

Прикладная биохимия и микробиология, 2022, T. 58, № 3, стр. 211-231
Гибернация рибосом – молекулярная стратегия выживания бактерий (обзор)
Е. А. Хаова 1, 2, Н. М. Кашеварова 1, А. Г. Ткаченко 1, 2, *
1 Институт экологии и генетики микроорганизмов ПФИЦ УрО РАН
614081 Пермь, Россия
2 Пермский государственный национальный исследовательский университет
614990 Пермь, Россия
* E-mail: agtkachenko@iegm.ru
Поступила в редакцию 09.09.2021
После доработки 03.01.2022
Принята к публикации 10.01.2022
- EDN: MYPTZK
- DOI: 10.31857/S0555109922030060
Аннотация
В обзоре рассмотрены механизмы поддержания жизнеспособности бактерий при неблагоприятных воздействиях, основанные на индукции факторов гибернации рибосом, вызывающих торможение синтеза белка посредством подавления функций рибосом. К факторам гибернации относятся рибосом-связывающие белки, инактивирующие 70S рибосомы (RaiA), формирующие неактивные 100S димеры рибосом (RMF, HPF) или действующие в различных фазах трансляционного цикла (RsfS, YqjD, SRA, EttA). Под воздействием факторов гибернации у части клеток популяции формируется дормантное состояние, характеризующееся низкой скоростью метаболических процессов. Это придает им свойство толерантности и, как следствие, способность к персистенции в присутствии антибиотиков и стрессов. Обсуждается роль метаболических факторов (полиамины, индолы) в регуляции уровня экспрессии генов гибернации.
Синтез белка представляет собой один из фундаментальных процессов, составляющих основу жизнедеятельности всех организмов. Несмотря на значительный прогресс, достигнутый в последнее время в понимании функционирования белоксинтезирующего аппарата, многие его аспекты все еще нуждаются в изучении. Это, в частности, касается ответной реакции со стороны рибосом на разнообразные изменения условий внутренней и внешней среды, воздействующих на бактерии и ведущих к остановке роста и переходу части клеток популяции в дормантное (“дремлющее”) состояние [1]. Покоящиеся дормантные клетки характеризуются замедлением процессов метаболизма, образования энергии, биосинтеза и пролиферации, на которые воздействует большинство антибиотиков. В таких условиях антибиотики теряют активность, в результате чего дормантные клетки приобретают толерантность и способность персистировать, то есть сохранять жизнеспособность при воздействии антибиотиков и стрессов. В отличие от антибиотикорезистентных клеток, имеющих генетическую природу устойчивости, персисторное состояние формируется как результат физиологических отклонений метаболизма.
Одним из наименее изученных аспектов образования дормантных клеток является роль факторов гибернации рибосом в регуляции функции рибосом в ответ на стрессы и действие антибиотиков [2]. Известно, что около 60% глобальной биомассы микроорганизмов, находящихся в естественных условиях, испытывают постоянное голодание и воздействие других стрессорных факторов, способствующих формированию у них состояния покоя [3, 4]. Когда бактерии подвергаются неблагоприятным воздействиям, происходит замедление или полная остановка роста за счет действия специализированных механизмов, обеспечивающих снижение скорости энергоемких биосинтетических процессов. При этом в клетках поддерживается минимально необходимый базовый уровень трансляции, за счет которого обеспечивается синтез связанных с рибосомами факторов, ответственных за снижение активности клеток. Часть из них вовлечена в процесс гибернации рибосом [5]. Факторы гибернации отвечают за ингибирование рибосом и снижение активности трансляции, которая восстанавливается при возврате клеток к оптимальным условиям. Структурно-функциональное разнообразие факторов гибернации и формируемых ими неактивных рибосомальных структур, по-видимому, имеет значение для специфической адаптации клеток к разнообразным стрессам и воздействию антибиотиков.
Первооткрыватели персистенции связывали ее формирование с замедлением скорости метаболических процессов и развитием дормантности без изменения генетических свойств бактерий [6]. Представления о природе персисторного состояния как следствия развития дормантности претерпевали изменения в связи с интенсивным развитием направления, связанного с изучением генетической природы адаптации бактерий, а также химической природы сигналов, формируемых в ответ на стресс [7, 8].
Концепция дормантности как фактора формирования персистенции. В настоящее время точка зрения о дормантном состоянии как основной причине образования персисторных клеток вновь становится преобладающей [9, 10]. Во время экспоненциального роста у незначительной части клеток (10–4–10–6) генетически гомогенной бактериальной популяции формируется дормантное состояние за счет случайных (стохастических) отклонений в уровне экспрессии некоторых генов, в частности, кодирующих глобальные регуляторы метаболических процессов [11–13]. В основе объяснения причины этих отклонений лежит концепция бистабильности генной экспрессии, согласно которой различные гены за счет случайных флуктуаций способны переходить из активного состояния в неактивное [13, 14]. При этом случайное сочетание отклонений метаболизма в различных дормантных клетках, по-видимому, является основной причиной формирования их фенотипической гетерогенности.
Постоянное присутствие в бактериальной популяции фенотипически гетерогеных дормантных клеток, способных переживать воздействие антибиотиков и стрессов, привело к формированию концепции уменьшения риска (bet-hedging) как эволюционной стратегии выживания генетически однородной популяции в динамически изменяемом окружении [15]. Таким образом, в популяции еще до начала летального воздействия присутствует некоторое количество дормантных клеток, способных к персистенции, что страхует ее от полного уничтожения.
Коллектив авторов, руководимый Balaban [16, 17], относит процесс формирования стохастических персисторов к спонтанной персистенции (spontaneous persistence), а образуемые при этом персисторы предлагают обозначать как персисторы Типа II. В отличие от них, персисторы Типа I образуются в результате так называемой индуцируемой персистенции (triggered persistence), например, в результате развития дормантного состояния в ответ на комплексное воздействие стрессов, формируемых при переходе клеток в стационарную фазу [18]. В реализации данного механизма участвуют такие компоненты стрессорного ответа, как активные формы кислорода, повреждающие ДНК и индуцирующие SOS ответ [19], RpoS (σS-субъединица РНК-полимеразы) [20, 21], (p)ppGpp как основной регулятор стринджент-ответа [22, 23] и другие [24]. Молекулярные механизмы, принимающие участие в образовании дормантных клеток, могут действовать кооперативно, что приводит к фенотипической гетерогенности групп дормантных клеток, обладающих толерантностью к различным стрессам [25–27]. Экспериментальное исследование персистенции часто связано с использованием модели перехода периодической культуры бактерий в стационарную фазу [28]. При этом сопутствующие стрессы индуцируют факторы, формирующие дормантное состояние, включая факторы гибернации рибосом, что приводит к возрастанию численности субпопуляции персисторных клеток на несколько порядков (индуцируемая персистенция).
В последнее время к идее самодостаточности дормантного состояния как причины развития персистенции стали возвращаться в связи с развитием методов изучения молекулярных механизмов на уровне индивидуальной клетки [29]. На основании результатов использования этих методов предложена концепция глубины дормантности (dormancy depth), от которой, как считают авторы, зависит способность бактериальной клетки формировать персисторное состояние [30].
Известно, что спустя некоторое время после переноса в оптимальные условия (лаг-период), дормантные клетки способны “оживать” (resuscitate) и возобновлять рост (regrowth). Продолжительность лаг-периода (Tregrowth) прямо пропорциональна глубине дормантности, которая зависит от степени выраженности метаболических изменений, обусловливающих уровень толерантности бактериальных клеток к антибиотикам и их способность к персистенции. Согласно данным, полученным на Escherichia coli, сравнительно низкие уровни дормантности имеют персисторные клетки, обозначенные как persister-FR (Fast Regrowth), то есть персисторы, быстро возобновляющие рост (Tregrowth < 12 ч), и persister-SR (Slow Regrowth) – персисторы, медленно возобновляющие рост (12 ч < Tregrowth < 40 ч). Максимальный уровень глубины дормантности характерен для жизнеспособных, но некультивируемых клеток (VBNC, viable but non-culturable), которые могут пребывать в этом состоянии практически неограниченное время, но имеют значение Tregrowth > 3 сут [30].
В модели перехода бактерий из экспоненциальной фазы в стационарную глубина дормантности возрастает пропорционально времени пребывания клеток в стационарной фазе, вплоть до формирования у них VBNC состояния [31–33]. Количество дормантных клеток в периодической культуре E. coli достигает максимума к 24 ч культивирования, после чего наблюдается снижение их численности. При этом после 48 ч пребывания клеток E. coli в стационарной фазе в популяции появляются VBNC клетки [34].
Механизм перехода бактерий в дормантное состояние связывают с образованием внутриклеточных структур, “агресом”, которые представляют собой высокомолекулярные белковые агрегаты, состоящие из белков, участвующих в основных метаболических путях. При этом активность катализирующих метаболические превращения ферментов в значительной степени утрачивается при их включении в структуру агресомы, что рассматривается как основная причина развития дормантности. Наряду с белковыми агрегатами, клетки E. coli, испытывающие стрессорные воздействия, характеризуются также способностью образовывать несколько типов ДНК агрегатов в комплексе с ДНК-связывающими белками DPS (DNA-binding protein of starvation) [35].
В результате процессов агрегации образуется популяция структурно и функционально гетерогенных дормантных клеток, способных выживать в специфических условиях меняющейся среды. Восстановление нормального вегетативного состояния клеток сопровождается переходом белков агресомы в растворенное состояние, условием которого является достаточный уровень энергии, необходимой для функционирования шаперонов и протеаз, ответственных за данный процесс [36].
В последнее время функция АТФ как источника энергии для осуществления переходов между дормантным и вегетативным состояниями бактериальных клеток существенно дополнена его свойствами как одного из наиболее эффективных биологических гидротропов, ответственных за поддержание нормальной обводненности белковых поверхностей и, соответственно, рефолдинга белков [37]. На основании данных о влиянии интенсивности метаболических процессов на вязкость и текучесть отдельных участков цитоплазмы считают, что уровень энергетического состояния клеток играет существенную роль в образовании и растворении белковых агрегатов [38]. При этом низкие энергетические потоки способствуют переходу макромолекул (белки, нуклеиновые кислоты) в агрегированное состояние и формированию дормантности.
Таким образом, персисторы представляют собой субпопуляцию фенотипически гетерогенных бактериальных клеток, временно утративших способность к пролиферации за счет перехода в дормантное состояние, что делает их способными переживать концентрации антибиотиков, летальные для основной части популяции, и возобновлять рост и размножение при переносе в оптимальные условия [39]. Это позволяет всей популяции поддерживать жизнеспособность на уровне, достаточном для появления и отбора более редких наследственно закрепленных мутаций резистентности к антибиотикам [40–42]. Резистентные клетки, в отличие от персисторов, способны размножаться в присутствии антибиотиков, что резко повышает возможность их распространения в окружающей среде и осложняет задачу борьбы с инфекцией.
Согласованное изменение транскрипционно-трансляционного аппарата бактериальной клетки – основа формирования персистенции. Существенную роль в персистообразовании играют метаболические процессы, которые изменяются в ответ на воздействие стрессов стационарной фазы. В этот период формируются стрессорные ответы на голодание, накопление токсических продуктов обмена, закисление, окислительный стресс и другие неблагоприятные факторы. Перечисленные процессы приводят к изменению типа генной экспрессии, уровень которой при этом снижается до 10% от значения, характерного для экспоненциального роста, с преобладанием экспрессии генов стационарной фазы [43] и реорганизацией генома внутри нуклеоида [44, 45].
В ответ на исчерпание сахаров, транспортируемых в клетку посредством фосфотрансферазной системы (ФТС сахаров), и стресс голодания, который сопровождается снижением синтеза аминокислот, в клетке формируется стринджент-ответ (рис. 1). Он представляет собой согласованное торможение биосинтеза белков, ДНК, РНК, пептидогликана, что сопровождается значительным замедлением скорости роста, приводящим к сбережению энергии в условиях голодания [46–48]. Ключевым регулятором стринджент-ответа является гуанозинтетра(пента)фосфат (p)ppGpp, который играет роль сигнала тревоги (alarm) и на этом основании отнесен к группе соединений алармонов (рис. 1а). Ответственными за поддержание определенного уровня (p)ppGpp в клетках E. coli являются стринджент-факторы – белки-ферменты RelA и SpoT. Первый из них синтезирует (p)ppGpp при взаимодействии тРНК, не связанной с аминокислотой, с рибосомой, т.е. в ответ на аминокислотное голодание, второй, в дополнение к менее выраженной (p)ppGpp-синтетазной, обладает также более значительной гидролазной активностью (рис. 1б, 1в). Аминокислотное голодание приводит к накоплению в цитоплазме клетки тРНК, не связанных с аминокислотами, которые взаимодействуют с А-сайтом рибосом. Такое состояние рибосомы распознается RelA белком, который, связываясь с 50S субъединицей рибосомы, инициирует синтез (p)ppGpp. После диссоциации RelA из рибосомального комплекса синтез может быть продолжен. Высокое содержание (p)ppGpp в клетках мобилизует метаболические ресурсы на восполнение клеточного пула аминокислот, что приводит к образованию ацилированных тРНК, их переносу с помощью фактора элонгации EF-Tu в А-сайт рибосом и возобновлению трансляции. Синтетазная активность SpoT изменяется при отклонении удельной скорости элонгации пептидной цепи от максимальной [49, 50]. В регуляции соотношения синтетазной и гидролазной активностей SpoT принимают участие также белки-регуляторы метаболизма, такие как ACP, Rsd и HPr [51–53].
Рис. 1.
Стринджент-ответ, структура и регуляция активности факторов, ответственных за его формирование в бактериальных клетках: а – синтез гуанозинтетрафосфата и гуанозинпентафосфата, (p)ppGpp, в реакциях, катализируемых семейством белков RSH (RelA-SpoT homologue); б – доменная структура SpoT и RelA. Домены белков представлены в порядке, отражающем их относительное расположение в белковой молекуле: гидролитический (HD), синтетический (SYNTH), регуляторные (TGS, CC, ACT). Перечеркиванием обозначено отсутствие гидролазной активности; в – механизм активности RelA; г – механизм регуляции (p)ppGpp-гидролазной и -синтетазной активностей SpoT при глюкозном голодании. Пояснения к рисунку приводятся в тексте.

В настоящее время показано, что, наряду с адаптацией к стрессу голодания, стринджент-ответ вносит существенный вклад в регуляцию многих внутриклеточных процессов, связанных с ростом, вторичным метаболизмом, вирулентностью, биопленкообразованием и персистенцией. Основной его регулятор, (p)ppGpp, вызывает глубокие изменения транскрипции, подавляя синтез стабильных РНК и индуцируя экспрессию генов, кодирующих ферменты биосинтеза аминокислот и факторы стрессорного ответа. В клетках E. coli идентифицировано более 50 мишеней для (p)ppGpp [54]. Действие этого алармона затрагивает около 500 генов и направлено на предотвращение бесполезной траты ресурсов при неблагоприятных воздействиях [50, 55, 56].
К наиболее энергоемким процессам метаболизма относится синтез белка. Поэтому одной из мишеней (p)ppGpp являются рибосомальные структуры на уровне регуляции экспрессии генов рибосомальных РНК, rrnB. Особенностью их промоторной области является присутствие богатой ГЦ основаниями “дискриминаторной” последовательности, связывание которой с (p)ppGpp приводит к дестабилизации открытия промоторов и ингибированию инициации транскрипции [57].
Реорганизация метаболических процессов при формировании дормантного и персисторного состояний в значительной степени обусловлена заменой основной вегетативной σD-субъединицы РНК-полимеразы (σ70, RpoD) на альтернативные, в первую очередь σS (σ38, RpoS – фактор общего стрессорного ответа), которая отвечает за избирательную транскрипцию генов адаптации клеток к многочисленным стрессам стационарной фазы [45]. Основным фактором, ответственным за происходящие замены, является антисигма-фактор Rsd (регулятор σD-субъединицы) [58]. Через него осуществляется регуляция не только транскрипционного профиля стационарных клеток, но и их метаболизма. Rsd способен образовывать комплекс с σD-субъединицей РНК-полимеразы, что приводит к ее инактивации и замене на специфическую для стационарной фазы σS-субъединицу в составе холофермента [59].
Наряду с заменой вегетативной σD-субъединицы РНК-полимеразы на альтернативные субъединицы, Rsd способен, взаимодействуя с SpoT, сдвигать баланс его (p)ppGpp-cинтетазной и гидролазной активностей в сторону последней (рис. 1г). Однако это может происходить только в условиях глюкозного голодания [51]. Во время же нормального роста Rsd присутствует в клетках в неактивном состоянии в связанной форме с гистидин-содержащим белком HPr, переносчиком фосфатной группы к ФТС сахарам. HPr способен также взаимодействовать с различными белками, оказывая регуляторный эффект на широкое разнообразие реакций углеродного и энергетического метаболизма [60]. В отсутствие глюкозного голодания HPr, находясь преимущественно в дефосфорилированной форме, способен связывать Rsd и удерживать его от взаимодействия с другими белками, в том числе с SpoT (рис. 1г). Преобладание фосфорилированного состояния HPr при исчерпании в среде глюкозы приводит к высвобождению Rsd, что открывает ему возможность, взаимодействуя с SpoT, сдвигать баланс активности в сторону гидролиза (p)ppGpp [51, 52, 61].
Обратный эффект описан для ацил-переносящего белка ACP (рис. 1г). Исчерпание глюкозы сопровождается снижением пула ацетил-CoA и жирнокислотным голоданием, во время которых ACP переходит в деацилированное состояние и при связывании с SpoT усиливает его синтетазную активность [52, 53]. Наоборот, в условиях экспоненциального роста ACP присутствует в ацилированной форме и, связываясь с SpoT, сдвигает баланс его активности в сторону гидролиза (p)ppGpp. Предполагается, что Rsd и ACP могут конкурировать между собой за связывание с SpoT во время голодания, оптимизируя при этом клеточный уровень (p)ppGpp [52].
Показано, что чрезмерно высокая концентрация алармона в клетке значительно ограничивает синтез рибосом, тогда как слишком низкое его содержание лимитирует уровень метаболических белков E. coli [62]. Действие Rsd, ACP, HPr и, возможно, других регуляторных белков в конечном итоге направлено на поддержание концентрации (p)ppGpp, оптимальной для обеспечения нормального уровня экспрессии генов, ответственных за адаптацию к стрессу, в том числе генов гибернации рибосом и, следовательно, персистообразования как механизма адаптации бактерий к разнообразным стрессам [10].
Таким образом, наряду со стрессорными ответами, белок-белковые взаимодействия между Rsd, HPr, ACP и SpoT способствуют сохранению согласованного действия процессов транскрипции и трансляции во время стационарной фазы [43].
Гибернация рибосом – основной механизм регуляции синтеза белка в формировании персистенции. Одним из наиболее действенных механизмов, приводящих к быстрой инактивации уже присутствующих в клетке рибосом при переходе бактерий в стационарную фазу, является индукция факторов гибернации рибосом, синтез которых положительно регулируется (p)ppGpp [2, 63]. Факторы гибернации формируют неактивные рибосомальные структуры, 70S мономеры и 100S димеры [43, 64, 65], а также осуществляют другие способы ингибирования рибосом на различных стадиях трансляции. В результате этого в клетках стационарной фазы образуются рибосомальные комплексы с разной трансляционной активностью. Это дает возможность бактериям оперативно реагировать на изменения условий окружающей среды.
Действие факторов гибернации рибосом приводит к остановке роста клетки, при этом бактерии сохраняют способность к его возобновлению в течение 1–2 мин после возврата клеток к нормальным условиям. Показано, что делеция генов, кодирующих факторы гибернации рибосом, приводит к уменьшению выживаемости клеток в стационарной фазе роста и при воздействии различных видов стресса, а также к снижению толерантности к антибиотикам [2]. Кроме того, максимальный уровень экспрессии генов гибернации рибосом наблюдается во время перехода периодической культуры E. coli в стационарную фазу [5, 21, 66]. Однако функциональная активность белков гибернации, отвечающих за их выход из комплекса с рибосомами и возобновление роста дормантных клеток при переносе культуры в свежую среду, возрастает пропорционально длительности их пребывания в стационарной фазе [5]. Это указывает на возможность того, что факторы гибернации рибосом могли бы участвовать в регуляции длительности периода “пробуждения”, а следовательно, и глубины дормантности бактериальных клеток [30].
Поскольку мишенью воздействия факторов гибернации являются рибосомы, знание структурно-функциональных особенностей этих органелл важно для понимания механизма действия факторов гибернации.
Как известно, рибосома представляет собой рибонуклеопротеидный комплекс, состоящий из двух субъединиц: большой 50S и малой 30S. Большая субъединица содержит около 30 рибосомальных белков, 23S рРНК и 5S рРНК, тогда как малая субъединица состоит из 21 рибосомального белка и 16S рРНК. На границе между двумя субъединицами локализованы участки рибосомальных РНК. Малая субъединица связывает мРНК благодаря расположению в ней фрагмента 16S рРНК, комплементарного последовательности Шайна-Дальгарно (SD) мРНК. Рибосома содержит 3 сайта связывания тРНК: А-, Р- и Е-сайты. В А-сайте связывается аминоацил-тРНК, комплементарная кодону мРНК.
Большая субъединица содержит в себе каталитический центр рибосомы, где происходит формирование пептидной связи между синтезирующейся пептидной цепочкой, связанной с тРНК в Р-сайте, и аминокислотным остатком, связанным с тРНК в А-сайте. После пептидилтрансферазной реакции происходит транслокация, в результате которой тРНК, не связанная с аминокислотой, временно находится в Е-сайте перед ее выходом из рибосомального комплекса.
Трансляция состоит из 3 основных стадий: инициации, элонгации, терминации (рис. 2). На первой стадии формируется 70S инициаторный комплекс, который состоит из большой и малой субъединиц, мРНК и инициаторной формил-метионил-тРНК, связанной в Р-сайте. В течение элонгации происходит включение аминокислотных остатков в растущую пептидную цепь. Рибосомальный комплекс, перемещающийся вдоль мРНК, в конечном итоге приходит в контакт со стоп-кодоном, индуцирующим терминацию, что приводит к высвобождению синтезированного полипептида.

Протекание всех стадий трансляции обеспечивается вспомогательными факторами (рис. 2). Первая стадия трансляции обслуживается тремя факторами инициации: IF1, IF2 (ГТФаза) и IF3, происходит за счет использования энергии гидролиза фосфатной группы ГТФ и завершается образованием комплекса 30S и 50S субъединиц на мРНК с загруженной инициаторной тРНК в P-сайте. Данные факторы модулируют аффинность Р-сайта по отношению к инициаторной формил-метионил-тРНК и настраивают сборку инициаторного комплекса для обеспечения точного начала синтеза белка. На стадии элонгации функционируют два фактора, являющиеся ГТФазами: EF-Tu и EF-G. Первый из них обеспечивает точный подбор аминоацил-тРНК и формирование пептидной связи, после чего второй способствует транслокации. Завершается трансляция благодаря функционированию факторов RF1 и RF2, которые стимулируются третьим фактором – ГТФазой RF3 и специфично распознают различные стоп-кодоны [2, 67]. Терминация завершается освобождением вновь синтезированного полипептида после распознавания стоп-кодона. Рециклирующий фактор рибосом RRF совместно с фактором элонгации EF-G подготавливают трансляционный аппарат для последующих процессов инициации (рециклирование).
Механизмы действия факторов гибернации рибосом. Гибернация рибосом почти повсеместно встречается у бактерий, а также в пластидах растений [68]. Стрессорные воздействия или переход бактериальных клеток в стационарную фазу сопровождаются прекращением роста и диссоциацией части рибосом на быстро деградирующие 30S и накапливающиеся по мере голодания 50S субъединицы [5]. Кроме того, значительные изменения трансляционного аппарата при стрессе обусловлены димеризацией 70S рибосом с образованием неактивных 100S комплексов (рис. 3) [69]. 100S димеры, отсутствующие в экспоненциальной фазе, формируются при переходе в стационарную фазу и далее сохраняются на всем ее протяжении [70–73]. Они могут составлять до 40–60% от общего содержания рибосом в клетке [2, 73]. 100S димеры способны быстро диссоциировать на отдельные 70S рибосомы в течение 1 мин после наступления благоприятных условий [71, 74, 75].
Рис. 3.
Механизмы действия факторов гибернации рибосом E. coli. Пояснения к рисунку приводятся в тексте.
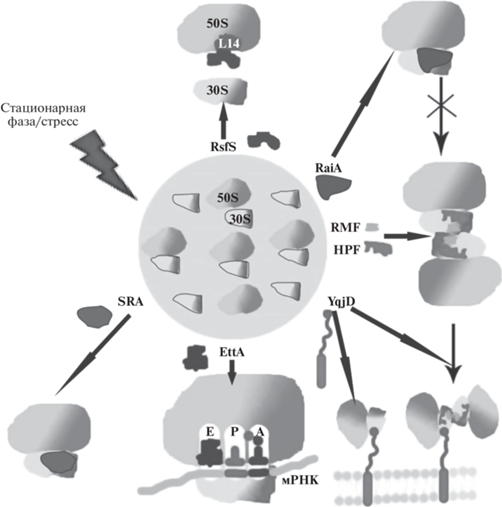
У E. coli (как и остальных представителей гаммапротеобактерий) 100S димеры формируются под воздействием двух факторов гибернации: RMF (Ribosome Modulation Factor) и HPF (Hibernation Promoting Factor) [76] (рис. 3). RMF формирует 90S промежуточный димер, который далее преобразуется, благодаря действию HPF, в зрелый 100S димер [77] (рис. 4). В составе 100S комплекса мономеры взаимодействуют друг с другом посредством 30S субъединиц, связанных при участии RMF, расположенного в непосредственной близости к рибосомальным белкам S13, L13, L2 и Р-сайту [78].
Рис. 4.
Модель формирования персисторов E. coli при участии факторов гибернации. Пояснения к рисунку приводятся в тексте.

Известно, что делеция гена rmf приводит к неспособности бактерий формировать 100S рибосомальные димеры [79]. Несмотря на кажущуюся вспомогательную роль HPF, делеционный мутант по данному фактору также не образует 100S димеры.
Действие третьего фактора гибернации рибосом RaiA (Ribosome-Associated Inhibitor A), прежние названия YfiA и pY, обусловлено его способностью связываться с 70S рибосомами и инактивировать их трансляционную функцию. Предполагается, что RaiA обладает антагонистическими свойствами по отношению к фактору HPF, препятствуя его связыванию с рибосомами, тем самым сдвигая баланс между 100S димерами и неактивными 70S мономерами в сторону последних. Как известно, HPF и RaiA на 40% гомологичны по аминокислотной последовательности [71], а также имеют общий сайт связывания с рибосомами [69], что может быть причиной конкурентных отношений между ними. При этом RaiA, взаимодействуя с рибосомами, блокирует их А- и Р-сайты [80], чему способствует его удлиненный С-терминальный домен, который, кроме того, препятствует связыванию RMF с рибосомой и, следовательно, подавляет образование димеров (рис. 3, 4). Кроме того, сайт связывания RaiA перекрывается с сайтами связывания факторов IF1, IF3, EF-G, тем самым нарушая нормальное протекание процессов инициации и элонгации [69]. Эти особенности структуры и локализации RaiA обусловливают его способность стабилизировать и инактивировать 70S рибосомы [81]. Это подтверждается тем, что в делеционных мутантах ∆raiA содержание 100S рибосом выше, чем в клетках дикого типа [71]. Таким образом, способность бактерий образовывать различные рибосомальные комплексы – RMF-HPF-100S или RaiA-70S – обусловливает присутствие в клетках гетерогенных неактивных рибосомальных структур, что, предположительно, делает их специфичными для формирования персисторов, толерантных к определенным антибиотикам и стрессам.
В отличие от представителей гаммапротеобактерий, имеющих факторы гибернации рибосом RMF, HPF и RaiA, большая часть бактерий, не относящихся к данной категории, а также пластиды растений содержат единственный фактор гибернации рибосом – lHPF (long HPF). Последний представляет собой гомолог фактора HPF, который, в отличие от него, имеет удлиненный С‑терминальный домен, что дает lHPF возможность формировать неактивные 70S и 100S рибосомы. При этом комплекс lHPF-70S является не только интермедиатом 100S димера, но и представляет собой зрелую неактивную форму, дополняющую пул неактивных рибосом, подобно комплексу RaiA-70S [65, 77, 82].
Наряду с этим, среди бактерий и эукариот распространены альтернативные способы инактивации рибосом, в том числе действующие при участии RsfS (Ribosome silencing factor S), еще одного из представителей факторов гибернации рибосом [83]. RsfS препятствует окончательной сборке рибосомы посредством связывания с 50S субъединицей через L14 рибосомальный белок (рис. 3). Данный механизм гибернации, наряду с другими, обеспечивает мобильную перестройку клеточного метаболизма, обратимую при возникновении благоприятных условий, когда комплекс RsfS c 50S субъединицей диссоциирует, а малые и большие субъединицы участвуют в сборке функциональных рибосом [83, 84].
Среди описанных факторов гибернации рибосом представляет также интерес YqjD, который посредством своего N-терминального домена связывается с 30S субъединицей в составе 70S или 100S рибосомальных комплексов. В то же время С-терминальный домен YqjD, содержащий трансмембранный мотив, участвует в интеграции рибосомальных комплексов во внутренней мембране грам-отрицательных бактерий и, таким образом, выводит их из клеточного метаболизма [85].
Фактор гибернации рибосом EttA (Energy-dependent translational throttle A) является АТФ-связывающим белком семейства АВС-F [86] (рис. 3). Его взаимодействие с рибосомой происходит в Е-сайте, что вызывает ингибирование трансляции в промежутке между формированием первой пептидной связи и первой транслокацией, индуцируемой фактором элонгации EF-G. Диссоциация EttA происходит за счет гидролиза АТФ [87, 88]. Таким образом, особенностью действия EttA является то, что он способен непосредственно воспринимать уровень АТФ в бактериальной клетке и ингибировать трансляцию в ответ на его снижение.
SRA (Stationary-phase-induced ribosome-associated) – небольшой белок 5 kDa, обнаруженный у энтеробактерий. Известно, что SRA связывается с 30S субъединицей рибосомы в стационарной фазе [89]. ơS-зависимая транскрипция sra в условиях стресса положительно регулируется сигнальной молекулой (p)ppGpp, а также гистоноподобными белками H-NS и FIS [89]. Поскольку SRA мало изучен, его роль до сих пор остается до конца не известной. Однако, основываясь на относительно малочисленных экспериментальных данных, можно предположить, что SRA имеет функцию, сходную с таковой для факторов гибернации рибосом (рис. 3).
Механизмы диссоциации факторов гибернации рибосом и снятия их ингибирующего действия мало исследованы. Известно лишь, что диссоциация 100S димеров у E. coli и Staphylococcus aureus ос-уществляется высоко консервативным белком, ГТФазой HflX, преимущественно при повышенной температуре. При нормальных же условиях культивирования более значительную роль в диссоциации 100S димеров играют трансляционные факторы RRF и EF-G [64, 82, 90].
На основании обобщения представленных в литературе данных о механизмах, регулирующих трансляционную активность рибосом, можно прийти к заключению, что “биологическая целесообразность” регуляторных механизмов в первую очередь обусловлена необходимостью избежать непроизводительных затрат энергии на один из самых энергоемких процессов – синтез белка – в условиях неблагоприятных воздействий, несовместимых с ростом. В соответствии с этим, каждая из рибосом, присутствующих в бактериальной клетке в данный момент времени, может находиться как в активном, так и неактивном состоянии, благодаря действию различных механизмов гибернации рибосом, формирующих, таким образом, гетерогенный набор их неактивных форм.
Таким образом, гетерогенность рибосомальных структур играет ключевую роль в выживании бактерий в меняющихся условиях окружающей среды. Структурно-функциональное разнообразие рибосом создает для бактериальных клеток ряд преимуществ:
1. Гетерогенность – это страхующий механизм. В случае, если по какой-либо причине не сработает один из механизмов гибернации рибосом, его функции выполнит другой механизм.
2. Различные виды неактивных рибосом имеют разное соотношение скоростей ассоциации-диссоциации с факторами гибернации, что позволяет предположить возможность разграничения действия разных факторов гибернации во времени.
3. Функциональная дифференциация различных факторов гибернации рибосом отражается на эффективности их участия в противостоянии стрессам разной природы.
Физиологическая роль факторов гибернации рибосом. Экспрессия факторов гибернации рибосом находится под контролем механизмов общего стрессорного ответа, включая rpoS регулон и стринджент-ответ (рис. 1). Эти механизмы универсальны и носят генерализованный характер, то есть функционируют при различных стрессах, и их действие охватывает значительную часть клеточных процессов. Итогом является изменение экспрессионного профиля клетки [91], в том числе за счет индукции факторов гибернации рибосом, экспрессия которых находится на базовом уровне в экспоненциальной фазе, но возрастает при переходе в стационарную фазу [74, 83, 87, 92].
Известно, что транскрипция генов rmf, hpf и raiA индуцируется сигналом стринджент-ответа, (p)ppGpp, и не зависит от RpoS [2, 93]. В то же время экспрессия yqjD находится под строгим контролем RpoS [21, 85]. rpoS-регулон представляет собой совокупность генов, промоторы которых имеют сродство к ơS-субъединице РНК-полимеразы. Продукты этих генов выполняют разнообразные функции в стационарной фазе, а также при адаптации к различным стрессам [94, 95]. Зависимость экспрессии факторов гибернации рибосом от разных стрессорных механизмов может быть связана с разделением функций между ними. Наряду с (p)ppGpp, транскрипция rmf и raiA зависит также от транскрипционного регулятора cAMP-CRP [96].
В результате регуляции экспрессии на транскрипционном уровне белок RMF присутствует в клетках только в течение стационарной фазы и деградирует в течение 1 мин после переноса клеток в свежую питательную среду.
Результаты исследований показывают, что транскрипция rmf индуцируется в ответ на разнообразные стрессы, включая аминокислотное голодание, тепловой и холодовой шок, воздействие этанола и бромистого этидия, осмотический шок, кислотный стресс [65, 97, 98]. Остальные факторы гибернации рибосом менее изучены, известно только, что содержание RaiA в клетке также возрастает в ответ на холодовой шок [80, 92].
Физиологическая роль факторов гибернации рибосом заключается в выживании бактериальной клетки при воздействии неблагоприятных условий. У большинства протеобактерий, в том числе E. coli, описаны различные механизмы гибернации, подробно рассмотренные в главе “Механизмы действия факторов гибернации рибосом” (рис. 3). Поскольку основные факторы гибернации (RMF, HPF и RaiA) индуцируются в присутствии (p)ppGpp и cAMP, их содержание в клетке возрастает в стационарной фазе и состоянии глюкозного голодания (рис. 4) [74, 99]. В то же время, определенный уровень RaiA регистрируется также в экспоненциальной фазе [75]. Это может играть существенную роль в тонкой настройке процесса рибосомальной гибернации посредством конкуренции белковых факторов за связывание с рибосомами. При этом обеспечивается максимальное предохранение рРНК от расщепления и быстрый возврат рибосом в активное состояние после снятия голодания и стресса.
Сходную роль играет также другой рибосом-связывающий белок SRA (рис. 3), взаимодействующий с рибосомами при лимитировании питательных компонентов и переходе клеток в стационарную фазу. SRA рассматривается как дополнительный рибосомальный белок S22 30S субъединицы, который функционирует как фактор гибернации рибосом совместно с другим рибосом-связывающим белком RimJ, играющим роль в созревании 30S субъединицы [100]. Взаимодействие всех рибосомальных белков с рРНК приводит к полной сборке и созреванию рибосом. Кроме того, совместные функции белков S22 и RimJ приводят к снижению синтеза DnaA и, таким образом, участвуют в тонкой настройке инициации репликации. Таким образом, данные факторы участвуют в синхронизации процессов транскрипции, трансляции и репликации в условиях стрессорных ситуаций.
Наиболее полная картина, дающая представление о физиологической роли факторов гибернации, получена с использованием делеционных мутантов по генам рибосомальной гибернации. В частности, показано, что у мутантов ∆rmf снижена выживаемость в стационарной фазе [72, 79] и наблюдается более высокая чувствительность к тепловому [101] и осмотическому шоку [102], а также кислотному стрессу [97]. Мутанты ∆rsfS проявляют сниженную выживаемость при переносе клеток в бедную питательную среду и демонстрируют своеобразную динамику роста. После смены питательной среды рост мутанта не отличается от дикого типа, но в середине экспоненциальной фазы наступает внезапная остановка роста, который восстанавливается только по истечении 12 ч дальнейшей инкубации [83]. На питательной среде LB показана пониженная выживаемость мутантов ∆ettA [87].
Кроме того, делеция генов, кодирующих факторы гибернации рибосом, снижает толерантность к аминогликозидным антибиотикам, мишенью которых являются рибосомы. Бактерицидное действие аминогликозидов, как известно, заключается в ингибировании трансляции и образовании аномальных белков, неспособных выполнять свои функции [103]. Мутант ∆hpf демонстрирует сниженную толерантность к гентамицину, ∆rmf – к гентамицину и нетилмицину [104], ∆yqjD – к нетилмицину [21].
В целом, анализируя результаты влияния генных делеций по факторам гибернации рибосом можно прийти к заключению об их вовлеченности в процессы выживания бактерий в условиях воздействия антибиотиков и стрессов.
Роль факторов гибернации рибосом в бактериальной персистенции. В последнее время внимание исследователей к факторам гибернации рибосом возросло в связи с их возможным участием в формировании персистенции. Условия среды обитания бактериальных популяций, как известно, переменчивы по ряду факторов, определяющих скорость роста и характер метаболизма. Неоптимальные значения любого фактора среды могут приводить к замедлению или полной остановке роста, поэтому нормальными для микроорганизмов в их естественном местообитании являются переходные состояния между ростом и его отсутствием. Одним из состояний, которые характеризуются торможением пролиферативных и метаболических процессов, является персистенция.
Наиболее полно бактериальная персистенция изучена в отношении действия антибиотиков. Известно, что персисторы – это малочисленная субпопуляция клеток, часто составляющая около 1% от общего их числа в популяции, которые находятся в дормантном состоянии и, благодаря этому, не имеют мишеней для воздействия антибиотиков, нарушающих ростовые процессы. Поэтому персисторы часто, хотя и не всегда, обладают множественной толерантностью к антибиотикам. После снятия воздействия антибактериального препарата персисторные клетки способны возобновить рост. В связи с этим персистенция представляет собой основную причину рецидивов инфекционных заболеваний [17, 27, 105–107]. Фенотипическая гетерогенность, лабильность, стохастический и индуцибельный характер формирования, полигенная природа, а также низкий уровень содержания персисторов в популяции делают их трудно исследуемым объектом [107–109].
Показано, что ключевую роль в персистенции играет (p)ppGpp – сигнальная молекула стринджент-ответа [46, 110]. Несмотря на то, что участие (p)ppGpp в персистенции не подвергается сомнению, окончательные механизмы его действия до сих пор не установлены. Ранее считалось, что алармон действует через токсин-антитоксиновые системы. Однако эта модель не приобрела достаточной доказательной базы [111].
На современном этапе одной из признанных моделей формирования персистенции является (p)ppGpp индукция персистообразования, опосредованная факторами гибернации рибосом (рис. 4) [63]. Данная модель, которую авторы обозначили аббревиатурой PRDP (ppGpp ribosome dimerization persister), основана на том, что ppGpp генерирует персисторные клетки путем индукции уровня экспрессии генов, кодирующих факторы гибернации рибосом (RMF, HPF, RaiA), связывание которых с 70S рибосомами (RaiA) или участие в образовании 100S димерных рибосомальных комплексов (RMF, HPF) приводит к инактивации функции рибосом. Эта гипотеза нашла экспериментальное подтверждение в том, что делеционные мутанты по генам гибернации рибосом демонстрируют существенное снижение частоты персистенции при воздействии ампициллина и ципрофлоксацина, наиболее выраженные в отношении ∆rmf мутанта [63].
Наряду с (p)ppGpp, цАМФ также индуцирует факторы гибернации RaiA, RMF, HPF в условиях стресса (рис. 4). При этом данные сигнальные молекулы ингибируют фактор HflX, ответственный за диссоциацию димеров рибосом. Это, в свою очередь, приводит к образованию неактивных форм рибосом и индукции персистенции. Возобновление поступления питательных веществ (глюкоза) сопровождается дефосфорилированием EIIA белка ФТС системы, что вызывает падение активности аденилатциклазы и, соответственно, содержания цАМФ в клетках (рис. 4). Это, в свою очередь, активирует ГТФазу HflX, расщепляющую 100S димеры на функциональные 70S рибосомы. Поэтому делеция гена, кодирующего ГТФазу HflX, блокирует разделение рибосомальных димеров и реактивацию рибосом, что делает невозможным возврат персисторов в вегетативное состояние и, таким образом, является еще одним прямым доказательством состоятельности представленной гипотезы [63].
Роль метаболических факторов в регуляции синтеза белков гибернации E. coli. Функции полиаминов в тонкой настройке экспрессии генов гибернации. Полиамины – продукты метаболизма аминокислот орнитина, аргинина, метионина – несмотря на сравнительную простоту химической структуры, характеризуются многообразием функциональных активностей и вовлечены в разнообразные процессы, связанные с ростом и регуляцией генной экспрессии. В клетках бактерий, в частности E. coli, они представлены главным образом путресцином (NH2(CH2)4NH2), спермидином (NH2(CH2)3NH(CH2)4NH2) и кадаверином (NH2CH2(CH2)3CH2NH2) [112].
Ранние работы, посвященные выяснению регуляторных функций полиаминов, проведенные с использованием in vitro систем белкового синтеза, показали, что полиамины в 1.5–2 раза стимулируют его скорость и повышают точность трансляции опосредованно через положительный эффект на сборку рибосом [113, 114]. Полиамины дифференцированно повышают синтез некоторых рибосомальных белков, среди которых белки S20 и L34 наиболее подвержены их воздействию на уровне транскрипции, участвуя в связывании рибосомальных субъединиц и 16S рРНК. Синтез же большинства рибосомальных белков сбалансирован и координированно регулируется в зависимости от скорости роста бактерий [115]. При этом сборка 30S субъединицы рибосомы положительно модулируется полиаминами посредством стимуляции ферментативного метилирования двух соседних остатков аденина, располагающихся на 3' конце 16S рРНК [116]. Кроме того, как уже сказано выше, стимулирующий эффект полиаминов осуществляется на уровне синтеза полипептидов на матрице мРНК, при этом действие спермидина усиливается в условиях возрастания содержания урациловых нуклеотидов в составе мРНК [117].
Участие полиаминов в фосфолипидном обмене способствует стабилизации мембранной организации при повреждающих условиях среды, что показано на примере воздействия на клетки E. coli процедуры замораживания-оттаивания [118]. Описано большое число примеров стабилизирующих эффектов полиаминов на различные клеточные компоненты in vitro [119]. Ускоренный рост требует пропорционального возрастания синтеза фосфолипидов как основных мембранных компонентов [120] и происходит за счет усиления их синтеза и снижения скорости катаболизма [121].
Таким образом, ранние работы, описывающие эффекты полиаминов, касались главным образом активно пролиферирующих клеток и связаны с ускорением синтеза белка, а также положительным эффектом на системы фосфолипидного обмена.
Последующие направления исследования функциональных активностей полиаминов получили свое развитие в связи с тем, что в клетках эти соединения присутствуют главным образом в виде комплексов с РНК, в том числе мРНК. Это послужило основанием для отнесения генов, экспрессия которых регулируется при участии полиаминов, в группу, обозначенную как “полиаминовый модулон” [122]. Данная функция полиаминов сохраняется даже в условиях остановки роста, вызванной исчерпанием источников питания или воздействия неблагоприятных факторов, включая антибиотики. При этом снижается не только эффективность генной экспрессии до 10% от уровня экспоненциального роста, но изменяется также генно-экспрессионный профиль бактерий [44, 45]. Благодаря этому, полиамины способны оказывать стимулирующий эффект на процесс трансляции белков, не только вовлеченных в пролиферацию, но и повышающих выживаемость бактериальных клеток в ответ на стрессорные воздействия [21, 123–125]. В настоящее время описано три различных типа механизмов положительной модуляции генной экспрессии полиаминами, действующими на уровне трансляции посредством взаимодействия с мРНК [122] (рис. 5). Механизм первого типа функционирует в мРНК с необычно большим расстоянием (12 нуклеотидов вместо обычных 7) между стартовым кодоном и последовательностью Шайна-Дальгарно (SD), ответственной за связывание мРНК с рибосомой. В присутствии полиаминов (1 мМ спермидина) это расстояние сокращается за счет образования вторичной структуры мРНК в данной области, что приводит к сильной стимуляции процесса инициации трансляции. Такой механизм действует для мРНК oppA, fecI (σ18), fis, rpoN (σ54), hns, rpoE (σ24) stpA, emrR, rmf, rpoZ (ω), cpxR и soxR [117].
Рис. 5.
Полиаминовый модулон. Три механизма стимуляции полиаминами белкового синтеза: 1 – преодоление ограничения трансляции генов с необычно длинным участком мРНК между стартовым кодоном и последовательностью Шайна–Дальгарно (SD) посредством образования вторичной структуры в данной области; 2 – увеличение трансляции генов с неэффективными кодонами инициации UUG или GUG; 3 – положительная модуляция генной экспрессии путем воздействия полиаминов на процесс терминации трансляции, сдвиг рамки считывания +1. Пояснения к рисунку приводятся в тексте.

Примером второго типа регуляции является положительный эффект полиаминов на трансляцию мРНК, где вместо AUG присутствует неэффективный кодон инициации, осуществляющий UUG- или GUG-зависимое связывание формилметионил-тРНК (fMet-tRNA) с cya, cra, spoT, uvrY, frr (RRF) или gshA в комплексе мРНК-рибосома (рис. 5).
Третий тип положительной модуляции генной экспрессии характеризуется воздействием полиаминов на процесс терминации трансляции, что сопровождается ускоренным освобождением завершенных белковых молекул или стимулирует +1 сдвиг рамки считывания на 26-м UGA кодоне мРНК prfB, кодирующем RF2 фактор (рис. 5). Этот механизм действует в отношении синтеза σ38 (RpoS), одной из альтернативных σ-субъединиц РНК-полимеразы, ответственной за транскрипцию генов, чьи продукты обусловливают адаптацию E. coli к переходу в стационарную фазу роста, голоданию, окислительному стрессу и другим факторам среды, характерным для стационарной фазы [126–128].
Стимулирующий эффект полиаминов на трансляцию данной субъединицы проявлялся только в отношении тех штаммов E. coli, у которых мРНК RpoS содержит необычный 33 кодон в области терминатора (UAG вместо обычного CAG) [78, 129]. Полиамины стимулируют считывание данного амбер-кодона терминации как за счет повышения уровня супрессорной тРНК, так и увеличения аффинности связывания Gln-tRNAsupE с рибосомами. Мутация в данном положении часто встречается в природных штаммах E. coli, чем обусловлена избирательность эффекта полиаминов. Их стимулирующее воздействие не проявляется в отношении других кодонов терминации (UGA, UAA), частота встречаемости которых в различных штаммах составляет 29.3 и 63.1% соответственно [130]. Вследствие того, что UAG используется в качестве кодона терминации в сравнительно небольшом числе генов, происходит избирательная стимуляция полиаминами трансляции мРНК RpoS, не затрагивающая экспрессию основной части генов (рис. 5).
Известно, что полиамины функционируют во многих клеточных процессах, в том числе в адаптации к стрессу [27, 131, 132]. Исходя из этого, логично выглядит возможность их вовлеченности в регуляцию персистенции через положительный эффект на экспрессию rpoS [66]. В то же время, до сих пор мало изученными остаются возможные функции полиаминов и других метаболических факторов в регуляции процессов гибернации, принимающих участие в формировании бактериальной персистенции. Исследование влияния генов гибернации рибосом rmf и yqjD на формирование персисторов, толерантных к нетилмицину, показало, что делеционные мутанты по данным генам имеют значительно более низкую частоту персистенции по сравнению с родительским штаммом. При этом наиболее существенный вклад в формирование персисторов вносит rmf, экспрессия которого стимулируется полиаминами в соответствии с концентрационной зависимостью [21, 133]. Сходная картина имеет место в медленно растущих клетках [73, 89]. Максимальная стимуляция rmf в контрольном родительском штамме совпадает по времени с наиболее значительным негативным эффектом делеционной мутации на уровень персистенции мутанта ∆rmf. Это свидетельствует об опосредованном через rmf участии полиаминов в формировании персисторных клеток [21].
Одним из важных аспектов снижения белок синтезирующей способности бактерий при воздействии стрессов является возрастание активности, приводящей к расщеплению 16S рРНК в 30S субъединицах рибосом [134–136]. Показано, что при переходе в стационарную фазу в клетках E. coli происходит фрагментация 16S рРНК, локализованная преимущественно на конце спирали 6 внутри 30S рибосомальной субъединицы [135]. Это сопровождается снижением белоксинтезирующей активности рибосомы [136] и может приводить к задержке ее восстановления после возврата клеток к оптимальным условиям роста, что само по себе является эффективным механизмом гибернации рибосом. Недавно показано, что деградация 16S рРНК обусловлена активностью эндорибонуклеазы YbeY и экзорибонуклеазы RNase R, вводящих разрыв на 3' конце 16S рРНК, локализованном между 30S и 50S субъединицами [5, 136]. В последнее время опубликованы данные, свидетельствующие, что накопление фрагментированных рРНК как в цельных рибосомах, так и в рибосомальных субъединицах значительно возрастает в отсутствие факторов гибернации рибосом [5]. Это указывает на возможность участия последних в защитных механизмах, направленных на поддержание целостности рибосомальной структуры.
В регуляции РНКазной активности принимают участие как факторы гибернации рибосом, так и полиамины, связывание которых с 16S рРНК в определенных локусах приводит к подавлению этой активности [5, 135, 136]. Описанный механизм действует во время формирования клетками ответа на стресс голодания, однако в отсутствие факторов гибернации у делеционных мутантов в местах их потенциального связывания происходит фрагментация 16S рРНК.
Важным фактором в стимуляции сборки 30S субъединицы рибосом E. coli, как известно, являются полиамины [137]. Исходя из этого, становится понятной их функция, направленная, наряду с рибосомальными белками и факторами гибернации, на сохранение целостности 16S рРНК и восстановление синтеза белка после снятия стрессов стационарной фазы.
В течение длительного времени накоплен значительный объем данных о полиаминах как факторах, способствующих осуществлению рибосомальных функций. Константы связывания полиаминов с рибосомальными РНК, как опубликовано, составляют 0.18 × 104 M–1 для спермина и 2.2 × 103 M–1 для спермидина с числом сайтов от 0,11 аминов на фосфат нуклеиновой кислоты для спермидина и в более широком диапазоне (0.082–0.133) для спермина [138]. Знание особенностей сайтов преимущественного связывания полиаминов с РНК важны для понимания механизмов их участия в настройке специфических функций рибосомальных, транспортных и информационных РНК. В настоящее время известно, что такие сайты представлены в структуре РНК главным образом остатками аденина и урацила и располагаются в слабо структурированных областях, включая одноцепочечные петли [135].
Одним из основных видов белковых мишеней для вторичного мессенджера (p)ppGpp, ответственного за развитие стринджент-ответа, являются пиридоксальфосфат-зависимые декарбоксилазы алифатических аминокислот, включая индуцибельную лизиндекарбоксилазу (LdcI/CadA), конститутивную лизиндекарбоксилазу (LdcC), индуцибельную аргининдекарбоксилазу (AdiA), идуцибельную орнитиндекарбоксилазу (SpeF) и конститутивную орнитиндекарбоксилазу (SpeC) [48, 139]. Эти мультидоменные ферменты формируют большие олигомерные комплексы, состоящие из димеров (SpeF, SpeC) или декамеров (LdcI, LdcC, AdiA). Индуцибельные ферменты вовлечены в стрессорные ответы (кислотный стресс), тогда как конститутивные, в частности SpeC, выполняют функции синтеза полиаминов.
Показано, что перечисленные ферменты способны с высокой аффинностью связывать (p)ppGpp, что сопровождается ингибированием их активности. Это объясняется необходимостью сохранения ресурсов аминокислот в условиях голодания и стресса. Однако падение внутриклеточного содержания полиаминов становится реальным скорее лишь в поздней стационарной фазе. В то же время, к моменту прекращения роста бактериальных клеток при переходе в стационарную фазу в культуре накапливается достаточно высокая их концентрация. В таких условиях полиамины способны осуществлять свои регуляторные функции применительно к РНК на уровнях транскрипции и трансляции, несмотря на ингибирование орнитиндекарбоксилаз как реакции на стринджент-ответ. В том числе, это, по-видимому, касается способности полиаминов оказывать положительное воздействие на содержание факторов гибернации. Кроме того, в присутствии ГТФ и ГДФ возможна активация ферментов синтеза полиаминов и возрастание содержания их продуктов [48].
Естественные уровни полиаминов в бактериальных клетках могут контролировать структурно-функциональные свойства РНК, связанные с синтезом белка. Кроме того, полиамины способны модулировать множество дополнительных процессов, обеспечивающих функции ДНК, таких как хеликазная активность. Следовательно, изучение процессов взаимодействия полиаминов с нуклеиновыми кислотами in vivo или in vitro может быть высоко информативным с точки зрения выяснения механизма гибернации рибосом [135].
Таким образом, полиамины обладают способностью осуществлять регуляцию клеточного метаболизма, положительно воздействуя на синтез рибосомальных белков, скорость сборки рибосом, общую скорость белкового синтеза, целостность 16S рРНК, а также генную экспрессию на уровнях транскрипции, трансляции и терминации. Анализ генов, входящих в группу полиаминового модулона, свидетельствует о том, что на сегодняшний день некоторые из генов гибернации рибосом отнесены к этой группе [117].
Функции индола в регуляции активности факторов гибернации рибосом и формировании персисторного состояния бактериальных клеток. Индол представляет собой гетероциклическое ароматическое соединение [140], которое продуцируется более чем 85 видами грам-отрицательных и грам-положительных бактерий, включая их патогенных и непатогенных представителей [141]. В последнее время индол привлекает внимание присущими ему свойствами сигнальной молекулы, используемой как средство внутри- и межвидовой коммуникации, причем не только между бактериальными видами, но и в их взаимодействии с клетками организма-хозяина [141–144]. Образование индола зависит исключительно от количества экзогенного триптофана [145]. При этом клетки E. coli конвертируют эту аминокислоту в равное количество индола, конечный уровень которого в среде может максимально достигать 5 мМ.
Несмотря на присутствие альтернативных транспортеров (AroP и Mtr), продукция индола зависит исключительно от белка TnaB, ответственного за транспорт триптофана из среды, который далее расщепляется триптофаназой TnaA до индола и пирувата аммония [146, 147]. Это обеспечивает условие, при котором в отсутствие экзогенного триптофана TnaA не может гидролизовать внутриклеточный анаболический пул этой аминокислоты и, таким образом, влиять на рост бактерий.
В настоящее время описано, по меньшей мере, два типа действия индола: длительный, но низкоуровневый “персистентный сигнал” и короткоживущий высокоуровневый “пульс сигнал” [148, 149]. Первый из них (0.5–1.0 мМ индола) участвует в формировании устойчивости к множественным стрессам, тогда как второй, обусловленный кратковременным возрастанием внутриклеточной концентрации индола до 60 мМ в момент перехода клеток в стационарную фазу, делает вклад в выживание E. coli при голодании и участвует в формировании персисторного состояния.
Индол, представляя собой межклеточный сигнал в микробных сообществах [150], оказывает многообразный спектр эффектов на физиологию и метаболизм бактерий, включая образование биопленок [143], антибиотикорезистентность [151], образование персисторных клеток [19], вирулентность [152] и другие. Однако молекулярные мишени и механизмы его действия все еще остаются неясными. В частности, это касается роли индола в формировании персисторного состояния у бактерий. Пульс индола в момент перехода бактерий в стационарную фазу рассматривается как механизм, вызывающий прекращение роста для сохранения ресурсов, обеспечивающих выживание в новых условиях [148, 149].
Гены триптофаназы TnaA и транспортного белка TnaB, входящие в tna оперон, находятся под контролем cAMP/CRP-зависимого механизма катаболитной репрессии и индуцируются на уровне транскрипции при исчерпании углеводов и переходе клеток в стационарную фазу [153]. Это приводит к накоплению индола в клетках за счет различий в скоростях его синтеза и выхода в среду. При этом ионофорные свойства индола, как считают, могут быть ответственны за рассеивание энергии трансмембранного потенциала протонов и преждевременную остановку роста [149, 154]. В то же время, низкое энергетическое состояние бактерий рассматривается как один из факторов формирования дормантности, способствующий развитию персистенции [155, 156].
Наряду со стринджент-ответом, который приводит к синтезу (p)ppGpp, снятие катаболитной репрессии при переходе в стационарную фазу роста или при воздействии стрессов сопровождается образованием цАМФ. Оба эти вторичных мессенджера совместно индуцируют факторы гибернации рибосом (RMF, HPF, RaiA), которые также отвечают за формирование персисторов (рис. 4) [2, 157].
В то же время, недавно показано, что субингибиторные (суб-МПК) (МПК – минимальная подавляющая концентрация) концентрации антибиотиков, в частности, аминогликозидов, могут выступать в качестве сигналов, повышающих продукцию индола [158]. В свою очередь, исследование отклика со стороны уровня экспрессии различных генов на добавку индола показало сильное возрастание уровня экспрессии фактора гибернации рибосом raiA, что сопровождалось 100-кратным увеличением числа персисторов, толерантных к аминогликозидам [158]. В то же время, Заркан с соавт. [159] показали участие индола в формировании специфических персисторов, толерантных к фторхинолонам, посредством взаимодействия с АТФ-связывающим сайтом ДНК-гиразы, мишени антибиотиков данного класса. Несмотря на это, на основе широкого анализа данных большинство авторов склонно считать, что в развитии персисторного состояния преобладающими являются генерализованные эффекты индола на метаболизм, приводящие к замедлению его скорости и формированию неспецифической толерантности к антибиотикам и стрессам [148]. К подобным воздействиям можно отнести такие, как: деэнергизация клеток и стимуляция экспрессии факторов гибернации рибосом.
Роль индола в формировании персисторного состояния недавно подтверждена на его естественных производных, индолокинах, представляющих собой нормальные продукты метаболизма бактерий [160]. Этот класс соединений в большом разнообразии представлен в различных видах бактерий, входящих в консорциум микрофлоры кишечника человека, прежде всего, в E. coli. Причем индолокины, как показано авторами, обладают свойствами межвидовых сигналов между бактериями, с одной стороны, и человеком и растениями – с другой [160]. В последнем случае они способны повышать врожденные иммунные ответы человека и растений.
Синтез индолокинов, в отличие от индола, катализируется не триптофаназой TnaA, а аминотрансферазой аминокислот AspC путем переноса аминогруппы от ароматических аминокислот, включая триптофан, на 2-кетоглутарат как преобладающий косубстрат [161], чем обусловлено большое разнообразие индолокинов. Содержание этих соединений возрастало в ответ на сублетальные воздействия антибиотиков, окислительного стресса и при переходе клеток в стационарную фазу, что сопровождалось формированием персисторов. В этот период активировалась функция факторов гибернации рибосом, положительно регулируемых индолами на транскрипционном уровне [2, 158].
Наряду с нормальными продуктами метаболизма, индолокинами, синтезированы и исследованы также разнообразные индольные производные (индигоиды) [157], функции которых авторы связывают с их способностью повреждать мембраны и вызывать прямо противоположный эффект, т.е. гибель персисторных клеток. Возможно, что противоположные эффекты (индукция персистенции [19, 160] либо киллерные функции в отношении персисторов [157]), описанные разными авторами для близких по структуре индольных соединений объясняются существованием тонкой грани между нарушением энергетической функции мембран и их необратимым изменением. При этом необходимо учитывать также различия между глубиной дормантного состояния, формируемого под воздействием различных индольных производных, от которой зависит уровень персистенции. Высшей степенью проявления глубины дормантности является формирование VBNC клеток, возрастание содержания которых в популяции может создавать иллюзию гибели персисторов, поскольку восстановление роста VBNC клеток характеризуется значительно более длительным лаг-периодом (Tregrowth) [30].
Получение эффективных средств на базе индольных производных, усиливающих действие антибиотиков, делает необходимым их углубленное изучение, в том числе применительно к действию на факторы гибернации рибосом и персистообразование.
* * *
Бактерии демонстрируют способность выживания в самых разнообразных условиях среды. Это обусловлено высокой мобильностью их метаболизма, которая достигается за счет согласованного действия регуляторных механизмов, участвующих в формировании адаптивных ответов на стресс. При неблагоприятных условиях среды механизмы общего стрессорного ответа (стринджент-ответ, rpoS-регулон, полиаминовый модулон) индуцируют факторы гибернации рибосом, которые переводят рибосомы в неактивное состояние. К факторам гибернации относятся рибосом-связывающие белки, инактивирующие 70S рибосомы (RaiA), формирующие неактивные 100S димеры (RMF, HPF) или действующие на других стадиях трансляционного цикла (RsfS, YqjD, SRA, EttA). Прекращение биосинтеза белков под действием факторов гибернации вызывает замедление клеточного метаболизма вплоть до полной остановки роста и деления. Это способствует формированию в бактериальной популяции дормантных клеток, обладающих свойствами фенотипической толерантности ко многим антибиотикам и стрессам и способных к персистенции. Механизм перехода бактерий в дормантное состояние связывают с их способностью образовывать внутриклеточные структуры, “агресомы”, которые представляют собой малоактивные высокомолекулярные белковые агрегаты, состоящие из ферментов, участвующих в формировании основных метаболических путей. Обсуждается концепция глубины дормантности, согласно которой персисторное состояние соответствует относительно более низким уровням ее глубины, тогда как состояние некультивируемости (VBNC) имеет максимальное значение глубины дормантности. В регуляции активности факторов гибернации рибосом принимают участие продукты метаболизма бактерий, включая полиамины и индолы. Обсуждаются механизмы воздействия этих соединений на функции факторов гибернации рибосом в процессе формирования персисторного состояния. При переносе дормантных клеток в оптимальные условия происходит освобождение рибосом от факторов гибернации, что приводит к восстановлению нормального роста исходных клеток. В случае патогенных видов бактерий это вызывает рецидивирующее течение инфекционного процесса. Поэтому исследование функции факторов гибернации рибосом становится крайне важным для практической медицины.
Работа поддержана грантом Российского Научного Фонда № 18-73-10156.
Список литературы
Sachidanandham R., Yew-Hoong Gin K. // Appl. Microbiol. Biotechnol. 2009. V. 81. № 5. P. 927–941.
Prossliner T., Winther K.S., Sorensen M.A., Gerdes K. // Annu. Rev. Genet. 2018. V. 52. № 1. P. 321–348.
Gray J.V., Petsko G.A., Johnston G.C., Ringe D., Singer R.A., Werner-Washburne M. // Microbiol. Mol. Biol. Rev. 2004. V. 68. № 2. P. 187–206.
Kolter R., Siegele D.A., Tormo A. // Annu. Rev. Microbiol. 1993. V. 47. P. 855–874.
Prossliner T., Gerdes K., Sørensen M.A., Winther K.S. // Nucleic Acids Res. 2021. V. 49. № 4. P. 2226–2239.
Bigger J. Treatment of Staphylococcal Infections with Penicillin by Intermittent Sterilisation. 1944. https://doi.org/10.1016/S0140-6736(00)74210-3
Nguyen D., Joshi-Datar A., Lepine F., Bauerle E., Olakanmi O., Beer K., McKay G., Siehnel R., Schafhauser J., Wang Y., Britigan B.E., Singh P.K. // Science. 2011. V. 334. № 6058. P. 982–986.
Orman M.A., Brynildsen M.P. // Antimicrob. Agents Chemother. 2013. V. 57. № 7. P. 3230–3239.
Wood T.K., Knabel S.J., Kwan B.W. // Appl. Environ. Microbiol. 2013. V. 79. № 23. P. 7116–7121.
Wood T.K., Song S. // Biofilm. 2020. V. 2. P. 100018. https://doi.org/10.1016/j.bioflm.2019.100018
Choi P.J., Cai L., Frieda K., Xie X.S. // Science. 2008. V. 322. № 5900. P. 442–446.
Dubnau D., Losick R. // Mol. Microbiol. 2006. V. 61. № 3. P. 564–572.
Buerger S., Spoering A., Gavrish E., Leslin C., Ling L., Epstein S.S. // Appl. Environ. Microbiol. 2012. V. 78. № 9. P. 3221–3228.
Kotte O., Volkmer B., Radzikowski J.L., Heinemann M. // Mol. Syst. Biol. 2014. V. 10. № 7. P. 736. https://doi.org/10.15252/msb.20135022
Kussell E., Leibler S. // Science. 2005. V. 309. № 5743. P. 2075–2078.
Balaban N.Q., Merrin J., Chait R., Kowalik L., Leibler S. // Science. 2004. V. 305. № 5690. P. 1622–1625.
Balaban N.Q., Helaine S., Lewis K., Ackermann M., Aldridge B., Andersson D.I., et al. // Nat. Rev. Microbiol. 2019. V. 17. № 7. P. 441–448.
Levin-Reisman I., Balaban N. Quantitative Measurements of Type I and Type II Persisters Using ScanLag. Bacterial Persistence. /Eds. J. Michiels, M. Fauvart. N.Y.: Springer, 2016. P. 75–81.
Vega N.M., Allison K.R., Khalil A.S., Collins J.J. // Nat. Chem. Biol. 2012. V. 8. № 5. P. 431–433.
Battesti A., Majdalani N., Gottesman S. // Proc. Natl Acad. Sci USA. 2015. V. 112. № 16. P. 5159–5164.
Tkachenko A.G., Kashevarova N.M., Tyuleneva E.A., Shumkov M.S. // FEMS Microbiol Lett. 2017. V. 364. № 9. https://doi.org/10.1093/femsle/fnx084
Pacios O., Blasco L., Bleriot I., Fernandez-Garcia L., Ambroa A., López M., et al. // Antimicrob. Agents Chemother. 2020. V. 64. № 10. P. e01283-20.
Tkachenko A.G., Kashevarova N.M., Sidorov R.Y., Nesterova L.Y., Akhova A.V., Tsyganov I.V. et al. // Cell Chem. Biol. 2021. V. 28. № 10. P. 1420–1432.e9.
Paul P., Sahu B.R., Suar M. // Mol. Oral Microbiol. 2019. V. 34. № 3. P. 97–107.
Fridman O., Goldberg A., Ronin I., Shoresh N., Balaban N.Q. // Nature. 2014. V. 513. № 7518. P. 418–421.
Pu Y., Zhao Z., Li Y., Zou J., Ma Q., Zhao Y., et al. // Mol. Cell. 2016. V. 62. № 2. P. 284–294.
Ткаченко А.Г. // Прикл. биохимия и микробиология. 2018. Т. 54. № 2. С. 110–133.
Keren I., Shah D., Spoering A., Kaldalu N., Lewis K. // J. Bacteriol. 2004. V. 186. № 24. P. 8172–8180.
Dewachter L., Bollen C., Wilmaerts D., Louwagie E., Herpels P., Matthay P.,et al. // mBio. 2021. V. 12. № 4. P. e0070321. https://doi.org/10.1128/mBio.00703-21
Pu Y., Li Y., Jin X., Tian T., Ma Q., Zhao Z., Lin S.Y., Chen Z., Li B., Yao G., Leake M.C., Lo C.J., Bai F. // Mol. Cell. 2019. V. 73. № 1. P. 143–156. https://doi.org/10.1016/j.molcel.2018.10.022
Shleeva M., Mukamolova G.V., Young M., Williams H.D., Kaprelyants A.S. // Microbiology. 2004. V. 150. № 6. P. 1687–1697.
Su X., Chen X., Hu J., Shen C., Ding L. // World J. Microbiol. Biotechnol. 2013. V. 29. № 12. P. 2213–2218.
Nystrom T. // Bioessays. 2003. V. 25. № 3. P. 204–211.
Desnues B., Cuny C., Gregori G., Dukan S., Aguilaniu H., Nystrom T. // EMBO Rep. 2003. V. 4. № 4. P. 400–404.
Loiko N., Danilova Y., Moiseenko A., Kovalenko V., Tereshkina K., El-Registan G.I., Sokolova O.S., Kru-pyanskii Y.F. // PLoS One. 2020. V. 15. № 10. e0231562. https://doi.org/10.1371/journal.pone.0231562
Seyffer F., Kummer E., Oguchi Y., Winkler J., Kumar M., Zahn R., Sourjik V., Bukau B., Mogk A. // Nat. Struct. Mol. Biol. 2012. V. 19. № 12. P. 1347–1355.
Patel A., Malinovska L., Saha S., Wang J., Alberti S., Krishnan Y., Hyman A.A. // Science. 2017. V. 356. № 6339. P. 753–756.
Parry B.R., Surovtsev I.V., Cabeen M.T., O’Hern C.S., Dufresne E.R., Jacobs-Wagner C. // Cell. 2014. V. 156. № 1. P. 183–194.
Maisonneuve E., Gerdes K. // Cell. 2014. V. 157. № 3. P. 539–548.
Baym M., Lieberman T.D., Kelsic E.D., Chait R., Gross R., Yelin I., Kishony R. // Science. 2016. V. 353. № 6304. P. 1147–1151.
Lee H.H., Molla M.N., Cantor C.R., Collins J.J. // Nature. 2010. V. 467. № 7311. P. 82–85.
Cohen N.R., Lobritz M.A., Collins J.J. // Cell Host Microbe. 2013. V. 13. № 6. P. 632–642.
Yoshida H., Wada A., Shimada T., Maki Y., Ishihama A. // Front. Genet. 2019. V. 10. P. 1153. https://doi.org/10.3389/fgene.2019.01153
Hengge-Aronis R. // Curr. Opin. Microbiol. 2002. V. 5. № 6. P. 591–595.
Ishihama A. // Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2012. V. 88. № 9. P. 485–508.
Hauryliuk V., Atkinson G.C., Murakami K.S., Tenson T., Gerdes K. // Nat. Rev. Microbiol. 2015. V. 13. № 5. P. 298–309.
Liu K., Bittner A.N., Wang J.D. // Curr. Opin. Microbiol. 2015. V. 24. P. 72–79.
Kanjee U., Ogata K., Houry W.A. // Mol. Microbiol. 2012. V. 85. № 6. P. 1029–1043.
Bremer H., Dennis P. // Biochimie. 2008. V. 90. № 3. P. 493–499.
Srivatsan A., Wang J.D. // Curr. Opin. Microbiol. 2008. V. 11. № 2. P. 100–105.
Lee J.W., Park Y.H., Seok Y.J. // Proc. Natl. Acad. Sci. USA. 2018. V. 115. № 29. P. E6845–E6854.
Fernández-Coll L., Cashel M. // Front. Microbiol. 2020. V. 11. P. 592718. https://doi.org/10.3389/fmicb.2020.592718
Battesti A., Bouveret E. // Mol. Microbiol. 2006. V. 62. № 4. P. 1048–1063.
Wang B., Dai P., Ding D., Del Rosario A., Grant R.A., Pentelute B.L., Laub M.T. //Nat. Chem. Biol. 2019. V. 15. № 2. P. 141–150.
Magnusson L.U., Farewell A., Nystrom T. // Trends Microbiol. 2005. V. 13. № 5. P. 236–242.
Potrykus K., Cashel M. // Annu. Rev. Microbiol. 2008. V. 62. № 1. P. 35–51.
Paul B.J., Ross W., Gaal T., Gourse R.L. // Annu. Rev. Genet. 2004. V. 38. P. 749–770.
Jishage M., Ishihama A. // J. Bacteriol. 1995. V. 177. № 23. P. 6832–6835.
Park Y.H., Lee C.R., Choe M., Seok Y.J. // Proc. Natl Acad. Sci. USA. 2013. V. 110. № 52. P. 21142–21147.
Rodionova I.A., Zhang Z., Mehla J., Goodacre N., Babu M., Emili A., Uetz P., Saier M.H.Jr. // J. Biol. Chem. 2017. V. 292. № 34. P. 14250–14257.
Fernandez-Coll L., Maciag-Dorszynska M., Tailor K., Vadia S., Levin P.A., Szalewska-Palasz A., Cashel M. // mBio. 2020. V. 11. № 2. e03223-19. https://doi.org/10.1128/mBio.03223-19
Zhu M., Dai X. // Nucleic Acids Res. 2019. V. 47. № 9. P. 4684–4693.
Song S., Wood T.K. // Biochem. Biophys. Res. Commun. 2020. V. 523. № 2. P. 281–286.
Gohara D.W., Yap M.F. // Curr. Genet. 2018. V. 64. № 4. P. 753–760.
Yoshida H., Wada A. // Wiley Interdiscip. Rev. RNA. 2014. V. 5. № 5. P. 723–732.
Tkachenko A.G., Kashevarova N.M., Karavaeva E.A., Shumkov M.S. // FEMS Microbiol Lett. 2014. V. 361. № 1. P. 25–33.
Marshall R.A., Aitken C.E., Dorywalska M., Puglisi J.D. // Annu. Rev. Biochem. 2008. V. 77. № 1. P. 177–203.
Trosch R., Willmund F. // Biol. Chem. 2019. V. 400. № 7. P. 879–893.
Polikanov Y.S., Blaha G.M., Steitz T.A. // Science. 2012. V. 336. № 6083. P. 915–918.
Shcherbakova K., Nakayama H., Shimamoto N. // Genes Cells. 2015. V. 20. № 10. P. 789–801.
Ueta M., Yoshida H., Wada C., Baba T., Mori H., Wada A. // Genes Cells. 2005. V. 10. № 12. P. 1103–1112.
Wada Y., Sambongi Y., Futai M. // Biochim. Biophys. Acta. 2000. V. 1459. № 2–3. P. 499–505.
Wada A., Yamazaki Y., Fujita N., Ishihama A. // Proc. Natl. Acad. Sci. USA. 1990. V. 87. № 7. P. 2657–2661.
Aiso T., Yoshida H., Wada A., Ohki R. // J. Bacteriol. 2005. V. 187. № 6. P. 1951–1958.
Wada A. // Genes Cells. 1998. V. 3. № 4. P. 203–208.
Maki Y., Yoshida H., Wada A. // Genes Cells. 2000. V. 5. № 12. P. 965–974.
Ueta M., Ohniwa R.L., Yoshida H., Maki Y., Wada C., Wada A. // J. Biochem. 2008. V. 143. № 3. P. 425–433.
Yoshida M., Kashiwagi K., Kawai G., Ishihama A., Igarashi K. // J. Biol. Chem. 2002. V. 277. № 40. P. 37139–37146.
Yamagishi M., Matsushima H., Wada A., Sakagami M., Fujita N., Ishihama A. // EMBO J. 1993. V. 12. № 2. P. 625–630.
Vila-Sanjurjo A., Schuwirth B.S., Hau C.W., Cate J.H. // Nat. Struct. Mol. Biol. 2004. V. 11. № 11. P. 1054–1059.
Agafonov D.E., Kolb V.A., Nazimov I.V., Spirin A.S. // Proc. Natl Acad. Sci. USA. 1999. V. 96. № 22. P. 12345–12349.
Basu A., Shields K.E., Eickhoff C.S., Hoft D.F., Yap M.-N.F. // J. Bacteriol. 2018. V. 200. № 24. e00426-18. https://doi.org/10.1128/JB.00426-18
Hauser R., Pech M., Kijek J., Yamamoto H., Titz B., Naeve F., et al.// PLoS Genet. 2012. V. 8. № 7. e1002815. https://doi.org/10.1371/journal.pgen.1002815
Li X., Sun Q., Jiang C., Yang K., Hung L.W., Zhang J., Sacchettini J.C. // Structure. 2015. V. 23. № 10. P. 1858–1865.
Yoshida H., Maki Y., Furuike S., Sakai A., Ueta M., Wada A. // J. Bacteriol. 2012. V. 194. № 16. P. 4178–4183.
Murina V., Kasari M., Takada H., Hinnu M., Saha C.K., Grimshaw J.W. et al. // J. Mol. Biol. 2019. V. 431. № 18. P. 3568–3590.
Boel G., Smith P.C., Ning W., Englander M.T., Chen B., Hashem Y. et al. // Nat. Struct. Mol. Biol. 2014. V. 21. № 2. P. 143–151.
Chen B., Boel G., Hashem Y., Ning W., Fei J., Wang C., Gonzalez R.L.Jr., Hunt J.F., Frank J. // Nat. Struct. Mol. Biol. 2014. V. 21. № 2. P. 152–159.
Izutsu K., Wada A., Wada C. // Genes Cells. 2001. V. 6. № 8. P. 665–676.
Zhang Y., Mandava C.S., Cao W., Li X., Zhang D., Li N., Zhang Y., Zhang X., Qin Y., Mi K., Lei J., Sanyal S., Gao N. // Nat. Struct. Mol. Biol. 2015. V. 22. № 11. P. 906–913.
Ткаченко А. Молекулярные механизмы стрессорных ответов у микроорганизмов./ Ред. В.А. Демаков. Екатеринбург: Изд-во УрО РАН, 2012. 237 с.
Agafonov D.E., Kolb V.A., Spirin A.S. // Cold Spring Harb. Symp. Quant. Biol. 2001. V. 66. P. 509–514.
Izutsu K., Wada C., Komine Y., Sako T., Ueguchi C., Nakura S., Wada A. // J. Bacteriol. 2001. V. 183. № 9. P. 2765–2773.
Battesti A., Majdalani N., Gottesman S. // Annu. Rev. Microbiol. 2011. V. 65. P. 189–213.
Hengge-Aronis R. // J. Mol. Microbiol. Biotechnol. 2002. V. 4. № 3. P. 341–346.
Shimada T., Yoshida H., Ishihama A. // J. Bacteriol. 2013. V. 195. № 10. P. 2212–2219.
El-Sharoud W.M., Niven G.W. // Microbiology. 2007. V. 153. № 1. P. 247–253.
Moen B., Janbu A.O., Langsrud S., Langsrud O., Kohler A., Rudi K. // Can. J. Microbiol. 2009. V. 55. № 6. P. 714–728.
Shimada T., Makinoshima H., Ogawa Y., Miki T., Maeda M., Ishihama A. // J. Bacteriol. 2004. V. 186. № 21. P. 7112–7122.
Zhang S., Wunier W., Yao Y., Morigen M. // J. Basic Microbiol. 2018. V. 58. № 12. P. 1091–1099.
Niven G.W. // Arch. Microbiol. 2004. V. 182. № 1. P. 60–66.
Garay-Arroyo A., Colmenero-Flores J.M., Garciarrubio A., Covarrubias A.A. // J. Biol. Chem. 2000. V. 275. № 8. P. 5668–5674.
Serio A.W., Keepers T., Andrews L., Krause K.M. // EcoSal Plus. 2018. V. 8. № 1. https://doi.org/10.1128/ecosalplus.ESP-0002-2018
McKay S.L., Portnoy D.A. // Antimicrob. Agents Chemother. 2015. V. 59. № 11. P. 6992–6999.
Van den Bergh B., Fauvart M., Michiels J. // FEMS Microbiol. Rev. 2017. V. 41. № 3. P. 219–251.
Fisher R.A., Gollan B., Helaine S. // Nat. Rev. Microbiol. 2017. V. 15. № 8. P. 453–464.
Lewis K. // Annu. Rev. Microbiol. 2010. V. 64. № 15. P. 357–372.
Dhar N., McKinney J.D. // Curr. Opin. Microbiol. 2007. V. 10. № 1. P. 30–38.
Zhang Y. // Emerg. Microbes Infect. 2014. V. 3. № 1. e3. https://doi.org/10.1038/emi.2014.3
Kushwaha G.S., Oyeyemi B.F., Bhavesh N.S. // Biochimie. 2019. V. 165. P. 67–75.
Maisonneuve E., Castro-Camargo M., Gerdes K. // Cell. 2018. V. 172. № 5. P. 1135. https://doi.org/10.1016/j.cell.2018.02.023
Tabor H., Tabor C.W. // Pharmacol. Rev. 1964. V. 16. № 3. P. 245–300.
Huang S.C., Panagiotidis C.A., Canellakis E.S. // Proc. Natl Acad. Sci. USA. 1990. V. 87. № 9. P. 3464–3468.
Igarashi K., Hashimoto S., Miyake A., Kashiwagi K., Hirose S. // Eur. J. Biochem. 1982. V. 128. № 2–3. P. 597–604.
Nomura M., Gourse R., Baughman G. // Annu. Rev. Biochem. 1984. V. 53. № P. 75–117.
Igarashi K., Kishida K., Kashiwagi K., Tatokoro I., Kakegawa T., Hirose S. // Eur. J. Biochem. 1981. V. 113. № 3. P. 587–593.
Igarashi K., Kashiwagi K. // J. Biol. Chem. 2018. V. 293. № 48. P. 18702–18709.
Souzu H. // Biochim. Biophys. Acta. 1986. V. 861. № 2. P. 353–360.
Tabor C.W., Tabor H. // Annu. Rev. Biochem. 1976. V. 45. № 1. P. 285–306.
Munro G.F., Bell C.A. // J. Bacteriol. 1973. V. 116. № 3. P. 1479–1481.
Peter H.W., Gunther T., Seiler N. // FEMS Microbiol. Lett. 1979. V. 5. № 6. P. 389–393.
Igarashi K., Kashiwagi K. // J. Biochem. 2006. V. 139. № 1. P. 11–16.
Sakamoto A., Terui Y., Yoshida T., Yamamoto T., Suzuki H., Yamamoto K., Ishihama A., Igarashi K., Kashiwagi K. // PLoS One. 2015. V. 10. № 4. P. e0124883. https://doi.org/10.1371/journal.pone.0124883
Schneider B.L., Hernandez V.J., Reitzer L. // Mol. Microbiol. 2013. V. 88. № 3. P. 537–550.
Tkachenko A.G., Akhova A.V., Shumkov M.S., Nesterova L.Y. // Res. Microbiol. 2012. V. 163. № 2. P. 83–91.
Jaishankar J., Srivastava P. // Front. Microbiol. 2017. V. 8. P. 2000. https://doi.org/10.3389/fmicb.2017.02000
Klauck E., Typas A., Hengge R. // Sci. Prog. 2007. V. 90. № 2–3. P. 103–127.
Hengge R. // Res. Microbiol. 2009. V. 160. № 9. P. 667–676.
Yoshida M., Kashiwagi K., Shigemasa A., Taniguchi S., Yamamoto K., Makinoshima H., Ishihama A., Igarashi K. // J. Biol. Chem. 2004. V. 279. № 44. P. 46008–46013.
Blattner F.R., Plunkett G.III, Bloch C.A., Perna N.T., Burland V., Riley M. et al. // Science. 1997. V. 277. № 5331. P. 1453–1462.
Igarashi K., Kashiwagi K. // Int. J. Biochem. Cell Biol. 2010. V. 42. № 1. P. 39–51.
Tabor C.W., Tabor H. // Microbiol. Rev. 1985. V. 49. № 1. P. 81–99.
Terui Y., Tabei Y., Akiyama M., Higashi K., Tomitori H., Yamamoto K., Ishihama A., Igarashi K., Kashiwagi K. // J. Biol. Chem. 2010. V. 285. № 37. P. 28698–28707.
Vourekas A., Stamatopoulou V., Toumpeki C., Tsitlaidou M., Drainas D. // IUBMB Life. 2008. V. 60. № 10. P. 669–683.
Lightfoot H.L., Hall J. // Nucleic Acids Res. 2014. V. 42. № 18. P. 11275–11290.
Luidalepp H., Berger S., Joss O., Tenson T., Polacek N. // J. Mol. Biol. 2016. V. 428. № 10. P. 2237–2247.
Igarashi K., Kashiwagi K. // IUBMB Life. 2015. V. 67. № 3. P. 160–169.
Kućan Z., Naranda T., Plohl M., Nöthig-Laslo V., Weygand-Durasević I. // Adv. Exp. Med. Biol. 1988. V. 250. P. 525–533.
Kanjee U., Gutsche I., Ramachandran S., Houry W.A. // Biochemistry. 2011. V. 50. № 43. P. 9388–9398.
Roychowdhury P., Basak B.S. // Acta Crystallogr. B. 1975. V. 31. № 6. P. 1559–1563.
Lee J.-H., Lee J. // FEMS Microbiol. Rev. 2010. V. 34. № 4. P. 426–444.
Wang D., Ding X., Rather P. // J. Bacteriol. 2001. V. 183. № 14. P. 4210–4216.
Lee J., Jayaraman A., Wood T. // BMC Microbiol. 2007. V. 7. № 1. P. 42. https://doi.org/10.1186/1471-2180-7-42
Spaepen S., Vanderleyden J., Remans R. // FEMS Microbiol. Rev. 2007. V. 31. № 4. P. 425–448.
Li G., Young K.D. // Microbiology. 2013. V. 159. Pt 2. P. 402–410.
Phillips R.S., Demidkina T.V., Faleev N.G. // Biochim. Biophys. Acta. 2003. V. 1647. № 1–2. P. 167–172.
Newton W.A., Morino Y., Snell E.E. // J. Biol. Chem. 1965. V. 240. P. 1211–1218.
Zarkan A., Liu J., Matuszewska M., Gaimster H., Summers D.K. // Trends Microbiol. 2020. V. 28. № 7. P. 566–577.
Gaimster H., Cama J., Hernández-Ainsa S., Keyser U.F., Summers D.K. // PLoS One. 2014. V. 9. № 4. e93168. https://doi.org/10.1371/journal.pone.0093168
Kim J., Park W. // J. Microbiol. 2015. V. 53. № 7. P. 421–428.
Hirakawa H., Inazumi Y., Masaki T., Hirata T., Yamaguchi A. // Mol. Microbiol. 2005. V. 55. № 4. P. 1113–1126.
Hirakawa H., Kodama T., Takumi-Kobayashi A., Honda T., Yamaguchi A. // Microbiology. 2009. V. 155. № 2. P. 541–550.
Stewart V., Yanofsky C. // J. Bacteriol. 1985. V. 164. № 2. P. 731–740.
Zarkan A., Caño-Muñiz S., Zhu J., Al Nahas K., Cama J., Keyser U.F., Summers D.K. // Sci. Rep. 2019. V. 9. № 1. P. 3868. https://doi.org/10.1038/s41598-019-40560-3
Chimerel C., Field C.M., Pinero-Fernandez S., Keyser U.F., Summers D.K. // Biochim. Biophys. Acta. 2012. V. 1818. № 7. P. 1590–1594.
Lewis K. // Nat. Rev. Microbiol. 2007. V. 5. № 1. P. 48–56.
Song S., Wood T.K. // Front. Microbiol. 2020. V. 11. P. 1565. https://doi.org/10.3389/fmicb.2020.01565
Lang M., Krin E., Korlowski C., Sismeiro O., Varet H., Coppée J.-Y., Mazel D., Baharoglu Z. // iScience. 2021. V. 24. № 10. P. 103128. https://doi.org/10.1016/j.isci.2021.103128
Zarkan A., Matuszewska M., Trigg S.B., Zhang M., Belgami D., Croft C., Liu J., El-Ouisi S., Greenhalgh J., Duboff J.S., Rahman T., Summers D.K. // Sci. Rep. 2020. V. 10. № 1. P. 11742. https://doi.org/10.1038/s41598-020-68693-w
Kim C.S., Li J.H., Barco B., Park H.B., Gatsios A., Damania A., Wang R., Wyche T.P., Piizzi G., Clay N.K., Crawford J.M. // Cell Chem. Biol. 2020. V. 27. № 6. P. 698–707.
Han Q., Fang J., Li J. // Biochem. J. 2001. V. 360. № 3. P. 617–623.
Дополнительные материалы отсутствуют.
Инструменты
Прикладная биохимия и микробиология