущественно входят в семейство AA10. При этом следует отметить, что многие из рассматриваемых ферментов не проявляют строгой субстратной специфичности. Например, некоторые ЛПМО семейства АА10 активны не только по отношению к хитину, но и к целлюлозе, тогда как ряд ЛПМО семейства АА9 способны расщеплять и гемицеллюлозы – ксилан, ксилоглюкан и др. [8–10].
Что касается собственно реакций, катализируемых ЛПМО “целлюлазного” типа, то все эти ферменты расщепляют полимерную цепь целлюлозы с окислением либо по С1 (КФ 1.14.99.54), либо по С4 атому гликозидного кольца (КФ 1.14.99.56) с образованием соответственно альдоновой кислоты (ее лактона) или 4-кетоальдозы [5, 11]. Довольно многочисленная третья группа ЛПМО смешанного типа генерирует продукты, окисленные как в положении С1, так и в положении С4 [4, 12, 13].
Несмотря на то, что имеется множество публикаций, демонстрирующих синергизм между ЛПМО и целлюлазным комплексом гидролаз в целом, включающим несколько различных ЭГ и ЦБГ [14–22], данных, описывающих взаимодействие ЛПМО с индивидуальными (очищенными) ЭГ и ЦБГ значительно меньше, и иногда они противоречивы [23–28].
Цель работы – исследование закономерностей кинетического взаимодействия трех грибных ЛПМО, различающихся по типу действия на полимерный субстрат, с индивидуальными ЦБГ I, ЦБГ II и ЭГ II из Penicillium verruculosum при деструкции микрокристаллической и аморфной целлюлозы. Две из трех ЛПМО (из Trichoderma reesei и P. verruculosum) относятся к ферментам смешанного (C1/C4) типа с точки зрения образуемых окисленных продуктов реакции [29, 30], тогда как третий фермент (из Thielavia terrestris) является ЛПМО C1-типа [20].
МЕТОДИКА
Ферменты. Выделение и очистку рекомбинантных ЛПМО из T. terrestris (TtLPMO, код GenBank AEO71030), T. reesei (TrLPMO, CAA71999) и P. verruculosum (PvLPMO, полноразмерная форма фермента, QBL14594) проводили, как описано ранее [19, 30]. Нативные очищенные ЦБГ I, ЦБГ II и ЭГ II P. verruculosum также были получены ранее [31, 32]. Рекомбинантную очищенную БГ из Aspergillus niger [33] использовали в качестве вспомогательного фермента в смесях, содержащих ЦБГ I или ЦБГ II.
Гидролиз целлюлозы смесями очищенных целлюлаз и ЛПМО. Обработку ферментами микрокристаллической целлюлозы (МКЦ, ООО “МК-Центр”, Россия) и аморфной целлюлозы, полученной из МКЦ регенерацией из раствора фосфорной кислоты по методу Вуда [34], проводили в пробирках объемом 1.5 мл в течение 24–48 ч при 40°С и рН 5.0, как подробно описано в предыдущей работе [19]. В качестве донора электронов для ЛПМО во всех случаях использовали 5 мМ аскорбиновую кислоту.
При составлении смесей на основе целлобиогидролаз использовали композицию ЦБГ I или ЦБГ II P. verruculosum с БГ A. niger в соотношении 9 : 1 по концентрации белка для того, чтобы конвертировать в глюкозу образующуюся при гидролизе МКЦ целлобиозу, являющуюся продуктом–ингибитором для ЦБГ. При исследовании взаимодействия между указанными целлюлазами и ЛПМО часть смеси ЦБГ/БГ эквивалентно замещали на 5, 10, 15, 20 или 25% ЛПМО по концентрации белка, поддерживая его общую дозировку на уровне 1 мг/г субстрата. Исходная концентрация МКЦ в реакционной смеси составляла 100 мг/мл. Отбор проб реакционной смеси (по 50 мкл) осуществляли после 1, 3, 6, 24 и 48 ч, после чего пробы центрифугировали, и в супернатанте определяли концентрацию восстанавливающих сахаров (ВС) методом Шомоди–Нельсона [35]. Поскольку находящаяся в реакционной смеси аскорбиновая кислота также давала ответ на используемые для анализа ВС реагенты, в качестве контроля (фона) использовали те же разведения исходной 5 мМ аскорбиновой кислоты, что и в соответствующих пробах, вычитая затем полученные фоновые значения оптической плотности из значений для проб на ВС. Стоит отметить, что заметный фон наблюдался только для начального периода ферментативной реакции (при малых разведениях проб).
В случае смесей на основе ЭГ II P. verruculosum и различных ЛПМО в качестве субстрата использовали аморфную целлюлозу (20 мг/мл). Эксперименты проводили, как описано выше для гидролиза МКЦ смесями на основе ЦБГ и ЛПМО, при той же общей концентрации белка (1 мг/г субстрата), однако пробы на ВС отбирали через 30 мин, 1, 3 и 24 ч.
Во всех случаях концентрацию белка определяли, используя модифицированный метод Лоури [36]. Каждый из экспериментов по гидролизу целлюлозных субстратов в отсутствие или в присутствии ЛПМО проводили в трех повторностях.
Коэффициент синергизма Ks между целлюлазами и ЛПМО рассчитывали по формуле:
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Синергизм и антагонизм между ЦБГ и ЛПМО. Для исследования закономерностей взаимодействия между целлобиогидролазами P. verruculosum и ЛПМО, различающимися по типу действия (C1 или C1/C4) на полимерный субстрат, проводили обработку МКЦ индивидуальными ЦБГ I или ЦБГ II (в присутствии небольшого количества БГ для снятия ингибирующего действия продукта реакции на ЦБГ), их смесями с ЛПМО, варьируя процентное содержание последних от 5 до 25% с шагом 5%, а также собственно ЛПМО (в отсутствие ЦБГ/БГ).
На рис. 1а, 1б показана концентрация полученных ВС для указанных смесей на основе ЦБГ I через 1 и 24 ч гидролиза МКЦ. Через 1 ч реакции для большинства смесей, содержащих ЛПМО из T. reesei и P. verruculosum (обозначенных как TrLPMO и PvLPMO, соответственно), относящихся к ферментам смешанного C1/C4-типа, наблюдали заметное увеличение концентрации образующихся сахаров по сравнению с таковой для ЦБГ I/БГ в отсутствие ЛПМО, несмотря на пониженное содержание ЦБГ I в смесях, что свидетельствует о синергизме между ферментами; наиболее заметный эффект проявлялся при содержании ЛПМО 10–15%. Коэффициент синергизма (Ks) достигал в максимальном случае значений 1.35–1.36 (рис. 2а, 2б). Однако в случае использования ЛПМО C1-типа из T. terrestris (TtLPMO), напротив, наблюдали проявление антагонизма между ферментами (рис. 1а, 2в), который усиливался при увеличении содержания TtLPMO в реакционной смеси (при этом Ks принимал значения меньше 0). При длительном времени гидролиза (24 ч) эффект увеличения концентрации ВС для смесей, содержащих TrLPMO и PvLPMO, становился менее заметным (рис. 1б), при этом также уменьшался и коэффициент синергизма по сравнению с начальным периодом проведения ферментативной реакции (рис. 2а, 2б). Что касается антагонизма между ЦБГ I и TtLPMO, то его степень была максимальной через 6 ч гидролиза МКЦ, уменьшаясь при большем времени реакции (24 ч). Стоит отметить, что сами по себе PvLPMO и TrLPMO в отсутствие целлюлаз образовывали незначительное количество растворимых ВС (0.2–0.3 мг/мл) даже через 24 ч реакции за счет разрывов полимерной цепи целлюлозы при ее окислении по C4 атому ангидроглюкозного остатка, тогда как в случае TtLPMO, где окислению подвергался исключительно C1 атом с образованием альдоновой кислоты, накопления детектируемых количеств ВС не наблюдали.
Рис. 1.
Выход ВС при действии на МКЦ (а-г) и аморфную целлюлозу (д, е) смесей различных ЛПМО и целлюлаз: ЦБГ I/БГ и ЛПМО через 1 (а) и 24 (б) ч; ЦБГ II/БГ и ЛПМО на МКЦ через 1 (в) и 24 (г) ч; ЭГ II и ЛПМО через 30 мин (д) и 3 ч (е) реакции. 1 – TtLPMO; 2 – PvLPMO; 3 – TrLPMO. 1', 2', 3' (штриховые линии) – соответствуют теоретически возможным концентрациям ВС, которые могли бы быть получены при отсутствии взаимодействия между ферментами, как арифметическая сумма действия индивидуальных компонентов смеси при данном соотношении компонентов.

Рис. 2.
Зависимости коэффициента синергизма (Ks) от времени реакции для смесей различных ЛПМО и целлюлазы: а, б, в – смеси ЦБГ I/БГ с PvLPMO (а), TrLPMO (б), TtLPMO (в); г, д, е – смеси ЦБГ II/БГ с PvLPMO (г), TrLPMO (д), TtLPMO (е); ж, з, и – смеси ЭГ II с PvLPMO (ж), TrLPMO (з), TtLPMO (и). Содержание ЛПМО в смесях (%): 1 – 5, 2 – 10, 3 – 15, 4 – 20, 5 – 25.
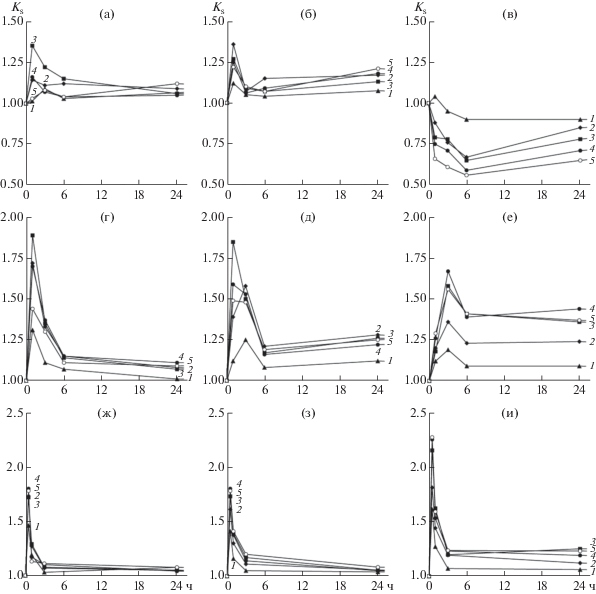
Подобные же эксперименты по обработке МКЦ смесями ЦБГ II/БГ с различными ЛПМО показали, что во всех случаях, в том числе и при использовании TtLPMO, наблюдался синергизм между ферментами (рис. 1в, 1г), причем степень синергизма была в целом существенно выше, чем в случае смесей на основе ЦБГ I/БГ. Коэффициент синергизма достигал в максимальном случае значений 1.67–1.89 (рис. 2г, 2д, 2е). Если в начальный период реакции (1 ч) наибольший синергизм и выход ВС обеспечивали TrLPMO и PvLPMO при содержании 15% в качестве вспомогательных ферментов (рис. 1в), то при длительном гидролизе (24 ч) эффективность применения TtLPMO при ее содержании в смеси 15–20% была сравнима с таковой для TrLPMO, однако используемой при меньшем соотношении (10%) с ЦБГ I/БГ (рис. 1 г).
Синергизм между ЭГ II и ЛПМО. Поскольку ЭГ II P. verruculosum, как и другие эндоглюканазы, не способна эффективно гидролизовать кристаллические формы целлюлозы, включая МКЦ [31], эксперименты со смесями ЭГ II и ЛПМО проводили, используя в качестве субстрата аморфную целлюлозу, при том же соотношении ферментов и субстрата (1 мг/г субстрата). Положительный эффект на выход ВС от замены части ЭГ II на любую из трех использованных ЛПМО в начальный период реакции (рис. 1д) оказался даже несколько выше, чем случае действия ЦБГ I или ЦБГ II на МКЦ. В зависимости от вида ЛПМО и времени реакции наибольший выход ВС наблюдали при содержании ЛПМО в смеси от 10 до 20% (рис. 1д, 1е), при этом коэффициент синергизма достигал в максимальном случае значений 1.78–2.27 (рис. 2ж, 2з, 2и). Как и в случае с ЦБГ, максимальный синергетический эффект проявлялся на начальной стадии процесса гидролиза целлюлозы.
Таким образом, эксперименты по обработке целлюлозных субстратов смесями индивидуальных целлюлаз (ЦБГ I/БГ, ЦБГ II/БГ, ЭГ II) и ЛПМО, различающихся по типу действия на полимерный субстрат, показали, что с точки зрения выхода образующихся ВС ферменты могут проявлять как синергизм, так и антагонизм.
В случае смесей на основе целлобиогидролаз результаты вполне объяснимы, учитывая специфику действия данных ферментов. ЦБГ I P. verruculosum, относящаяся к семейству 7 гликозидгидролаз, последовательно отщепляет остатки целлобиозы с восстанавливающего конца полимерной цепи целлюлозы по так называемому процессивному механизму [1, 31]. Когда в смеси находилась TtLPMO C1-типа, при разрыве молекулы полисахарида вместо дополнительного восстанавливающего конца (ВС) – субстрата для ЦБГ I – образовывался исключительно остаток альдоновой кислоты, который существует в равновесии с соответствующим лактоном [37]. Вероятно, этот остаток выступал в качестве ингибитора ЦБГ I, когда фермент мог связываться с данным участком полимерного субстрата, однако не был способен осуществлять расщепление соответствующей гликозидной связи, что привело к кажущемуся кинетическому антагонизму с TtLPMO, выражающемуся в уменьшении выхода ВС по сравнению с теоретически возможным при отсутствии взаимодействия между ферментами, показанным пунктирной линией на рис. 1а и 1б. Действительно, недавно было обнаружено, что целлобионовая кислота (целлобионолактон), один из растворимых продуктов катализа ЛПМО C1-типа, является конкурентным ингибитором ЦБГ I, хотя и более слабым, чем целлобиоза – основной продукт действия самой ЦБГ I [38]. Учитывая, что при атаке на нерастворимую целлюлозу ЛПМО окисляют внутренние связи в полимере, в результате образуются в основном новые окисленные концы полисахарида, не выходящие в раствор, тогда как окисленные растворимые олигосахариды, включая целлобионовую кислоту (целлобионолактон), являются минорными продуктами, особенно в начальный период ферментативной реакции. Маловероятно, что наблюдаемый значительный антагонизм между ЦБГ I и TtLPMO (рис. 1а) был вызван ингибированием ЦБГ I именно целлобионовой кислотой (не исключая частичного ингибирования ею), и скорее новые окисленные концы целлюлозы, остающиеся в составе нерастворимого субстрата в форме лактона или альдоновой кислоты, оказывали ингибирующее действие на ЦБГ I. В случае же использования в смеси TrLPMO или PvLPMO, относящихся к смешанному C1/C4-типу, при окислительной деструкции полимерной цепи целлюлозы образовывались как дополнительные восстанавливающие концы для связывания и катализа ЦБГ I по процессивному механизму, так и “концы–ингибиторы” этого фермента. По-видимому, вклад первых из указанных продуктов действия ЛПМО, стимулирующих образование ВС при катализе ЦБГ I, был более существенным, чем вторых (ингибирующих), в результате чего наблюдался умеренный синергизм между ЦБГ I и TrLPMO (либо PvLPMO).
Когда же в смеси ферментов находилась ЦБГ II P. verruculosum из семейства 6 гликозидгидролаз, действующая на целлюлозу подобно ЦБГ I, однако с невосстанавливающего конца ее молекулы, то во всех случаях использованных ЛПМО образовывались либо дополнительные интактные невосстанавливающие концы полисахарида, являющиеся субстратом для ЦБГ II (при использовании TtLPMO C1-типа), либо частично, наряду с интактными концами, окисленные по C4 атому гликозидного кольца 4-кетоальдозы (в случае TrLPMO или PvLPMO), которые, вероятно, не являлись ингибиторами ЦБГ II, поскольку во всех смесях, содержащих указанную целлюлазу и любую из ЛПМО наблюдали значительное проявление синергизма (рис. 1в, 1г).
В целом, механизм кинетического синергизма между ЛПМО и ЦБГ сходен с таковым для пары ЭГ – ЦБГ, когда действие первого из ферментов увеличивает концентрацию субстрата (концов полимерной цепи) для второго фермента [1].
Что касается смесей, содержащих ЭГ II и ЛПМО, то во всех случаях при их действии на аморфную целлюлозу также проявлялся эффект синергизма между ферментами, особенно в начальный период ферментативной реакции (рис. 1д), который был выражен даже в несколько большей степени, чем для смесей ЦБГ II/БГ с ЛПМО (рис. 1в). Это результат оказался довольно неожиданным, поскольку в данном случае в качестве субстрата использовали аморфную целлюлозу, обладающую высокой реакционной способностью по отношению к практически любым целлюлазам, включая эндоглюканазы. Считается, что, помимо образования новых концов полисахарида в результате действия ЛПМО на МКЦ и другие кристаллические формы целлюлозы, данные ферменты способны нарушать упорядоченную структуру субстрата, приводя к ее аморфизации и увеличению удельной площади поверхности [23, 39, 40]. Однако, в случае действия ЭГ II на аморфную целлюлозу ферменту не нужны ни новые дополнительные концы молекул целлюлозы, поскольку эндоглюканазы расщепляют внутренние гликозидные связи в полисахариде, ни, вероятно, дополнительные нарушения его надмолекулярной структуры, поскольку аморфная целлюлоза, как уже отмечалось выше, исходно является высокореакционным субстратом для целлюлаз. Для выявления причин, по которым наблюдался значительный синергизм между ЭГ II и ЛПМО, требуются дополнительные исследования.
При действии на МКЦ смесей на основе ЦБГ I или ЦБГ II в начальный период реакции наибольшую эффективность демонстрировали TrLPMO и PvLPMO (рис. 1а, 1в), тогда как при длительном времени вторая из использованных ЛПМО стала заметно уступать первой (рис. 1б, 1г). По-видимому, это было связано с более низкой стабильностью PvLPMO, при этом потеря активности фермента могла происходить как вследствие термоинактивации (PvLPMO действительно обладает более низкой термостабильностью, чем TrLPMO и TtLPMO [30]), так и в результате влияния на белок пероксида водорода, являющегося побочным продуктом катализа ЛПМО [41].
Анализ литературных данных показывает, что, как и в наших экспериментах, различные ЛПМО при взаимодействии с эндоглюканазами увеличивают выход сахаров при гидролизе различных форм целлюлозы [24, 26, 27, 29]. Так, похожие величины синергизма были получены при совместном действии TrLPMO и бактериальной ЭГ семейства 5 гликозидгидролаз (в соотношении 1 : 5) на предобработанные остатки сои (Ks варьировал 1.07 до 1.27 в зависимости от времени реакции: 8–72 ч) [29], а также при действии комбинации ферментов из Myceliophthora thermophila (MtEG5A и MtLPMO9) в соотношении 10 : 1 и 10 : 2 на аморфную целлюлозу (Ks варьировал от 1.2 до 1.6 при времени реакции 30–60 мин) [24].
ЛПМО, как C1-, так и смешанного C1/C4-типа, как правило, проявляли синергизм также и с целлобиогидролазами 6 семейства гликозидгидролаз (ЦБГ II) при действии на аморфную и кристаллическую целлюлозу [25–28]. При этом отмечалось, что степень синергизма выше для аморфной целлюлозы, достигая в начальный период реакции значений 1.9–2.6 для смесей ЦБГ II T. reesei с ЛПМО из Lentinus similis, Thermoascus aurantiacus и T. terrestris, тогда как в случае МКЦ Ks варьировал в диапазоне 1.3–2.0 для тех же ферментов [28], что хорошо согласуется с данными, полученными в настоящей работе.
Что же касается литературных данных относительно взаимодействия ЛПМО разного типа с целлобиогидролазами 7 семейства (ЦБГ I), то они довольно противоречивы, причем расхождения, как правило, касаются ЛПМО C1-типа. Так, в работе [25] было показано, что при совместном действии MtLPMO9L C1-типа из M. thermophila и ЦБГ I в соотношении 1 : 10 или 1 : 1 на аморфную целлюлозу наблюдался умеренный синергизм, тогда как в случае использования МКЦ в качестве субстрата ферменты проявляли антагонизм. Напротив, ЛПМО из Neurospora crassa, обладающая сходным (C1) механизмом катализа, оказывала ингибирующее действие на ЦБГ I T. reesei при гидролизе целлюлозы, содержащей в значительном количестве аморфные зоны, однако проявляла заметный синергизм с ЦБГ I при действии на МКЦ и нанокристаллическую целлюлозу [23]. Авторы указанной работы [23] объясняли наблюдаемые эффекты различиями в морфологии целлюлозных субстратов. Как и в настоящей работе, TtLPMO C1-типа ингибировала действие ЦБГ I (однако, из другого источника – T. reesei) независимо от типа субстрата – аморфной целлюлозы или МКЦ, тогда как ЛПМО смешанного типа из L. similis и T. aurantiacus проявляли синергизм с ЦБГ I [28].
В заключение необходимо отметить, что полученные в данной работе результаты будут полезными при разработке современных ферментных препаратов на основе целлюлаз и вспомогательных ферментов, обладающих повышенной осахаривающей способностью в процессах биоконверсии возобновляемого лигноцеллюлозного сырья.
Работа выполнена в рамках НИР “Молекулярный дизайн, структурно-функциональный анализ и регуляция ферментных систем, клеточных конструкций, бионаноматериалов: фундаментальные основы и приложения в технологии, медицине, охране окружающей среды”, номер госрегистрации АААА-А21-121011290089-4.
Конфликт интересов. Авторы заявляют об отсутствии конфликта интересов.
Соблюдение этических норм. Настоящая статья не содержит описания каких-либо исследований с участием людей или животных в качестве объектов.
Список литературы
Lynd L.R., Weimer P.J., van Zyl W.H., Pretorius I.S. // Microbiol. Molec. Biol. Rev. 2002. V. 66. № 3. P. 506–577. https://doi.org/10.1128/MMBR.66.3.506-577.2002
Tiwari R., Nain L., Labrou N.E., Shukla P. // Crit. Rev. Microbiol. 2018. V. 44. № 2. P. 244–257. https://doi.org/10.1080/1040841X.2017.1337713
Vaaje-Kolstad G., Westereng B., Horn S.J., Liu Z., Zgai H., Sørlie M., Eijsink V.G.H. // Science. 2010. V. 330. P. 219–222. https://doi.org/10.1126/science.1192231
Quinlan R.J., Sweeney M.D., Leggio L.L., Otten H., Poulsen J.-C.N., Johansen K.S., Krogh K.B.R.M., Jørgensen C.I., Tovborg M., Anthonsen A., Tryfona T., Walter C.P., Dupree P., Xu F., Davies G.J., Walton P.H. // Proc. Natl. Acad. Sci. USA. 2011. V. 108. № 37. P. 15079–15084. https://doi.org/10.1073/pnas.1105776108
Horn S.J., Vaaje-Kolstad G., Westereng B., Eijsink V.G.H. // Biotechnol. Biofuels. 2012. V. 5. Article № 45. https://doi.org/10.1186/1754-6834-5-45
Pollegioni L., Tonin F., Rosini E. // FEBS J. 2015. V. 282. № 7. P. 1190–1213. https://doi.org/10.1111/febs.13224
Levasseur A., Drula E., Lombard V., Coutinho P.M., Henrissat B. // Biotechnol. Biofuels. 2013. V. 6. Article № 41. https://doi.org/10.1186/1754-6834-6-41
Agger J.W., Isaksen T., Várnai A., Vidal-Melgosa S., Willats W.G.T., Ludwig R., Horn S.J., Eijsink V.G.H., Westereng B. // Proc. Natl. Acad. Sci. USA. 2014. V. 111. № 17. P. 6287–6292. https://doi.org/10.1073/pnas.1323629111
Frommhagen M., Sforza S., Westphal A.H., Visser J., Hinz S.W.A., Koetsier M.J., van Berkel W.J.H., Gruppen H., Kabel M.A. // Biotechnol. Biofuels. 2015. V. 8. Article № 101. https://doi.org/10.1186/s13068-015-0284-1
Jensen M.S., Klinkenberg G., Bissaro B., Chylenski P., Vaaje-Kolstad G., Kvitvang H.F., Nærdal G.K., Sletta H., Forsberg Z., Eijsink V.G.H. // J. Biol. Chem. 2019. V. 294. № 50. P. 19349–19364. https://doi.org/10.1074/jbc.RA119.010056
Phillips C.M., Beeson W.T., Cate J.H., Marletta M.A. // ACS Chem. Biol. 2011. V. 6. № 12. P. 1399–1406. https://doi.org/10.1021/cb200351y
Li X., Beeson W.T., Phillips C.M., Marletta M.A., Cate J.H.D. // Structure. 2012. V. 20. № 6. P. 1051–1061. https://doi.org/10.1016/j.str.2012.04.002
Hemsworth G.R., Davies G.J., Walton P.H. // Curr. Opin. Struct. Biol. 2013. V. 23. № 5. P. 660–668. https://doi.org/10.1016/j.sbi.2013.05.006
Müller G., Várnai A., Johansen K.S., Eijsink V.G.H., Horn S.J. // Biotechnol. Biofuels. 2015. V. 8. Article № 187. https://doi.org/10.1186/s13068-015-0376-y
Kim I.J., Nam K.H., Yun E.J., Kim S., Youn H.J., Lee H.J., Choi I.-G., Kim K.H. // Appl. Microbiol. Biotechnol. 2015. V. 99. P. 8537–8547. https://doi.org/10.1007/s00253-015-6592-3
Sun F.F., Hong J., Hu J., Saddler J.N., Fang X., Zhang Z., Shen S. // Enzyme Microb. Technol. 2015. V. 79–80. P. 42–48. https://doi.org/10.1016/j.enzmictec.2015.06.020
Ghatge S.S., Telke A.A., Waghmode T.R., Lee Y., Lee K.-W., Oh D.-B., Shin H.-D., Kim S.-W. // Appl. Microbiol. Biotechnol. 2015. V. 99. P. 3041–3055. https://doi.org/10.1007/s00253-014-6116-6
Hu J., Chandra R., Arantes V., Gourlay K., van Dyk J.S., Saddler, J. // Biores. Technol. 2015. V. 186. P. 149–153. https://doi.org/10.1016/j.biortech.2015.03.055
Булахов А.Г., Гусаков А.В., Чекушина А.В., Сатрутдинов А.Д., Кошелев А.В., Матыс В.Ю., Синицын А.П. // Биохимия. 2016. Т. 81. № 5. С. 701–709.
Kim I.J., Seo N., An H.J., Kim J.-H., Harris P.V., Kim K.H. // Biotechnol. Biofuels. 2017. V. 10. Article № 46. https://doi.org/10.1186/s13068-017-0721-4
Long L., Yang H., Ren H., Liu R., Sun F.F., Xiao Z., Hu J., Xu Z. // ACS Sustainable Chem. Eng. 2020. V. 8. № 32. P. 11986–11993. https://doi.org/10.1021/acssuschemeng.0c02564
Guo X., Sang J., Chai C., An Y., Wei Z., Zhang H., Ma L., Dai Y., Lu F., Liu F. // Biochem. Eng. J. 2020. V. 162. Article № 107712. https://doi.org/10.1016/j.bej.2020.107712
Eibinger M., Ganner T., Bubner P., Rosker S., Kracher D., Haltrich D., Ludwig R., Plank H., Nidetzky B. // J. Biol. Chem. 2014. V. 289. № 52. P. 35929–35938. https://doi.org/10.1074/jbc.M114.602227
Karnaouri A., Muraleedharan M.N., Dimarogona M., Topakas E., Rova U., Sandren M., Christakopoulos P. // Biotechnol. Biofuels. 2017. V. 10. Article № 126. https://doi.org/10.1186/s13068-017-0813-1
Zhou H., Li T., Yu Z., Ju J., Zhang H., Tan H., Li K., Yin H. // Int. J. Biol. Macromol. 2019. V. 139. P. 570–576. https://doi.org/10.1016/j.ijbiomac.2019.08.004
Garrido M.M., Landoni M., Sabbadin F., Valacco M.P., Couto A., Bruce N.C., Wirth S.A., Campos, E. // Appl. Microbiol. Biotechnol. 2020. V. 104. P. 9631–9643. https://doi.org/10.1007/s00253-020-10911-6
Keller M.B., Badino S.F., Blossom B.M., McBrayer B., Borch K., Westh P. // ACS Sustainable Chem. Eng. 2020. V. 8. № 37. P. 14117–14126. https://doi.org/10.1021/acssuschemeng.0c04779
Tokin R., Ipsen J.O., Westh P., Johansen K.S. // Biotechnol. Lett. 2020. V. 42. P. 1975–1984. https://doi.org/10.1007/s10529-020-02922-0
Pierce B.C., Agger J.W., Wichmann J., Meyer A.S. // Enzyme Microb. Technol. 2017. V. 98. P. 58–66. https://doi.org/10.1016/j.enzmictec.2016.12.007
Semenova M.V., Gusakov A.V., Telitsin V.D., Rozhkova A.M., Kondratyeva E.G., Sinitsyn A.P. // Biochim. Biophys. Acta – Proteins and Proteomics. 2020. V. 1868. № 1. Article № 140297. https://doi.org/10.1016/j.bbapap.2019.140297
Morozova V.V., Gusakov A.V., Andrianov R.M., Pravilnikov A.G., Osipov D.O., Sinitsyn A.P. // Biotechnol. J. 2010. V. 5. № 8. P. 871–880. https://doi.org/10.1002/biot.201000050
Dotsenko A.S., Rozhkova A.M., Gusakov A.V. // Moscow Univ. Chem. Bull. 2015. V. 70. № 6. P. 283–286. https://doi.org/10.3103/S0027131415060024
Korotkova O.G., Semenova M.V., Morozova V.V., Zorov I.N., Sokolova L.M., Bubnova T.M., Okunev O.N., Sinitsyn A.P. // Biochemistry (Moscow). 2009. V. 74. № 5. P. 569–577. https://doi.org/10.1134/S0006297909050137
Wood T.M. // Methods Enzymol. 1988. V. 160. P. 19–25. https://doi.org/10.1016/0076-6879(88)60103-0
Nelson N. // J. Biol. Chem. 1944. V. 153. P. 375–380.
Peterson G.L. // Anal. Biochem. 1979. V. 100. № 2. P. 201–220. https://doi.org/10.1016/0003-2697(79)90222-7
Westereng B., Agger J.W., Horn S.J., Vaaje-Kolstad G., Aachmann F.L., Stenstrøm Y.H., Eijsink V.G.H. // J. Chromatogr. A. 2013. V. 1271. № 1. P. 144–152. https://doi.org/10.1016/j.chroma.2012.11.048
Hildebrand A., Addison J.B., Kasugo T., Fan Z. // Biochem. Eng. J. 2016. V. 109. P. 236–242. https://doi.org/10.1016/j.bej.2016.01.024
Villares A., Moreau C., Bennati-Granier C., Garajova S., Foucat L., Falourd X., Saake B., Berrin J.-G., Cathala B. // Sci. Rep. 2017. V. 7. Article № 40262. https://doi.org/10.1038/srep40262
Song B., Li B., Wang X., Shen W., Park S., Collings C., Feng A., Smith S.J., Walton J.D., Ding S.-Y. // Biotechnol. Biofuels. 2018. V. 11. Article № 41. https://doi.org/10.1186/s13068-018-1023-1
Forsberg Z., Sørlie M., Petrović D., Courtade G., Aachmann F.L., Vaaje-Kolstad G., Bissaro B., Røhr A.K., Eijsink V.G.H. // Curr. Opin. Struct. Biol. 2019. V. 59. P. 54–64. https://doi.org/10.1016/j.sbi.2019.02.015
Дополнительные материалы отсутствуют.
Инструменты
Прикладная биохимия и микробиология