

Прикладная биохимия и микробиология, 2022, T. 58, № 3, стр. 232-243
Функционирование мембранного белка-транспортера Р-гликопротеина в условиях ингибирования синтеза глутатиона
Ю. В. Абаленихина 1, *, П. Д. Ерохина 1, П. Ю. Мыльников 1, А. В. Щулькин 1, Е. Н. Якушева 1
1 Рязанский государственный медицинский университет им. акад. И.П. Павлова Минздрава России
390026 Рязань, Россия
* E-mail: abalenihina88@mail.ru
Поступила в редакцию 10.11.2021
После доработки 10.12.2021
Принята к публикации 10.01.2022
- EDN: RQIEHP
- DOI: 10.31857/S0555109922030023
Аннотация
Изучены функционирование и механизм регуляции P-гликопротеина (Pgp) в условиях ингибирования синтеза глутатиона в клетках аденокарциномы ободочной кишки человека (Сасо-2), являющимися моделью абсорбции ксенобиотиков in vitro. Синтез глутатиона ингибировали действием D,L-бутионинсульфоксимина (БСО, ингибитор γ-глутамилцистеинсинтетазы). Установлено, что БСО в концентрациях 50–500 мкМ и инкубации 3 ч не влиял на уровень глутатиона и снижал активность Pgp. Увеличение срока экспозиции с БСО в концентрациях 10–100 мкМ до 24 ч приводило к развитию умеренного окислительного стресса и повышению количества Pgp (эффект реализовался через транскрипционный фактор Nrf2). При этом активность Pgp увеличивалась при воздействии БСО в концентрации 10 мкМ и снижалась при концентрациях 100–500 мкМ. Развитие выраженного окислительного стресса при воздействии БСО в концентрации 500 мкМ в течение 72 ч, сопровождалось снижением количества и активности Pgp. Таким образом, БСО является прямым ингибитором Pgp, однако снижение уровня SH-групп, вызываемое данным веществом, приводило к повышению уровня Nrf2, который, в свою очередь, вызывал повышение количества и активности данного белка-транспортера. Полученные результаты можно использовать в клинической практике для оптимизации дозирования субстратов белка-транспортера при терапии заболеваний, сопровождающихся развитием окислительного стресса в кишечнике.
Мембранные транспортные белки могут быть разделены на 4 типа: ионные каналы, транспортеры, аквапорины и АТФ-зависимые насосы. АТФ-зависимые насосы используют энергию АТФ для перемещения субстратов через мембраны против электрохимического градиента и среди них выделяют [1]: Р-тип (транспортирует ионы через цитоплазматическую мембрану), F-тип (обеспечивает синтез АТФ из АДФ во внутренней мембране митохондрий и обладает АТФазной активностью), V-тип (транспортирует Н+ через цитоплазматическую мембрану вакуолей растительных клеток и лизосом животных клеток) и АВС-транспортеры.
АВС-транспортеры (АTP-binding cassette) – АТФ-зависимые белки-транспортеры, обеспечивающие энергозависимый перенос веществ через цитоплазматические мембраны.
Самым изученным белком из суперсемейства АВС-транспортеров является Р-гликопротеин (Pgp, ABCB1-белок, MDR1) – мембранный АТФ-зависимый эффлюксный белок с молекулярной массой 170 кДа, обладающий широкой субстратной специфичностью.
Молекула белка-транспортера представлена двумя гомологичными частями, которые соединены между собой подвижным линкерным полипептидом [2]. Каждая часть молекулы Pgp состоит из гидрофобного трансмембранного домена (TMD) и консервативного цитоплазматического домена, включающего АТФ-связывающий сайт (NBD) [3]. Благодаря особенностям своего строения Pgp способен выполнять транспорт экзогенных и эндогенных субстратов. К субстратам Pgp относят преимущественно липофильные вещества с молекулярной массой 330–4000 Да (холестерин, фосфатиды) [4], а также лекарственные средства: противоопухолевые, гипотензивные, антигистаминные фармакологические препараты, сердечные гликозиды, антиагреганты, антикоагулянты, стероидные и тиреоидные гормоны, антибиотики, ингибиторы ВИЧ-протеиназы, иммунодепрессанты и др. [5].
Pgp изначально изучался в опухолевых клетках, где, как выяснилось, он обеспечивает механизмы резистентности опухолей к химиотерапии [6]. Однако экспрессия Pgp обнаружена и при физиологических состояниях организма. В настоящее время известно, что белок-транспортер Pgp локализован в энтероцитах кишечника, гепатоцитах, эпителиальных клетках почечных канальцев и участвует в транспорте эндогенных и экзогенных веществ, фармакокинетике лекарственных препаратов [7]. Кроме этого, из-за своей локализации в эндотелиальных клетках гистогематических барьеров (гематоэнцефалический барьер, гематоплацентарный барьер) Pgp участвует в защите забарьерных органов (мозг, тестикулы, плод) [8, 9]. В настоящее время доказано, для экспериментальных исследований классической модельной системой абсорбции веществ в тонком кишечнике являются клетки линии Сасо-2 (клетки аденокарциномы ободочной кишки человека), гиперэкспрессирующие белок-транспортер Pgp при культивировании 21 сут.
В ряде исследований показано, что повышение количества и активности Pgp отмечается при патологических состояниях, например, болезни Альцгеймера [10], болезни Паркинсона [11], эпилепсии [12], онкологических заболеваниях (карцинома, саркома, лимфома) [13] и др., но механизмы данных изменений на данный момент окончательно не установлены. При указанных патологиях окислительный стресс (ОС) является одним из ключевых звеньев патогенеза.
ОС представляет собой дисбаланс между продукцией активных форм кислорода (АФК) и способностью клеток защититься от них. Решающую роль в механизмах антиоксидантной защиты играет глутатион (GSH) – тиолсодержащий трипептид. Глутатион обладает способностью связывать свободные радикалы, модулирует реакцию клеток на окислительно-восстановительные изменения, участвует в детоксикации метаболитов лекарственных препаратов [14], регулирует экспрессию генов и апоптоз [15], участвует в трансмембранном транспорте органических растворенных веществ [16].
Глутатион-зависимая регуляция Pgp является недостаточно изученным вопросом. В литературе имеются противоречивые данные о влиянии ингибитора синтеза глутатиона – бутионинсульфоксимина (БСО) на экспрессию Pgp. Известно, что БСО в зависимости от времени экспозиции и концентрации может как снижать, так и повышать количество Pgp в эндотелиальных клетках сосудов головного мозга. Однако, механизм действия БСО и влияние изменения концентрации глутатиона на экспрессию и активность Pgp не описаны.
Изучение влияния истощения глутатиона на количество и функциональную активность Pgp представляется актуальным и имеет важное практическое значение. Благодаря барьерной роли Pgp в ограничении всасывания в кишечнике лекарственных веществ, ингибирование или активация белка-транспортера может приводить к существенному изменению биодоступности его субстратов, что, в свою очередь, может приводить к снижению эффективности проводимой терапии (при повышении активности Pgp и снижении всасывания) или развитию побочных эффектов (при снижении активности белка-транспортера и повышении всасывания).
Учитывая то обстоятельство, что широкий спектр патологии кишечника сопровождается гиперпродукцией свободных радикалов, актуально исследование функционирования Pgp в условиях ОС, что может повысить эффективность и безопасность проводимой терапии.
Цель исследования – изучение функционирования и механизма регуляции активности мембранного белка-транспортера Pgp в условиях ингибирования синтеза глутатиона – модели эндогенного окислительного стресса.
МЕТОДИКА
Культивирование клеток линии Caco-2. Исследование выполнено на линии клеток аденокарциномы ободочной кишки человека (Caco-2) (ЦКП “Коллекция культур клеток позвоночных”, Санкт-Петербург, Россия). Клетки культивировали при 37°С и 5%-ном содержании СО2 в инкубаторе WS-189C (“World Science”, Корея) в среде Игла, модифицированной Дульбекко (DMEM), с высоким содержанием глюкозы (4.5 г/л) (“Sigma-Aldrich”, США), содержащей L-глутамин (4 мМ) (“Sigma-Aldrich”, США), 15% эмбриональной бычьей сыворотки (“Sigma-Aldrich”, США), 100 Ед./мл и 100 мкг/мл пенициллина и стрептомицина (“Sigma-Aldrich”, США) соответственно.
При степени покрытия клетками культуральной поверхности (конфлюентность) 70–90%, клетки снимали с флакона добавлением раствора трипсин-ЭДТА (0.25% трипсина и 0.2% ЭДТА, “Sigma-Aldrich”, США), а затем высевали: в 96-луночный планшет (“Сorning”, США) для оценки цитотоксичности DL-бутионинсульфоксимина; в 6-луночные планшеты (“Сorning”, США) – для оценки влияния БСО на количество Pgp, Nrf2 и концентрацию карбонильных производных белков, содержание безбелковых и белковых SH-групп. Кроме того высевали в специальные трансвелл-системы (12 mm Transwell® with 0.4 µm Pore Polycarbonate Membrane Insert, Sterile, “Corning”, США) для оценки влияния БСО на проницаемость клеточных мембран для субстрата Pgp – фексофенадина. Клетки линии Caco-2 культивировали в течение 21 сут, поскольку за это время происходит их спонтанная дифференцировка в клетки, подобные энтероцитам тонкого кишечника, гиперэкспрессирующие Pgp.
Эндогенный окислительный стресс моделировали добавлением в культуральную среду ингибитора синтеза глутатиона – D,L-бутионинсульфоксимин (БСО, “Sigma-Aldrich”, США) в конечных концентрациях 1, 5, 10, 50, 100 и 500 мкМ и инкубацией в течение 3, 24 и 72 ч. Смену среды проводили каждые 24 ч. На каждый эксперимент было выполнено по 3 повторения.
Тест на выживаемость (цитотоксический тест). Клетки засевали в 96-луночный планшет из расчета 104 кл. на каждую лунку и культивировали в течение 21 сут, затем добавляли питательную среду с БСО. После окончания инкубации в каждую лунку добавляли по 20 мкл 0.5%-ного раствора бромида 3-(4,5-диметилтиазол-2-ил)-2,5-дифенил тетразолия (МТТ) и инкубировали в течение 2 ч, затем раствор МТТ удаляли и добавляли 200 мкл 1%-ного раствора диметилсульфоксида (“ПанЭко”, Россия). Поглощение измеряли через 10 мин при 530 нм на спектрофотометре для планшетов Stat Fax 2100 (“Awareness Technology”, США) [17].
Выживаемость клеток Сасо-2 в присутствии БСО рассчитывали по формуле: (ОП опытных лунок – ОП среды)/(ОП контрольных лунок – ОП среды) × 100%, где ОП — оптическая плотность.
Приготовление клеточных лизатов для анализа. Клетки в количестве 3 × 106 по окончании эксперимента снимали с 6-луночных планшетов раствором трипсин-ЭДТА.
Часть клеток (2 × 106) трехкратно промывали фосфатным буфером, рН 7.4 (“ПанЭко”, Россия), лизировали трехкратным проведением цикла замораживания-размораживания в 200 мкл буфера при –20°С и использовали для проведения иммуно-ферментного анализа (ИФА).
Вторую часть клеток (1 × 106) промывали изотоническим раствором натрия хлорида (“Медпро”, Россия), добавляли 150 мкл лизирующего буфера, (50 мМ, трис-HCl, pH 7.4, 150 мМ KCl, 0.5% тритон X-100, смесь ингибиторов протеиназ (аминоэтилбензенсульфонилфлуорида гидрохлорид, AEBSF, 2 мМ, апротинин 0.3 мкМ, бестатин 130 мкМ, ЭДТА 1 мМ, эпоксисукциниллейцингуанидинобутиламид (Е-64) 14 мкМ, лейпептин 1 мкМ, “Sigma-Aldrich”, США), встряхивали на шейкере и инкубировали на льду в течение 10 мин. Затем центрифугировали в течение 10 мин при 5000 g (СМ-50, “Eppendorf”, Германия). Цитоплазматическую (экстраядерную) фракцию переносили в отдельные пробирки и использовали для определения концентрации небелковых и белковых SH-групп.
Аналогичным образом лизировали клетки, которые параллельно инкубировали в 6-луночных планшетах, для определения концентрации карбонильных производных белков из расчета 3 × 106 кл. и 450 мкл лизирующего буфера.
Анализ содержания Pgp и Nrf2 в клетках линии Caco-2. В полученном лизате клеток методом ИФА определяли количество Pgp (Human Permeability glycoprotein ELISA kit, “Blue gene”, Китай) и Nrf2 (Human Nuclear factor erythroid 2-related factor 2 ELISA kit, “Blue gene”, Китай). Светопоглощение измеряли при 450 нм на спектрофотометре для планшетов Stat Fax 2100 (“Awareness Technology”, США). Количество белка в пробах анализировали методом Бредфорда (Pierce Coomassie Plus (Bradford) Assay Kit, “ThermoFisher”, США).
Определение концентрации небелковых и белковых SH-групп. Концентрацию белковых SH-групп определяли по разнице между уровнем общих и низкомолекулярных (небелковых) SH-групп. Анализ содержания общих SH-групп в лизате клеток проводили по методу Эллмана с 5,5'-дитиобис(2-нитро)-бензоатом (DTNB) в неденатурирующих условиях [18]. К 100 мкл образца добавляли 100 мкл 2 мМ DTNB (“Serva”, Германия) в 1 М трис-HCl буфере (рН 8.0) и 1000 мкл дистиллированной воды, после экспозиции 30 мин количественно оценивали содержание 5-тио-2-нитробензойной кислоты при 412 нм на плашетном фотометре Stat Fax 2100 (“Awareness Technology”, США). Концентрацию SH-групп рассчитывали исходя из коэффициента экстинкции ε412 = 13.6 мМ–1 см–1 [19]. Для определения содержания низкомолекулярных SH-групп пробу предварительно смешивали с охлажденной 5%-ной трихлоруксусной кислотой (“Химмед”, Россия), инкубировали на льду 15 мин, затем центрифугировали при 11 000 g (СМ-50, “Eppendorf”, Германия) 5 мин при 4°С. Полученный супернатант нейтрализовали с помощью 1%-ного NaOH и использовали для определения низкомолекулярных SH-групп по реакции с DTNB.
Определение концентрации карбонильных производных белков. В полученном лизате клеток с помощью коммерческого набора определяли концентрацию продуктов карбонильных производных белков (“Sigma-Aldrich”, США).
Метод определения продуктов карбонильных производных белков основан на их взаимодействии с 2,4-динитрофенилгидразином с образованием 2,4-динитрофенилгидразонов, которые регистрировали при длине волны 375 нм. Концентрацию карбонильных производных белков рассчитывали исходя из коэффициента экстинкции ε375 = 22 мМ–1 см–1 [20].
Анализ проводили на спектрофотометре для планшетов Stat Fax 2100.
Оценка активности Pgp. Активность Pgp оценивали по транспорту его маркерного субстрата – фексофенадина через монослой клеток линии Caco-2. Для этого клетки культивировали в специальных трансвелл-системах (рис. 1). Трансвелл-система представлена двумя камерами: апикальной и базолатеральной. Дно апикальной камеры является полупроницаемой мембраной, на которую высевали клетки линии Caco-2 с плотностью 105/см2 и культивировали в течение 21 сут.
Рис. 1.
Структура трансвелл-системы. Трансвелл-система представлена двумя камерами: апикальной и базолатеральной. Дно апикальной камеры является полупроницаемой мембраной, на которую высеивали клетки линии Caco-2 с плотностью 105/см2. *p < 0.05 по сравнению с контролем (критерий Ньюмена–Кейлса).

Целостность клеточного монослоя оценивали по величине трансэпителиального сопротивления, которое определяли с помощью вольтметра Millicell ERS-2 (“Millipore”, США). При его значении выше 500 мОм/см2 выполняли эксперименты. Для этого в лунки трансвелл-системы добавляли питательную среду с БСО в тестируемых концентрациях. После окончания инкубации питательную среду заменяли на транспортную среду, представляющую собой раствор Хэнкса (“Sigma-Aldrich”, США) с 25 мМ Хепес (“Sigma-Aldrich”, США) и 1% диметилсульфоксида (“ПанЭко”, Россия). Затем добавляли субстрат Pgp – фексофенадин (“Sigma-Aldrich”, США) в апикальную камеру в конечной концентрации 150 мкМ [21]. Через 1, 2 и 3 ч забирали образцы из базолатеральной камеры-реципиента для определения концентрации субстрата (a-b транспорт, обусловленный пассивной диффузией против функционирования Pgp).
В аналогичных трансвелл-системах оценивали транспорт фексофенадина из базолатеральной камеры в апикальную (b-a транспорт, обусловленный пассивной диффузией и функционированием Pgp). Для этого субстрат в той же концентрации добавляли в базолатеральную камеру, а затем через 1, 2 и 3 ч забирали образцы из апикальной камеры для определения концентрации фексофенадина.
Транспорт маркерного субстрата рассчитывали по формуле [22]:
где Рарр – коэффициент кажущейся проницаемости (apparent permeability coefficient, см/сек), dQ/dt – изменение количества субстрата в камере реципиенте за время инкубации (мкМ · мл/с), A – площадь полупроницаемой мембраны лунки в трансвелл-системе (см2), C0 – начальная концентрация субстрата в камере-доноре, мкМ.Затем рассчитывали отношение коэффициентов кажущейся проницаемости: b-a к a-b.
Данный параметр является интегральным и оценивает общий вклад Pgp в транспорт фексофенадина через билипидную мембрану.
Определение концентрации фексофенадина. Концентрации фексофенадина в транспортной среде определяли методом высокоэффективной жидкостной хроматографии (ВЭЖХ) с УФ-детектированием при длине волны 220 нм. Исследование выполнялось на ВЭЖХ хроматографе “Стайер” (Россия) по оригинальной методике [23]. Полученная проба транспортной среды (50 мкл), содержащая фексофенадин, разводилась до 150 мкл подвижной фазы, и 100 мкл полученного раствора вводились в хроматограф.
При анализе использовалась хроматографическая колонка Phenomenex Synergi 4u Polar-RP 80A (250 × 4.6) (США) с зернением 4 мкм. Температура разделения – 45°С, скорость потока – 1 мл/мин. Состав подвижной фазы: 128 мл ацетонитрила (“PanReac AppliChem”, Испания), 267.4 мл воды деионизированной, 6.33 мл кислоты уксусной ледяной (“PanReac AppliChem”, Испания), с добавлением триэтиламина (“PanReac AppliChem”, Испания) до pH 6.7. Время удерживания фексофенадина в данных условиях составляло 12.8 мин. Количественное определение проводилось методом абсолютной калибровки по площади пиков. Аналитический диапазон методики составлял 1.2–57.4 мкМ.
Статистический анализ. Полученные результаты анализировали с помощью программ “StatSoft Statistica 13.0” и Microsoft Excel. Результаты представлены в виде M ± SD. Для оценки статистической значимости различий использовали дисперсионный анализ (ANOVA), попарные сравнения выполняли с помощью критерия Ньюмена-Кейлса, корреляционный анализ проводили с помощью критерия Пирсона. Статистически значимыми считали различия при p < 0.05.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Клетки линии Caco-2 – это клетки аденокарциномы ободочной кишки человека. При культивировании в виде монослоя данные клетки обладают способностью спонтанно дифференцироваться в клетки, подобные тонкокишечным энтероцитам, гиперэкспрессирующим белки-транспортеры, в том числе и Pgp [24]. Данная клеточная линия широко используется для исследования абсорбции лекарственных веществ и функционировании Pgp in vitro.
Известно, что Pgp локализован на апикальной мембране энтероцитов, что обуславливает его участие в ограничении проникновения лекарств из просвета кишечника в эпителиальные клетки и далее в системный кровоток.
Ключевую роль в синтезе и поддержании клеточного уровня глутатиона играет фермент γ-глутамилцистеинсинтетаза (γ-GCS), ингибитором которого является БСО [25]. Использование БСО в качестве специфического ингибитора синтеза глутатиона имеет преимущество относительно других глутатион-истощающих агентов (N-этилмалеимид, йодацетат, 2-йодацетамид), так как он не влияет на активность цитохрома P-450 и не изменяет оксидазную активность ферментов (поэтому метаболизм субстрата Pgp будет неизменным) [26].
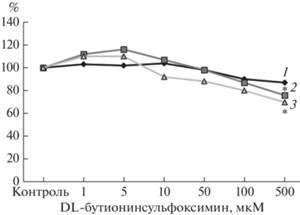
Цитотоксическое действие БСО на клетки линии Caco-2. Цитотоксическое действие БСО оценивалось по результатам МТТ-теста. В контрольной группе клеток выживаемость составила 100%. При воздействии БСО во всех используемых концентрациях жизнеспособность клеток линии С-асо-2 не изменялась при времени инкубации 3 ч. При концентрациях БСО 1–100 мкМ и инкубации 24 и 72 ч жизнеспособность клеток также не отличалась от значений контрольной группы, а при концентрации 500 мкМ снижалась на 23.9% (р = 0.003) и 30.1% (р = 0.001) соответственно (рис. 2).
Рис. 2.
Жизнеспособность клеток линии Сасо-2 (%) в зависимости от концентрации DL-бутионинсульфоксимина при инкубации 3 (1), 24 (2) и 72 (3) ч. *p < 0.05 по сравнению с контролем (критерий Ньюмена–Кейлса).
Оценка развития окислительного стресса в клетках линии Сасо-2 под действием БСО. В условиях окислительного стресса наиболее чувствительна к действию АФК аминокислота – цистеин. Цистеин может входить как в состав белков (белковые тиолы), так и небелковых сульфгидрилов (небелковые тиолы).
Небелковые SH-группы в основном представлены глутатионом [27], поэтому оценку степени ингибирования синтеза глутатиона проводили по изменению их уровня.
При воздействии БСО в течение 3 ч в концентрациях 1–500 мкМ уровень небелковых SH-групп не изменялся. При увеличение длительности экспозиции до 24 и 72 ч и концентрации БСО 1 и 5 мкМ уровень небелковых SH-групп также не изменялся, однако при концентрациях БСО 10, 50, 100, и 500 мкМ отмечалось снижение уровня небелковых SH-групп на 40.7% (р = 0.002) и 31.9% (р = 0.007); 57.6% (р = 0.0004) и 48.4% (р = 0.0006); 67.8% (р = 0.0002) и 62.3% (р = 0.0002); 55.9% (р = = 0.0004) и 57.4% (р = 0.0002) соответственно (рис. 3а).
Рис. 3.
Концентрация небелковых SH-групп (а, мкмоль/мг белка) и белковых SH-групп (б, мкмоль/мг белка) в клетках линии Сасо-2 под действием DL-бутионинсульфоксимина в концентрациях 1–500 мкМ в течение 3 (1), 24 (2) и 72 (3) ч (M ± SD, n = 3). *p < 0.05 по сравнению с контролем (критерий Ньюмена–Кейлса).

При окислительном стрессе АФК взаимодействуют с простыми белками, в результате чего происходит модификация полипептидной цепи по аминокислотным остаткам [28]. Одной из наиболее важных и изученных является модификация цистеина. Атом серы тиольной группы (-SH) имеет несколько состояний окисления, поэтому модификация боковой цепи, содержащей цистеин, происходит до различных продуктов [29, 30]. Важность оценки окисления данной аминокислоты обусловлена тем, что цистеин встречается в виде консервативного остатка в функциональных участках белков, что позволяет рассматривать реакционноспособные SH-группы протеинов в качестве регуляторных центров [31, 32].
В ходе выполненного исследования было показано, что при воздействии БСО в течении 3 ч их концентрация не изменялась. При увеличении времени инкубации до 24 ч отмечалось снижение содержания белковых SH-групп на 25.5% (р = 0.02) при концентрации БСО 10 мкМ, на 32.2% (р = = 0.01) – при 50 мкМ, на 50.0% (р = 0.001) – при 100 мкМ и на 44.7% (р = 0.002) – при 500 мкМ, а при концентрациях 1 и 5 мкМ достоверных отличий от контроля получено не было (рис. 3б).
При инкубации в течение 72 ч наблюдалось снижение уровня белковых SH-групп при воздействии на клетки линии Сасо-2 БСО в концентрациях 50; 100 и 500 мкМ на 37.1% (р = 0.03); 64.9% (р = 0.008) и 54.3% (р = 0.003) соответственно, а при концентрациях 1–10 мкМ изменений не отмечалось (рис. 3б).
Необратимыми белковыми продуктами окислительного стресса являются карбонильные производные белков, которые формируются за счет окисления нескольких аминокислотных остатков, а также взаимодействия с продуктами перекисного окисления липидов и редуцирующими сахарами, вследствие чего могут рассматриваться в качестве конечного продукта окислительного повреждения [28, 33].
Уровень карбонильных производных белков не изменялся при инкубации клеток с БСО в концентрациях 1–500 мкМ и времени экспозиции 3 ч, а также при концентрациях 1 и 5 мкМ и длительности воздействия 24 и 72 ч.
Инкубация клеток линии Сасо-2 с БСО в концентрациях 10, 50, 100 и 500 мкМ в течение 24 и 72 ч приводила к увеличению уровня карбонильных производных белков на 15.5% (р = 0.01) и 36.6% (р = 0.004); 19.8% (р = 0.003) и 37.6% (р = 0.005); 21.2% (р = 0.002) и 63.1% (р = 0.0002); 150.0% (р = = 0.0002) и 69.1% (р = 0.0002) соответственно (рис. 4а).
Рис. 4.
Концентрация карбонильных производных белков (а, нмоль/мг белка) и изменение количества Pgp (б, нмоль/мг белка) в клетках линии Сасо-2 под действием DL-бутионинсульфоксимина в концентрациях 1–500 мкМ в течение 3 (1), 24 (2) и 72 (3) ч (M ± SD, n = 3). *p < 0.05 по сравнению с контролем (критерий Ньюмена–Кейлса).

Таким образом, в условиях воздействия БСО в концентрациях 10–500 мкМ в течение 24 и 72 ч на клетки линии Сасо-2 наблюдалось снижение уровня небелковых SH-группы (за счет глутатиона), что приводило к развитию эндогенного окислительного стресса, характеризующегося повреждением белков по SH-группам и формированием карбонильных производных белков.
Влияние БСО на количество и функциональную активность Pgp. При воздействии БСО в течение 3 ч и концентрациях 1–500 мкМ количество Pgp статистически значимо не изменялось. Уровень Pgp при воздействии БСО в течение 24 ч и концентрациях 1, 5 и 500 мкМ не изменялся, но возрастал при концентрациях 10, 50 и 100 мкМ на 71.6% (р = = 0.002); 51.6% (р = 0.007) и 25.4% (р = 0.05) соответственно. При воздействии БСО в течение 72 ч и концентрациях 1–100 мкМ количество Pgp не изменялось, а при 500 мкМ снижалось на 35.6% (р = 0.002) (рис. 4б).
Для оценки активности Pgp клетки культивировали в трансвелл-системе, состоящей из двух камер – апикальной и базолатеральной. В ходе исследования оценивался транспорт фексофенадина – субстрата белка-транспортера из базолатеральной камеры в апикальную (Pappb-a), обусловленный пассивной диффузией и функционированием Pgp (модель транспорта веществ из кишечного эпителия в просвет кишечника). Также анализировали транспорт фексофенадина из апикальной камеры в базолатеральную (Pappa-b), обусловленный пассивной диффузией против работы Pgp (модель транспорта веществ из просвета кишки в кишечный эпителий, то есть всасывание). Кроме этого, рассчитывали отношение коэффициентов кажущейся проницаемости (b-a)/(a-b) для субстратов, который превышал значение “2”.
Коэффициент кажущейся проницаемости b-a фексофенадина составил 2.94 × 10–6 ± 0.54 × × 10–6 см/с, коэффициент кажущейся проницаемости a-b – 0.93 × 10–6 ± 0.26 × 10–6 см/с. Из представленных данных видно, что транспорт обусловленный Pgp в 3.16 раз выше транспорта, происходящего за счет пассивной диффузии, против работы белка-транспортера (р = 0.013) (табл. 1).
Таблица 1.
Влияние БСО на транспорт субстрата Pgp (фексофенадина) через билипидную мембрану клеток Caco-2 (M ± SD, n = 3)
| БСО, мкМ | Раррb-a ×10–6 см/с |
Рарра-b ×10–6 см/с |
Раррb-a ×10–6 см/с |
Рарра-b ×10–6 см/с |
Раррb-a ×10–6 см/с |
Рарра-b ×10–6 см/с |
|---|---|---|---|---|---|---|
| 3 ч | 24 ч | 72 ч | ||||
| Контроль | 3.02 ± 0.12 | 1.11 ± 0.37 | 2.81 ± 0.77 | 0.76 ± 0.17 | 2.98 ± 0.72 | 0.92 ± 0.17 |
| 1 | 3.18 ± 1.43 | 1.44 ± 0.42 | 2.83 ± 0.47 | 0.72 ± 0.16 | 3.27 ± 0.31 | 1.25 ± 0.44 |
| 5 | 3.08 ± 0.54 | 1.34 ± 0.77 | 2.76 ± 0.06 | 0.71 ± 0.11 | 1.95 ± 0.90 | 0.79 ± 0.12 |
| 10 | 2.72 ± 0.41 | 1.41 ± 0.38 | 4.09 ± 0.13* | 0.76 ± 0.03 | 2.05 ± 0.72 | 0.74 ± 0.29 |
| 50 | 1.88 ± 0.45* | 1.33 ± 0.33 | 2.24 ± 0.64 | 1.09 ± 0.27 | 1.14 ± 0.27* | 0.92 ± 0.38 |
| 100 | 1.59 ± 0.28* | 1.47 ± 0.29 | 1.52 ± 0.52* | 1.09 ± 0.35 | 1.04 ± 0.10* | 0.97 ± 0.26 |
| 500 | 1.29 ± 0.16* | 1.31 ± 0.19 | 3.72 ± 0.11* | 2.77 ± 0.25* | 4.11 ± 0.49* | 3.69 ± 0.87* |
Воздействие БСО в концентрациях 50, 100 и 500 мкМ в течение 3 ч приводило к снижению коэффициента кажущейся проницаемости b-a на 37.7% (р = 0.001), 47.4% (р = 0.001) и 57.3% (р = = 0.005) (табл. 1) соответственно и отношения коэффициентов кажущейся проницаемости (b-a)/(a-b) на 48.9% (р = 0.048), 62.4% (р = 0.03) и 65.9% (р = = 0.03) соответственно (рис. 5), что свидетельствовало о снижении активности белка-транспортера. Таким образом, учитывая отсутствие изменений количества Pgp при данном времени воздействия, полученные результаты свидетельствуют о том, что БСО является прямым ингибитором активности Pgp (за счет непосредственного взаимодействия с молекулой белка-транспортера).
Рис. 5.
Изменение отношения коэффициентов кажущейся проницаемости (b-a)/(a-b) в клетках линии Сасо-2 под действием DL-бутионинсульфоксимина в концентрациях 1–500 мкМ в течение 3 (1), 24 (2) и 72 (3) ч (M ± SD, n = 3).
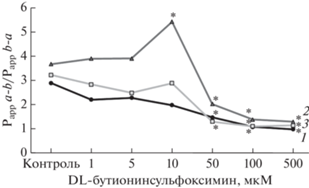
При увеличении длительности экспозиции до 24 ч было показано, что БСО в концентрации 10 мкМ вызывал увеличение коэффициента кажущейся проницаемости b-a на 45.6% (р = 0.028) и отношения коэффициентов кажущейся проницаемости (b-a)/(a-b) на 47.2% (р = 0.02), что и являлось проявлением повышения активности Pgp. На 24 ч инкубации отмечалось увеличение количества Pgp, что может указывать индукцию активации Pgp за счет синтеза белка, а не его активации.
При концентрации 50 мкМ отмечалось снижение отношения коэффициентов кажущейся проницаемости (b-a)/(a-b) на 44.8% (р = 0.01), а в концентрации 100 мкМ уменьшение коэффициента кажущейся проницаемости b-a на 45.9% (р = = 0.029) и отношения коэффициентов кажущейся проницаемости (b-a)/(a-b) на 62.2% (р = 0.002), что свидетельствовало о снижении активности белка-транспортера, несмотря на увеличение его количества. Полученные результаты могут быть обусловлены повреждением молекулы белка-транспортера за счет усиления выраженности окислительного стресса, что подтверждалось увеличением концентрации карбонильных производных белков, а также прямой ингибирующей активностью БСО.
Дальнейшее повышение концентрации БСО до 500 мкМ приводило к увеличению коэффициента кажущейся проницаемости b-a и a-b на 32.4% (р = 0.05) и 264.5% (р = 0.0003). При этом отношение коэффициентов кажущейся проницаемости (b-a)/(a-b) снижалось по сравнению с контролем на 64.7% (р = 0.0002), что свидетельствовало о снижении активности Pgp, повреждении клеточного монослоя и повышении транспорта фексофенадина – субстрата белка-транспортера по обе стороны мембраны.
Воздействие БСО в концентрациях 50 и 100 мкМ в течение 72 ч приводило к снижению коэффициента кажущейся проницаемости b-a на 61.7% (р = = 0.001) и 65.1% (р = 0.002) и отношения коэффициентов кажущейся проницаемости (b-a)/(a-b) на 59.4% (р = 0.002) и 65.6% (р = 0.002), что указывало на снижение активности белка-транспортера.
При увеличении концентрации БСО до 500 мкМ отмечалось увеличение коэффициента кажущейся проницаемости b-a и a-b на 37.9 (р = 0.02) и 301.1% (р = 0.0002) При этом отношения коэффициентов кажущейся проницаемости (b-a)/(a-b) снижалось по сравнению с контролем на 64.4% (р = = 0.002), что свидетельствовало о снижении активности Pgp, повреждении клеточного монослоя и повышении транспорта фексофенадина – субстрата белка-транспортера по обе стороны мембраны.
Таким образом, при кратковременном воздействии БСО (3 ч) отмечалось его ингибирующее действие на Pgp, не связанное с изменением его количества (прямое ингибирование). При увеличении времени инкубации до 24 ч и концентрации БСО до 10 мкМ активность Pgp возрастала в соответствии с увеличением его количества. При увеличении концентрации БСО до 50 и 100 мкМ и длительности воздействия 24 и 72 ч отмечалось снижение активности Pgp, что могло быть результатом повреждения молекулы белка-транспортера за счет усиления выраженности окислительного стресса (снижение концентрации белковых SH-групп и повышение карбонильных производных белков), а также прямой ингибирующей активности БСО.
При концентрации БСО 500 мкМ и длительности воздействия 24 и 72 ч отмечалось усиление окислительного стресса, повреждение целостности клеточного монослоя и повышении транспорта фексофенадина – субстрата белка-транспортера по обе стороны мембраны.
Механизмы регуляции Pgp в условиях моделирования ингибирования синтеза глутатиона. В настоящее время известны несколько механизмов регуляции Pgp – это изменение экспрессии гена MDR1 и его полиморфизм, стабилизация мРНК гена MDR1, изменение активности синтезированного белка-транспортера и изменение свойств цитоплазматических мембран [34].
В условиях ОС отмечалась активация редокс-чувствительного транскрипционного фактора Nrf2 (NF-E2-related factor 2), который защищает клетку от воздействия свободных радикалов и может вносить существенный вклад в регуляцию активности Pgp.
В условиях воздействия БСО в концентрациях 1–500 мкМ в течение 3 ч количество транскрипционного фактора Nrf2 не изменялось. При воздействии БСО в течение 24 ч и концентрациях 1, 5 и 500 мкМ количество Nrf2 также не изменялось, но возрастало при концентрации 10 мкМ на 157.8% (р = 0.005), 50 мкМ – 146.7% (р = 0.0003), 100 мкМ – 164.4% (р = 0.006) соответственно. Воздействие БСО в течение 72 ч приводило к увеличению количества Nrf2 при концентрации ингибитора 50 мкМ на 195.7% (р = 0.001), 100 мкМ – 210.8% (р = 0.001), а при концентрациях 1, 5, 10 и 500 мкМ статистически значимого эффекта не оказывало (рис. 6).
Рис. 6.
Изменение количества Nrf2 в клетках линии Сасо-2 под действием DL-бутионинсульфоксимина в концентрациях 1–500 мкМ в течение 3 (1), 24 (2) и 72 (3) ч (M ± SD, n = 3). *p < 0.05 по сравнению с контролем (критерий Ньюмена–Кейлса).

В ходе исследования установлено, что в диапазоне концентраций от 1 до 500 мкМ и времени инкубации 3–72 ч уровень Nrf2 был обратно пропорционален содержанию белковых SH-групп (рис. 7) (r = –0.56, р = 0.00001), что свидетельствовало об обратной зависимости между концентрацией восстановленных тиолов и экспрессией транскрипционного фактора Nrf2. Полученные результаты указывали на классический механизм регуляции транскрипции Nrf2 [31] в условиях воздействия ингибитора фермента γ-глутамилцистеинсинтетазы БСО.
Рис. 7.
Корреляционная зависимость между концентрацией белковых SH-групп (мкмоль/мг белка) и количеством Nrf2 (нг/г белка) под действием DL-бутионинсульфоксимина в концентрациях 1–500 мкМ в течение 3–72 ч. (r – коэффициент корреляции Пирсона; р – статистическая значимость результата по критерию Ньюмена–Кейлса).

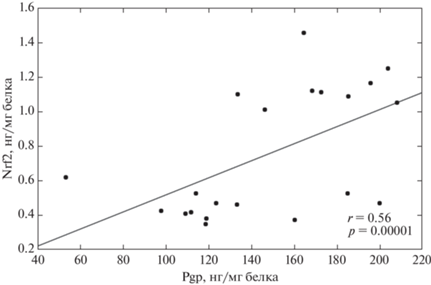
Белковые SH-группы представляют собой молекулярные переключатели активности протеинов [32, 35]. Keap1 – белок репрессор транскрипционного фактора Nrf2, в своем составе имеет два остатка цистеина и является мишенью для окислительной модификации. В физиологическом состоянии Nrf2 находится в комплексе с белком-репрессором Keap1, который функционирует как адаптерный белок. Keap1 способствует убиквитинированию и протеосомальной деградации Nrf2, а также предотвращает его проникновение из цитоплазмы в ядро [36]. После активации (окисления остатков цистеина в молекуле Keap1) комплекс Keap1-Nrf2 диссоциирует, и Nrf2 транслоцируется в ядро, где связывается с элементом антиоксидантного ответа (antioxidant-response elements, ARE). В свою очередь Nrf2 может участвовать в регуляции количества Pgp, поэтому проведен анализ взаимосвязи между количеством Nrf2 и количеством Pgp.
Анализ полученных результатов показал, что концентрация Pgp в лизате клеток Сасо-2 была прямо пропорциональна содержанию Nrf2 в экспериментах с БСО только при инкубации в течение 24 ч в диапазоне концентраций 1–500 мкМ (рис. 8).
Рис. 8.
Корреляционная зависимость между количеством Nrf2 (нг/мг белка) и Pgp (нг/мг белка) под действием DL-бутионин-сульфоксимина в концентрациях 1–500 мкМ в течение 24 ч. (r – коэффициент корреляции Пирсона; р – статистическая значимость результата по критерию Ньюмена–Кейлса).
Таким образом, в ходе настоящего исследования было показано, что БСО является прямым ингибитором Pgp, что расширяет имеющиеся представления о структуре ингибиторов и может быть использовано для предсказания наличия ингибирующей активности у веществ с аналогичной структурой.
С другой стороны, установлено, что развитие умеренного окислительного стресса приводит к повышению количества и активности Pgp в энтероцитах кишечника, что может приводить к уменьшению абсорбции субстратов белка-транспортера и, следовательно, к снижению эффективности проводимой терапии. Развитие выраженного ОС, наоборот, приводило к снижению количества и активности Pgp, увеличению проницаемости мембран клеток линии Caco-2 и могло сопровождаться повышением абсорбции субстратов белка-транспортера, увеличением их концентрации в плазме крови и, в конечном итоге, проявляться развитием нежелательных лекарственных реакций.
Настоящая статья не содержит описания каких-либо исследований с участием людей или животных в качестве объектов.
Исследование выполнено при финансовой поддержке гранта Президента Российской Федерации для государственной поддержки молодых российских ученых – кандидатов наук МК-1856.2020.7.
Авторы декларируют отсутствие явных и потенциальных конфликтов интересов, связанных с публикацией настоящей статьи.
Список литературы
Pedersen P.L. // J. Bioenerg. Biomembr. 2005. V. 37. № 6. P. 349–357.
Esser L., Zhou F., Pluchino K.M., Shiloach J., Ma J., Tang W., Gutierrez C., Zhang A., Shukla S., Madigan J.P., Zhou T., Kwong P.D., Ambudkar S.V., Gottesman M.M., Xia D. // J. Biol. Chem. 2017. V. 292. № 2. P. 446–461.
Sharom F.J. // Essays in Biochemistry. 2011. T. 50. C. 161–178.
Якушева Е.Н., Титов Д.С., Правкин С.К. // Успехи физиол. наук. 2017. Т. 48. № 4. С. 70–87.
Кукес В.Г., Грачев С.В., Сычев Д.А., Раменская Г.В. Метаболизм лекарственных средств. Научные основы персонализированной медицины: руководство для врачей. М.: Гэотар-Медиа, 2008. 304 с.
Dyson J., Foll F.L., Magal P., Noussair A., Pasquier J. // J. Theor Biol. 2019. V. 14. № 461. P. 239–253.
Ieiri I., Takane H., Otsubo K. // Clin. Pharmacokinet. 2004. V. 43. № 9. P. 553–576.
Черных И.В., Щулькин А.В., Гацанога М.В., Попова Н.М., Есенина А.С., Градинарь М.М., Якуше-ва Е.Н. // Наука молодых (Eruditio Juvenium). 2019. Т. 7. № 1. С. 46–52.
Han L.W., Gao C., Mao Q. // Expert Opinion on Drug Metabolism & Toxicology. 2018. V. 14. № 8. P. 817–829.
Chai A.B., Leung G.K.F., Callaghan R., Gelissen I.C. // FEBS J. 2020. V. 287. № 4. P. 612–625.
Furuno T., Landi M.T., Ceroni M., Caporaso N., Bernucci I., Nappi G., Martignoni E., Schaeffeler E., Eichelbaum M., Schwab M., Zanger U.M. // Pharmacogenetics. 2002. V. 12. № 7. P. 529–534.
Wang G.X., Wang D.W., Liu Y., Ma Y.H. // Int. J. Neurosci. 2016. V. 126. № 5. P. 385–392.
Muñoz-Pérez M.J., Casco S., Garza-González M.D.C., Soto-Vega E. // J. Pediatr. Hematol. Oncol. 2018. V. 40. № 8. P. 490–494.
Forman H.J., Zhang H., Rinna A. // Mol. Aspects Med. 2009. V. 30. № 1–2. P. 1–12.
Jefferies H., Coster J., Khalil A., Bot J., McCauley R.D., Hall J.C. // ANZ J. Surg. 2003 V. 73. № 7. P. 517–522.
Oestreicher J., Morgan B. // Biochem. Cell Biol. 2019. V. 97. № 3. P. 270–289.
Tolosa L., Donato M.T., Gómez-Lechón M. J. // Methods Mol. Biol. 2015. № 1250. P. 333–348.
Boschi-Muller S., Azza S., Sanglier-Cianferani S., Talfournier F., Dorsselear A.V., Branlant G. // J. Biol. Chem. 2000. № 275. P. 35908–35913.
Ellman L.G. // Arch. Biochem. Biophys. 1959. № 82. P. 70–77.
Weber D., Davies M.J., Grunea T. // Redox Biol. 2015. № 5. P. 367–380.
Petri N., Tannergren C., Rungstad D., Lennernäs H. // Pharmac. Res. 2004. V. 21. № 8. P. 1398–1404.
Elsby R., Surry D.D., Smith V.N., Gray A.J. // Xenobiotic. 2008. № 38. P. 1140–1164.
Ерохина П.Д., Абаленихина Ю.В., Щулькин А.В., Черных И.В., Попова Н.М., Слепнев А.А., Якушева Е.Н. // Российский медико-биологический вестник им. акад. И.П. Павлова. 2020. Т. 28. № 2. Р. 135–142.
Müller J., Keiser M., Drozdzik M., Oswald S. // Biol. Chem. 2017. V. 398. № 2. P. 175–192.
Haddad J.J. // Eur. Cytokine Netw. 2001. V. 12. № 4. P. 614–624.
Drew R., Miners J.O. // Biochem. Pharmacol. 1984. V. 33. № 19. P. 2989–2994.
Nagy L., Nagata M., Szabo S. // World J. Gastroenterol. 2007. V. 13. № 14. P. 2053–2060.
Лущак В.И. // Биохимия. 2007. Т. 72. № 8. С. 995–1017.
Reddie K.G. // Curr. Opin. Chem. Biol. 2008. V. 12. № 6. P. 746–754.
Forman H.J. Measuring Oxidants and Oxidative Stress in Biological Systems [Internet]. / Ed. L.J. Berliner, N.L. Parinandi. Cham (CH): Springer, 2020. Chapter 1. 251 p.
Poole L.B., Nelson K.J. // Curr. Opin. Chem. Biol. 2008. V. 12. № 1. P. 18–24.
Kosmachevskaya O.V., Topunov A.F. // Appl. Biochem. Microbiol. 2021. V. 57. № 5. P. 543–555.
Spickett C.M., Pitt A.R. // Essays Biochem. 2020. V. 64. № 1. P. 19–31.
Якушева Е.Н., Черных И.В., Щулькин А.В., Попова Н.М. // Успехи физиол. наук. 2014. Т. 45. № 4. С. 89–98.
Lennicke. C., Rahn J., Lichtenfels R., Wessjohann L.A., Seliger B. // Cell Communication and Signaling. 2015. № 13. e39.
Wen Zh., Liu W., Li X., Chen W., Liu J., Wen Zh., Liu Zh. // Oxid. Med. Cell Longev. 2019. Article ID 1759149. https://doi.org/10.1155/2019/1759149
Дополнительные материалы отсутствуют.
Инструменты
Прикладная биохимия и микробиология