

Прикладная биохимия и микробиология, 2022, T. 58, № 5, стр. 446-458
Модуляция каталитических свойств иммобилизованной рекомбинантной липазы из Тhermomyces lanuginosus в реакции этерификации путем выбора адсорбента
Г. А. Коваленко 1, *, Л. В. Перминова 1, А. Б. Беклемишев 1, 2, М. Б. Пыхтина 2, М. Г. Холявка 3, 4, В. А. Бучельникова 4, В. Г. Артюхов 3
1 Федеральный исследовательский центр “Институт катализа им. Г.К. Борескова Сибирского отделения
Российской академии наук”
630090 Новосибирск, Россия
2 Федеральный исследовательский центр фундаментальной и трансляционной медицины (ФИЦ ФТМ),
НИИ биохимии ФИЦ ФТМ
630117 Новосибирск, Россия
3 Воронежский государственный университет
394018 Воронеж, Россия
4 Севастопольский государственный университет
299053 Севастополь, Россия
* E-mail: galina@catalysis.ru
Поступила в редакцию 30.01.2022
После доработки 05.04.2022
Принята к публикации 26.04.2022
- EDN: HCFJHH
- DOI: 10.31857/S0555109922050099
Аннотация
Биокатализаторы с активностью липазы (БКЛ) были приготовлены путем адсорбционной иммобилизации рекомбинантной липазы (rPichia/lip) из термофильных микроскопических грибов Тhermomyces lanuginosus, продуцируемой генно-инженерным штаммом метилотрофных дрожжей Komagataella phaffii (Pichia pastoris). В качестве адсорбентов использовали различные по физико-химическим свойствам носители: мезопористый гидрофильный силикагель (SiO2) и макропористый гидрофобный аэрогель (МУА). Ферментативная активность, субстратная специфичность и операционная стабильность БКЛ были исследованы в реакции этерификации жирных насыщенных кислот и алифатических спиртов с количеством атомов углерода в молекуле от 2 до 18. Были составлены матрицы относительных активностей для более 60 пар субстратов – кислоты и спирта, путем сравнения скоростей реакции этерификации в идентичных условиях, что позволило выявить различия в специфичности БКЛ в зависимости от химической природы носителя. Было обнаружено, что для обоих типов биокатализаторов, “rPichia/lip на SiO2” (PLSi) и “rPichia/lip на МУА” (PLC) максимальная скорость реакции наблюдалась при этерификации гептановой кислоты (С7) бутиловым спиртом (С4). В одинаковых условиях синтеза сложных эфиров (20 ± 2°C, 1 бар, органический растворитель – смесь гексана и диэтилового эфира), в том числе, бутилгептаноата, rPichia/lip, адсорбированная на силикагеле, проявляла на порядок меньшую активность, чем липаза, адсорбированная на углеродном аэрогеле. Каталитические константы, равные соответственно 3.7 c–1 и 1.1 × 102 c–1, различались в 30 раз. Было обнаружено, что эфиры короткоцепочечных жирных кислот C4–C7 и этилового спирта синтезировались в 2–3 раза быстрее с помощью гидрофобного БКЛ типа PLC, чем с участием гидрофильного БКЛ типа PLSi. В то же время, эфиры высокомолекулярных С9, С10, С18 кислот и С8–С16 спиртов, обладающих выраженной гидрофобностью, синтезировались в 1.5–2 раза быстрее с участием БКЛ типа PLSi. Операционная стабильность приготовленных биокатализаторов была достаточно высокой: приготовленные БКЛ сохраняли 82–99% первоначальной активности после проведения более 30 реакционных циклов, при этом продолжительность каждого цикла до достижения конверсии кислоты выше 85% составила несколько часов (4–6 ч).
Функциональные свойства иммобилизованных ферментов, такие как каталитическая активность, субстратная специфичность и стабильность, определяются, как показали исследования, физико-химическими свойствами носителей-адсорбентов, используемых для их иммобилизации, например, текстурой и полярностью/гидрофобностью поверхности. В последние десятилетия интенсивно развивается направление под названием “ферментная инженерия путем выбора способа иммобилизации”, которое изучает изменение (модуляцию) функциональных свойств ферментов при иммобилизации. По мнению авторов обзора [1] “ферментная инженерия” полностью совместима с другими химическими и/или биологическими подходами, используемыми для улучшения функциональных свойств ферментов, и успех данной “инженерной” работы определяется доступностью широкого набора протоколов (способов) иммобилизации. Несомненно, управление (модуляция) функциональными свойствами ферментов с помощью ферментной инженерии является интересным и перспективным направлением в развитии гетерогенного биокатализа.
В своих ранних работах авторы, обобщив многочисленные результаты по адсорбционной иммобилизации ферментов класса оксидоредуктаз (лактат- и алкогольдегидрогеназы, глюкозооксидаза, тирозиназа) на пироуглерод-содержащих неорганических носителях, в том числе, мезопористом θ-оксиде алюминия, сделали вывод, что для приготовления активных и стабильных биокатализаторов необходимым условием являлось взаимное геометрическое и химическое соответствие физико-химических свойств фермента и поверхности адсорбента [2]. Например, геометрическое соответствие подразумевало соответствие размеров пор, преобладающих в текстуре адсорбента, размеру гидратированной молекулы фермента (в среднем 10 нм). Это значило, что мезопоры, размером выше 10–15 нм являлись оптимальными: в таких порах фермент взаимодействовал со стенками поры многоточечно, его конформация становилась более жесткой и, как результат, повышалась его стабильность. Химическое соответствие подразумевало, прежде всего, наличие оптимального гидрофильно-гидрофобного баланса между свойствами фермента и поверхностью адсорбента. Например, для глюкозооксидазы максимальный стабилизирующий эффект наблюдали при адсорбции данного фермента на мезопористом θ-Al2O3, содержащем 7–15 мас. % пироуглерода. Поверхность такого адсорбента напоминала “шахматную доску”: черные кластеры гидрофобного пироуглерода образовывались на сильных кислотно-основных центрах оксида алюминия и блокировали их. Оставшиеся участки незауглероженного Al2O3 белого цвета обладали слабокислыми полярными (гидрофильными) свойствами [2]. С другой стороны, при адсорбции на данных носителях алкогольдегидрогеназы и тирозиназы, наблюдали практически полную инактивацию этих ферментов, оптимальные носители содержали 0.5–3.0 мас. % углерода [2]. Задача выбора оптимального адсорбента решалась индивидуально для каждого конкретного фермента.
Липазы (гидролазы эфиров глицерина, КФ 3.1.1.3) катализируют разнообразные реакции – как прямые (гидролиз триглицеридов), так и обратные (синтез эфиров), последние протекают в неводных средах, где содержание воды не превышает 1 об. %. Биокатализаторы с активностью липазы (БКЛ) как в гомогенном (растворимом), так и в гетерогенном (иммобилизованном) состоянии находят широкое применение в различных отраслях промышленности, таких как:
1. производство “умных” стиральных порошков, удаляющих масляные и жировые пятна путем ферментативного гидролиза триглицеридов;
2. получение метиловых/этиловых эфиров жирных кислот (ЖК), входящих в состав растительных масел и отработанных кулинарных отходов, для производства биодизеля в качестве добавки к моторному топливу путем ферментативного алкоголиза (метанолиза, реже этанолиза) триглицеридов [3–5];
3. крупномасштабное производство ценных пищевых ингредиентов для получения спредов и маргаринов, заменителей масла какао и молочных жиров, не содержащих нежелательных транс-изомеров ЖК и обладающих заданными органолептическими и физико-химическими свойствами (температурой плавления, пластичностью, консистенцией, сливочным вкусом) путем ферментативной переэтерификации масложировых смесей при повышенных температурах (60–80°С) [6–9];
4. синтез сложных эфиров (СЭ) путем ферментативной этерификации в очень мягких условиях (20–50°С, 1 бар).
Известно, что СЭ пользуются значительным спросом на рынке ароматизаторов, смягчающих и увлажняющих компонентов (эмоллиентов), поверхностно-активных веществ и эмульгаторов в пищевой и косметической промышленности. Биокатализаторы, приготовленные на основе иммобилизованных липаз, интенсивно исследуются для проведения “зеленых” процессов в органической химии [9–12].
Возможность управлять функциональными свойствами липаз с помощью ферментной инженерии, а именно, путем выбора носителя для иммобилизации, можно продемонстрировать несколькими примерами. Так, каталитические свойства липазы B из Candida antarctica в реакции гидролитического разделения R- и S-изомеров (±)-2-O-бутирил-2-фенилуксусной кислоты изменяли (модулировали) иммобилизацией на носителях, обладающих гидрофобными свойствами, таких как бутил-(C4)-агароза, октил-(C8)-агароза и октадецил-(C18)-Sepabeads [13]. С ростом гидрофобности модифицирующих фрагментов (C4 → C18) скорость реакции увеличивалась приблизительно в 2 раза, при этом также изменялась стереоспецифичность биокатализа [13].
В работе [14] авторы исследовали свойства липазы из Penicillium sp., иммобилизованной на описанных выше носителях. Для данного фермента также было обнаружено, что с ростом гидрофобности модифицирующих фрагментов начальная скорость гидролиза p-нитрофенил-(С16)-пальмитата в буферном растворе и масла сардины в двухфазной системе с циклогексаном многократно увеличивались (в 4.1 и 2.3 раза соответственно) [14]. Авторы предположили, что носители, различающиеся по гидрофобности, оказывают влияние на конформацию активного центра фермента и, как результат, обусловливают гиперактивацию липазы и все наблюдаемые различия в функциональных свойствах фермента при его иммобилизации. В работе [15] было показано, что скорость этанолиза масла и региоселективность реакции в полностью безводной среде зависели от природы носителей, используемых для иммобилизации липазы из Тhermomyces lanuginosus. Так, при использовании C18-Sepabeads и C18-Purolite были получены неселективные биокатализаторы, тогда как при использовании носителей, модифицированных дивинил бензольными группами, наблюдалась 1,3-селективность по отношению к триглицеридам [15].
Современным направлением в биокатализе является компьютерное моделирование (КМ) 3D-структуры молекул ферментов, в том числе TLL (Тhermomyces Lanuginosus Lipase), расчет и предсказание влияния мутаций, вносимых в первичную структуру на функциональные свойства ферментов, прежде всего, на термостабильность [16]. Последующее проведение генно-инженерного конструирования штаммов-продуцентов и получение рекомбинантых ферментов с измененными функциональными свойствами является логичным продолжением таких работ. Очевидно, что дополнение экспериментальных методик современными методами КМ позволяет решать не только практические, но и фундаментальные задачи: выявлять механизмы процесса иммобилизации фермента на твердых носителях; проводить глубокий анализ возможных конформационных перестроек в молекуле фермента при его иммобилизации, а также структурно-функциональных особенностей комплекса фермент-носитель.
Авторы данной статьи ранее провели исследования БКЛ, приготовленных путем адсорбции рекомбинантной липазы rPichia/lip на неорганических носителях, в том числе, на различных углеродных нанотрубках (УНТ) [17], а также изучили процессы ферментативного низкотемпературного синтеза сложных эфиров с участием приготовленных биокатализаторов, включая выбор органического растворителя для реакционной среды [18–23]. В настоящей работе авторы продолжили систематическое изучение БКЛ и реакций этерификации ЖК, проанализировали все полученные результаты, дополнив работу современными методами компьютерного моделирования для анализа структурно-функциональных особенностей комплекса липаза-носитель.
Цель работы – продолжить сравнительные исследования функциональных свойств рекомбинантной липазы TLL (активности, субстратной специфичности, стабильности) в реакции синтеза высокомолекулярных СЭ, в зависимости от химической природы неорганических адсорбентов, а также проанализировать полученные результаты с целью поиска корреляционных зависимостей и закономерностей иммобилизации. Для выяснения механизма взаимодействия молекулы липазы TLL с углеродной нанотрубкой провести компьютерное моделирование с помощью жесткого и гибкого молекулярного докинга.
МЕТОДИКА
Рекомбинантная липаза из Тhermomyces lanuginosus (обознач. rPichia/lip) продуцировалась штаммом метилотрофных дрожжей Pichia pastoris X-33, специально сконструированным с помощью генно-инженерных манипуляций, как описано в работе [18]. Для приготовления БКЛ использовали буферные растворы rPichia/lip (0.02 М фосфатный буфер, pH 7.0) с концентрацией белка 2–15 мг/мл.
Концентрацию белка в растворе измеряли методом [24] с использованием красителя Coomassie G-250 (“Sigma”, США). Растворы бычьего сывороточного альбумина (“Sigma”, США) использовали для построения градуировочного графика.
Коммерческий силикагель (SiO2) марки КСК-Г (АО “Химический завод им. Л.Я. Карпова, Россия) имел следующие текстурные параметры: удельная поверхность (SудБЭТ) − 160 м2 · г–1, объем пор (VΣ) – 0.76 см3 · г–1. Пористость составляла 58%, мезопоры диаметром 20 нм преобладали в пористой структуре силикагеля. Размер гранул SiO2, взятых для приготовления БКЛ, составил 0.5–2 мм. Макропористый углеродный аэрогель (МУА) был получен в результате высокотемпературного пиролиза этилена на нанесенном катализаторе Fe:Co/CaCO3 [25, 26]. Насыпной вес составил 0.06 г · см–3. Гранулы МУА представляли собой легкие шарики черного цвета, образованные в результате хаотичного переплетения УНТ [19, 25]. МУА имел следующие текстурные параметры: SудБЭТ – 100 ± 20 м2 · г–1, VΣ – 12 ± 3 см3 · г–1, пористость составила 98%. Макропоры диаметром более 1 мкм преобладали в пористой структуре МУА, объем мезопор диаметром 2–20 нм не превышал 0.2% от VΣ. Гранулы МУА диаметром 3–4 мм использовали для приготовления БКЛ. Для измерения текстурных параметров носителей использовали методы азотной и ртутной порометрии на оборудовании AutoPore 9200 и ASAP 2400 V3.07 (“Micromeritics Instrument Corporation”, США). Для проведения электронно-микроскопических исследований МУА и биокатализаторов PLC типа использовали сканирующий электронный микроскоп (СЭМ) JSM 6460 LV (“JEOL”, Япония) и просвечивающий электронный микроскоп высокого разрешения (ПЭМ) JSM 2010 (“JEOL”, Япония).
Все реагенты: субстраты липазы (жирные кислоты и спирты), органические растворители (гексан, диэтиловый эфир) – реактивы производства России. Исходные реагенты и продукты реакции анализировали методом газовой хроматомасс-спектрометрии (ГХ-МС) на приборе Agilent 7000B GC/MS (“Agilent”, США) [23]. Для всех анализов была использована высокополярная колонка на основе ионной жидкости N-пропил-6-метил-хинолиний-бис(трифторметил-сульфонил)имида (10 м × × 0.25 мм × 0.2 мкм). Условия хроматографического анализа были следующими: сначала колонки выдерживались 3 мин при 100°С, затем температура повышалась со скоростью 10°С/мин до 280°С; температура испарителя − 280°С. Скорость потока газа-носителя (гелия) составляла 1 мл/мин. Условия работы масс-спектрометра: электронная ионизация – 70 эВ, температура источника ионизации – 230°С, температура переходной линии – 250°С. Спектр регистрировали в режиме сканирования в диапазоне 40–450 m/z.
Биокатализаторы с активностью липазы “rPichia/lip на силикагеле” (PLSi) и “rPichia/lip на углеродном аэрогеле” (PLC) получали путем адсорционной иммобилизации рекомбинантной липазы на соответствующих носителях. Для приготовления биокатализаторов типа PLSi, гранулы силикагеля предварительно высушивали при 115°C в течение 4 ч. Высушенные гранулы (1.0 г) пропитывали по влагоемкости буферным раствором липазы (рН 7.0) с концентраций белка 10 ± 4 мг · мл–1, помещали в плотно закрытый бюкс и выдерживали 5 ч при комнатной температуре (20 ± 2°C). Затем пропитанные раствором липазы гранулы высушивали в течение 1–2 сут в условиях окружающей среды до суховоздушного состояния. Количество иммобилизованной липазы (в мг · г–1) рассчитывали с учетом концентрации белка и объема пропиточного раствора, равного VΣ силикагеля (0.8 см3 · г–1).
Биокатализаторы типа PLC были приготовлены следующим образом. Гранулы МУА размером 3–4 мм заливали раствором липазы в буфере (pH 7.0) с концентраций белка 2 ± 1 мг · мл–1, в соотношении вес носителя : об. раствора = 1 : 100 и выдерживали в течение 1 сут при 20 ± 2°С при периодическом перемешивании. Затем раствор липазы декантировали, биокатализаторы многократно промывали буферным раствором (0.02 М фосфатный буфер, pH 7.0), помещали на бумажный фильтр для удаления избытка воды, и высушивали в течение 1–2 сут в условиях окружающей среды до суховоздушного состояния. Величину адсорбции (в мг · г–1) рассчитывали с учетом концентрации белка до адсорбции и после ее завершения.
Приготовленные БКЛ исследовали в реакции этерификации различных пар субстратов – насыщенной монокарбоновой кислоты (S1) и первичного алифатического спирта (S2), различающихся длиной углеродного скелета с количеством атомов углерода от 2 до 18. Этерификацию субстратов (более 60 пар S1 + S2) проводили в следующих условиях: 22 ± 2°С; 1 бар; начальные концентрации S1 и S2 соответственно 0.25 ± 0.03 моль · л–1 и 0.50 ± 0.04 моль · л–1; растворитель – смесь гексана и диэтилового эфира в соотношении 1 : 1 (об./об.). Навески биокатализаторов заливали раствором S1 и определяли начальную концентрацию кислоты, затем в реакционную среду добавляли S2 в двукратном молярном избытке. Содержание биокатализаторов в реакционной среде составило 20.8 мас. % для “rPichia/lip на SiO2” и 0.8 мас. % для “rPichia/lip на МУА”. Начальную скорость реакции определяли по линейному участку кинетической кривой убыли слабой органической кислоты (S1), концентрацию которой анализировали методом титриметрии с помощью этанольного раствора NaOH с известной молярностью (0.0256 ± ± 0.0006 моль · л–1) с применением фенолфталеина как индикатора точки эквивалентности. Скорость реакции и ферментативную активность биокатализаторов рассчитывали и выражали, соответственно, в мкмоль · л–1 · с–1 и в единицах активности (ЕА) на 1 г сухого биокатализатора (1 ЕА = = мкмоль · мин–1). Результаты титриметрического и хроматографического анализов совпадали в пределах экспериментальной ошибки, равной 10%.
Активность биокатализаторов измеряли в периодических реакционных циклах, продолжительность каждого определялась временем достижения конверсии кислоты не ниже 85%. В первом цикле биокатализаторы проходили стадию кондиционирования, при этом их активность увеличивалась в 2–4 раза. Это было обусловлено тем, что внутри биокатализатора аккумулировалась вода, образующаяся в ходе этерификации, и формировалось благоприятное водное микроокружение для адсорбированной липазы, поддерживающее структуру молекул фермента в функциональной гидратированной форме. После окончания этерификации реакционную смесь, содержащую сложный эфир, а также небольшое количество субстратов S1 и S2, декантировали. Биокатализаторы многократно промывали растворителем – смесью гексана и диэтилового эфира, затем дополнительно выдерживали в этом же растворителе в течение не менее 20 ч. Следующий реакционный цикл проводили с отмытым биокатализатором в условиях, описанных выше.
Статистическую обработку результатов проводили по критерию Стьюдента c доверительной вероятностью 0.95, количество измерений n = 3–6. Относительная ошибка не превышала 10% (минимум 6%).
Для расчета геометрии молекул субстратов липазы S1 и S2, использовали метод молекулярной механики (Силовое поле ММ+) в пакете программ HyperChem 7 (Hypercube Inc., США).
Компьютерное моделирование взаимодействия молекулы TLL с одностенной углеродной нанотрубкой было проведено с помощью молекулярного докинга. Кристаллическая структура TLL с разрешением 2.3 Å была взята из базы данных PDB (PDB ID: 4ZGB). Для моделирования использовали форму липазы, у которой активный центр находился в закрытой конформации [27]. Структуры УНТ различного размера были получены в программе Nanotube Modeler и оптимизированы в Avogadro. Молекулярная стыковка мономера и димера липазы с нанотрубками различной длины и диаметра была проведена с помощью программного пакета AutoDock Vina 1.1.2, позволяющего провести гибкий докинг (flexible docking) с учетом подвижности боковых остатков аминокислот, в частности, GLU1A, LEU6A, LYS24A, LYS24B, ASN26B, PRO29A, ARG84A, ARG84B, PRO136A, ASP137A, LYS237A. Боковые остатки этих аминокислот считались “гибкими” после проведения жесткого докинга, во время которого были оценены наиболее вероятные места связывания УНТ с липазой. Размер бокса, в который была помещена липаза, составил: x = 126; y = 118; z = 126. Пространственный центр липазы располагался в точке с координатами: x = 20.489; y = 49.005; z = 0.215. Расчeт был сведен к автоматическому перебору пространственных конформаций и ориентаций нанотрубок в сайте связывания с липазой, чтобы получить комплекс липаза-УНТ с минимальной свободной энергией системы. Визуализация результатов моделирования проводилась в программах MGLTools и PyMol.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Исследования биокатализаторов PLC типа методом сканирующей электронной микроскопии показали, что морфология поверхности исходного МУА и БКЛ были практически одинаковыми: отчетливо наблюдались хаотически переплетенные УНТ, аналогично описанному в работе [19]. После адсорбции липазы “ажурность” 3D-структуры углеродного аэрогеля уменьшалась; переплетение УНТ, более утолщенных по сравнению с исходным носителем, уплотнялось; образовывались гладкие пленки, вероятно, белкового происхождения. Как видно из изображений, полученных с помощью просвечивающего электронного микроскопа (ПЭМ), на стыках от переплетения УНТ наблюдались аморфные отложения, очевидно, образованные адсорбированной липазой rPichia/lip (рис. 1). Гранулы биокатализаторов PLC после высушивания уменьшались в диаметре 1.5–2 раза по сравнению с исходными гранулами МУА, полученными в каталитической установке высокотемпературного пиролиза этилена. После проведения первых двух-трех реакционных циклов этерификации гранулы БКЛ набухали из-за образования и аккумулирования воды, образующейся в ходе реакции, внутри данных биокатализаторов.
Рис. 1.
ПЭМ изображения участков биокатализатора PLC типа при различных увеличениях ×20 (а), ×10 нм (б). Стрелками показаны аморфные отложения белковой природы (адсорбированной липазы).

Учитывая текстурные параметры носителей – силикагеля и углеродного аэрогеля, а также размер гидратированной молекулы липазы, были рассчитаны величины поверхности, доступной для адсорбции rPichia/lip, с учетом того, что поры диаметром 20 нм и выше доступны для иммобилизации. Величина доступной поверхности (Sдоступ) силикагеля, равная 77 м2 · г–1, составила 51% от SудБЭТ. Для макропористого углеродного аэрогеля, как видно из текстурных параметров, вся поверхность SудБЭТ, равная 100 м2 · г–1, доступна для адсорбции липазы. Несмотря на близкие величины Sдоступ, величины адсорбции липазы на изученных носителях различались на порядок. Биокатализаторы PLSi содержали от 6.2 – 14.0 мг · г–1 (количество мг белка на 1 г носителя). Величина адсорбции rPichia/lip на МУА составила 100 ± 20 мг · г–1.
Каталитические свойства иммобилизованной липазы, рассчитанные ранее из полной кинетической кривой Михаэлиса-Ментен с помощью программного обеспечения Origin® (OriginLab Corp., США), такие как максимальная скорость реакции, константы Михаэлиса, каталитические константы (k, с–1), зависели от химической природы носителей: для липазы, иммобилизованной на силикагеле, каталитическая константа, была в ~30 раз меньше, чем для rPichia/lip, адсорбированной на МУА, а именно, значения k были равны 3.7 c–1 и 1.1 × 102 c–1 соответственно. Максимальные активности приготовленных БКЛ, измеренные в реакции этерификации гептановой кислоты н-бутанолом, составили 22.3 ± 2.0 и 513.9 ± 44.4 ЕА · г–1 для биокатализаторов PLSi и PLC соответственно [18].
Результаты систематического исследования субстратной специфичности адсорбированной rPichia/lip в зависимости от химической природы носителя – гидрофильного силикагеля и гидрофобного углеродного аэрогеля, представлены в виде матриц относительных активностей (Аотн) в работах [20, 21], а также в настоящей работе, в которой наиболее подробно и полно изучена субстратная специфичность биокатализаторов типа PLC по отношению к высокомолекулярным субстратам S1 (С7 и выше). Было показано, что для обоих типов БКЛ характерной является широкая субстратная специфичность в реакции синтеза широкого набора сложных эфиров насыщенных монокарбоновых (жирных) кислот и первичных алифатических спиртов в очень мягких условиях (20 ± 2°C, 1 бар), при этом за 2–4 ч достигалась конверсия кислоты не ниже 85%.
Сравнительный анализ полученных результатов также показал, что общими для двух типов БКЛ были следующие свойства: 1) скорость синтеза эфира гептановой кислоты с n- или iso-бутанолом (бутилгептаноата) была максимальной и в расчетах Аотн принималась за 1 ед.; 2) в большинстве случаев скорость синтеза этиловых эфиров (S2 – этанол, С2) была минимальной по сравнению со скоростью этерификации с участием более высокомолекулярных спиртов, в том числе, пропанола, С3 и бутанола, С4; 3) скорость синтеза сложных эфиров изомеров iso-С4–С5 кислот была на 1–2 порядка ниже по сравнению с их линейными молекулами, в то же время изомерия С4–С5 спиртов практически не влияла на скорость этерификации [22]; 4) скорость реакции этерификации с участием вторичных спиртов была на порядок ниже по сравнению с первичными; третичные спирты в реакции этерификации не участвовали [20, 21].
Масштабные сравнительные исследования двух типов приготовленных биокатализаторов с использованием широкого набора пар субстратов S1 и S2 (более 60 пар) позволили получить многочисленные экспериментальные результаты, провести их анализ и сделать вывод о том, как химическая природа носителей изменяет (модулирует) субстратную специфичность иммобилизованной рекомбинантной липазы. Поскольку иммобилизованная липаза более “чувствительна” к субстрату S1, то жирные кислоты C7, C9, C10, С12, C18 рассматривались индивидуально, а субстраты S2 (спирты) были разделены на три группы в зависимости от их полярности, характеристикой которой является logP [28]: группа 1 (logP < 1) – это С2 (этанол), С3 (пропанол), C4 (бутанол), группа 2 (1 < logP < 4) – это C5 (пентанол), C8 (октанол), группа 3 (logP > 4) – это C10 (деканол), C11 (ундеканол), C12 (додеканол), C16 (гексадеканол).
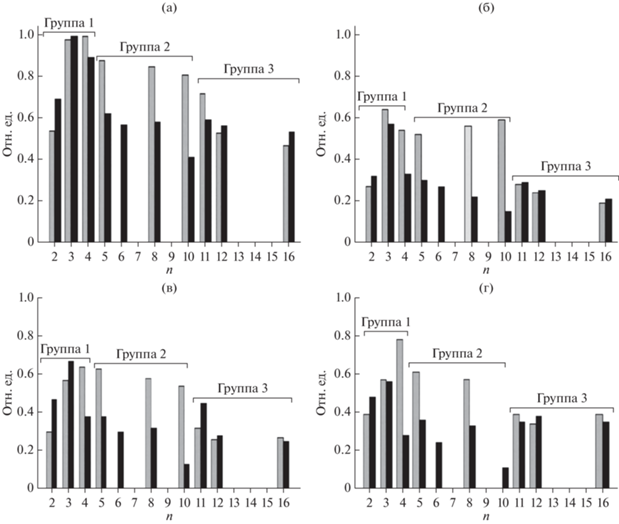
Наблюдаемые различия в субстратной специфичности БКЛ в зависимости от химической природы (гидрофильности/гидрофобности) носителей, используемых для адсорбционной иммобилизации rPichia/lip, представлены на рис. 2, где серые колонки соответствуют – биокатализаторам типа PLSi, черные колонки – типа PLC. Сравнение высоты колонок, соответствующих Аотн, позволило обнаружить следующее. Значительные различия между биокатализаторами разных типов наблюдались при синтезе этиловых эфиров (S2 – этанол, С2Н5ОН) различных по длине ЖК: скорости реакций этерификации были значительно выше для биокатализаторов PLC типа (“rPichia/lip на МУА”), а именно, в 2.4–2.8 раза – для С4–6 кислот [21] и 1.2–1.6 раз – для С7–18 кислот (рис. 2). Известно, что этанол способен инактивировать различные ферменты, в том числе, иммобилизованную rPichia/lip [29]. Поскольку гидрофобность исходного МУА в присутствии этанола значительно уменьшалась за счет адсорбции молекул этилового спирта на УНТ, то, по-видимому, этот адсорбент обладал способностью проявлять “защитный” эффект, уменьшая локальную концентрацию С2Н5ОН в микроокружении липазы. Для другого спирта из группы 1 – бутанола, такого защитного эффекта уже не наблюдали. При сравнении величин Аотн, например, в реакции синтеза бутиловых эфиров гептановой С7 (рис. 2а) и стеариновой С18 кислот (рис. 2г), оказалось, что относительная активность БКЛ типов PLSi и PLC уменьшилась в 1.3 (для С7) и 3.5 (для С18). Если S2 относился к группе 2, то относительная скорость этерификации С7–18 кислот с участием БКЛ типа PLC была также существенно, в 1.5–2 раза, ниже (рис. 2), что было весьма неожиданным результатом. Если S2 относился к группе 3, то особых различий в относительных скоростях синтеза СЭ, в большинстве случаев, не наблюдалось (рис. 2), что также являлось неочевидным фактом. Общеизвестно, что гидрофобные высокомолекулярные субстраты S1 и S2 обладают более высоким сродством к углеродной поверхности УНТ по сравнению с SiO2, их приповерхностная концентрация увеличивалась за счет гидрофобных взаимодействий с поверхностью, что могло приводить к увеличению скорости их этерификации. Однако эти же взаимодействия могли уменьшить локальную концентрацию субстратов вблизи иммобилизованной липазы или затруднять распад фермент-субстратного комплекса, или препятствовать десорбции СЭ как более высокомолекулярного и более гидрофобного по сравнению с S1 и S2 продукта реакции. В результате, скорость процесса этерификации в целом не увеличивалась. Таким образом, гидрофильный PLSi являлся более эффективным биокатализатором синтеза высокомолекулярных СЭ по сравнению с гидрофобным PLC.
Рис. 2.
Относительная активность (отн. ед.) биокатализаторов типа PLSi (серые колонки) и PLC (черные колонки) и в реакции этерификации насыщенных жирных кислот: а – гептановой (энантовой, C7), б – нонановой, пеларгоновой, C9), в – декановой (каприновой, C10), г –октадекановой (стеариновой, C18) алифатическими спиртами с различной длиной углеродного скелета (n – количество атомов углерода).
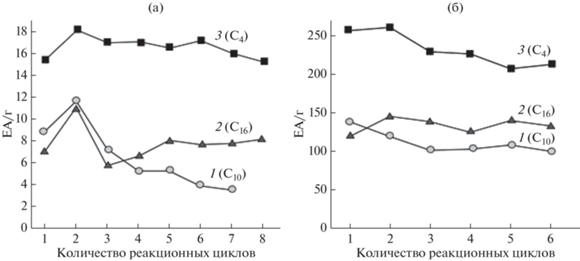
В настоящей работе были сделаны попытки обнаружить корреляции между этерифицирующей активностью БКЛ и физико-химическими характеристиками субстратов липазы. Как видно на рис. 3а, полярность алифатических спиртов (S2), характеризуемая величиной logP, линейно возрастала по мере удлинения углеродной цепи их молекул. В то же время, для изученных С7, С9 и С12 кислот (S1) монотонной или корреляционной зависимости активности биокатализатора от полярности S2 не наблюдалось (рис. 3б). В результате многократных экспериментов было обнаружено, что скорость синтеза эфиров бутанола С4 для всех изученных кислот была максимальной, а эфиров деканола С10 – воспроизводимо минимальной (рис. 3б). Было также показано, что деканол (дециловый спирт, С10) оказывал негативное влияние как на активность, так и на стабильность биокатализаторов типа PLSi: так, в течение семи реакционных циклов этерификации гептановой С7 кислоты деканолом первоначальная активность БКЛ типа PLSi упала в среднем в 3 раза (рис. 4а), тогда как активность биокатализатора типа PLC за шесть реакционных циклов уменьшилась в 1.4 раза (рис. 4б). Биокатализаторы обоих типов проявляли высокую активность и стабильность в реакции синтеза бутилгептаноата (рис. 4).
Рис. 3.
Полярность (logP) спиртовых субстратов S2 в зависимости от количества атомов углерода в молекуле (а), зависимость активности биокатализатора “rPichia/lip на МУА” от параметра logP (б) в реакции синтеза сложных эфиров: 1 – нонановой (пеларгоновой, С9), 2 – додекановой (лауриновой, С12), 3 – гептановой (энантовой, С7) кислот.
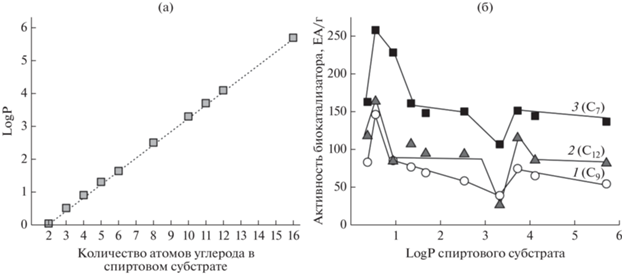
Рис. 4.
Зависимость активности (ЕА · г) биокатализаторов “rPichia/lip на силикагеле” (PLSi) (а) и “rPichia/lip на МУА” (PLC) (б) от количества реакционных циклов этерификации гептановой С7 кислоты спиртами: 1 – дециловым (С10), 2 – гексадециловым (цетиловым, С16), 3 – бутиловым (С4).
Выраженное негативное влияние деканола (S2) на функциональные свойства иммобилизованной липазы rPichia/lip не зависело ни от природы субстрата-кислоты (S1), ни от физико-химических свойств адсорбентов: для БКЛ обоих типов наблюдались аналогичные зависимости, включая, минимумы в скоростях реакции этерификации с участием деканола С10 (рис. 3б). Наиболее “глубокий” минимум был характерен для реакции этерификации додекановой кислоты (лауриновой, С12) деканолом С10: так, этерифицирующая активность БКЛ в реакции синтеза децил додеканоата (рис. 3б, 2) была в 3.2 раза ниже по сравнению со скоростью синтеза бутиловых эфиров этой кислоты. Для энантовой (гептановой, С7) и нонановой (пеларгоновой, С9) кислот (рис. 3б, 1 и 3), различия в скоростях синтеза дециловых эфиров этих кислот составило 2.2 раза. Интересным показался тот факт, что добавление одной СН2–группы в молекулу деканола приводит к 2–4-кратному увеличению активности биокатализатора (рис. 3б). Эти данные можно попытаться объяснить топологией активного центра фермента (Е), а также процессами образования и распада фермент-субстратного комплекса (ЕS1S2). Расчеты с помощью программы HyperChem® показали, что молекулы деканола и додекановой кислоты в “растянутом” виде имеют близкие по величине размеры, равные 1.3 ± 0.2 нм. По-видимому, углеродные цепи этих субстратов взаимодействуют друг с другом наиболее эффективно за счет дисперсионных (гидрофобных) взаимодействий между метиленовыми (–СН2–) группами. Это может приводить как к специфической блокировке активного центра, так и к затруднениям распада комплекса ЕS1S2. Как показали исследования, максимальные скорости этерификации наблюдали для гептановой кислоты и бутанола, возможно, из-за небольших размеров их молекул, 0.95 и 0.6 нм соответственно. При увеличении длины молекул субстратов липазы эффективность биокатализа падала. Более широкая специфичность иммобилизованной липазы по отношению к спиртовому субстрату указывала на то, что S2-связывающий сайт активного центра. липазы по размеру более широкий, чем S1-связывающий, что позволило подтвердить выводы о строении активного центра липазы из T. lanuginosus, сделанные в работах [27, 30–33]: активный центр липазы типа RML, к которому относится TLL, расположен близко к поверхности глобулы фермента и имеет узкую щель для S1 (кислоты), и более широкую –для S2 (спирта).
С целью выяснения механизма взаимодействия липазы из T. lanuginosus с УНТ было проведено компьютерное моделирование. Для моделирования были выбраны следующие объекты: мономер и димер TLL, одностенные углеродные нанотрубки диаметром 6.785 и 7.834 Å, длиной 50 и 100 Å. Были получены следующие результаты: 1) липаза TLL взаимодействовала с УНТ посредством гидрофобных взаимодействий остатков следующих аминокислот – лизина, аргинина, аспарагина, пролина, и π-катионных взаимодействий с остатком лизина (табл. 1, рис. 5); 2) энергия взаимодействия между димером липазы и нанотрубкой не зависела от длины УНТ и составила –28 ккал · моль–1; 3) энергия взаимодействия мономера TLL c углеродной нанотрубкой в 2 раза меньше, чем димера, поскольку число связей уменьшается пропорционально; 4) учет in silico подвижности боковых остатков аминокислот приводил к снижению энергии на 3 ккал · моль–1 (в расчете на мономер); 5) УНТ связывалась в щели между двумя мономерами липазы (рис. 6); 6) активные центры димера TLL, состоящего из мономеров А и Б, находились на расстоянии на 6.6 и 25.8 Å (0.66 и 2.58 нм) от УНТ (рис. 6). В этом случае для объяснения наблюдаемых максимума и минимума на кривых рис. 3б, можно также предположить, что для низкомолекулярных С4–7 субстратов доступны оба активных центра иммобилизованной липазы, тогда как для высокомолекулярных субстратов с количеством атомов углерода выше 10, доступен преимущественно один активный центр, находящийся на расстоянии 25.8 нм от УНТ (рис. 6а).
Таблица 1.
Взаимодействия между димером TLL, состоящим из двух мономеров А и В, и одностенной УНТ*
| Аминокислотный остаток | Длина связи | Атом УНТ | Атом белка | |
|---|---|---|---|---|
| Гидрофобные взаимодействия | ||||
| 24A | LYS | 3.43 | 5195 | 226 |
| 24A | LYS | 3.22 | 5150 | 227 |
| 24B | LYS | 3.34 | 5117 | 2763 |
| 29A | PRO | 3.67 | 5508 | 277 |
| 84A | ARG | 3.83 | 5226 | 761 |
| 84B | ARG | 3.54 | 5437 | 3295 |
| Взаимодействие с аспарагином | ||||
| 26B | ASN | 3.92 | 5428 | 2786 |
| π-Катионные взаимодействия | ||||
| 24B | LYS | 5.38 | 5118, 5119, 5120, 5121, 5163, 5165 | |
Рис. 5.
Докинг димера TLL с одностенной УНТ длиной 50 Å, диаметром 7.834 Å (n = 10, m = 0). Указаны атомы, которые непосредственно взаимодействуют с УНТ. Атомам 226, 227, 761, 2763, 2786, 3295 (из табл. 1) соответствуют атомы углерода в CH2, 277– атом углерода в CH.

Рис. 6.
Докинг димера TLL с одностенной УНТ длиной 50 Å, диаметром 7.834Å (n = 10, m = 0). Стрелка указывает на активный центр фермента. Указаны расстояния между активным центром липазы (Asp96-His110-Ser115) и УНТ.

Таким образом, в данной работе было установлено, что функциональные свойства иммобилизованной рекомбинантной липазы rPichia/lip, такие как активность и субстратная специфичность, изменялись (модулировались) в зависимости от химической природы носителя. Приготовленные БКЛ существенно различались по каталитическим свойствам: активность и каталитическая константа для биокатализатора “rPichia/lip на SiO2” (тип PLSi) были в 20–30 раз ниже, чем для "rPichia/lip на МУА” (тип PLC). Несмотря на широкую субстратную специфичность обоих типов БКЛ, синтез этиловых эфиров насыщенных монокарбоновых кислот протекал более эффективно с участием гидрофобного БКЛ типа PLC, тогда как синтез высокомолекулярных эфиров с выраженной гидрофобностью – с участием гидрофильного типа PLSi. Для обоих типов приготовленных биокатализаторов максимальная скорость реакции наблюдалась при этерификации гептановой С7 кислоты бутиловым спиртом, а минимальная – бутановой кислоты дециловым С10 спиртом. Приготовленные активные и стабильные биокатализаторы PLSi и PLC типов, несомненно, обладают высоким практическим потенциалом для “зеленых” процессов низкотемпературного синтеза разнообразных сложных эфиров.
Авторы благодарят Кузнецова В.Л. за предоставленные образцы МУА, Рудину Н.А. и Ищенко А.В. за проведение электронно-микроскопических исследований биокатализаторов PLC типа.
Работа выполнена при финансовой поддержке Министерства науки и высшего образования РФ в рамках государственного задания Института катализа СО РАН (проект НИР 0239-2021-0005).
Работы по молекулярному моделированию проведены в рамках государственного задания Севастопольского государственного университета (проект НИР FEFM-2020-0003).
Список литературы
Mateo C., Palomo J.M., Fernandez-Lorente G., Guisan J.M., Fernandez-Lafuente R. // Enzyme and Microbial Technology. 2007. V. 40. P. 1451–1463.
Sokolovskii V.D., Kovalenko G.A. // Biotechnol. Bioeng. 1988. V. 32. P. 916–919.
Stoytcheva M., Montero G., Toscano L., Gochev V., Valdez B. / Ed. Stoytcheva M., Montero G. // Biodiesel – Feedstocks and Processing Technol. InTech, 2011. P. 397–410.
Luna C., Garson-Perez V., Lopez-Tenllado F.J., Baustista F.M., Verdugo-Escamilla C., Aguado-Dedlas L. et al. // Catalysts. 2021. V. 11. P. 1350–1362.https://doi.org/10.3390/catal11111350
Hernandez-Martin E., Otero C. // Bioresource Technol. 2008. V.99. P. 277–286.
Li D., Adhikari P., Shin J.-A., Lee J.-H., Kim Y.-J., Zhu X.-M.,et al. // Food Sci. Technol. 2010. V. 43. P. 458–464. https://doi.org/10.1016/j.lwt.2009.09.013
Lopez-Hernandez A., Otero C., Hernandez-Martin E., Garcia H. S., Hill C. G., Jr. // Eur. J. Lipid Sci. Technol. 2007. V. 109. P. 1147–1159.
Osorio N.M., Gusmao J.H., Manuela da Fonseca M., Ferreira-Dias S. // Eur. J. Lipid Sci. Technol. 2005. V. 107. P. 455–463.
Handbook of Industrial Catalysis. /Ed. C.T. Hou. Boca Raton: CRC Press, 2005. 500 p.
Bucholdz K., Kasche V., Bornscheuer U.T. Biocatalysts and Enzyme Technology. Weinheim: Wiley-VCH Verlag, 2005. 447 p.
Biocatalysis for Green Chemistry and Chemical Process Development. Ed. J. Tao, R. Kazlauskas. John Wiley & Sons, 2011. 479 p.
Безбородов А.М., Загустина Н.А., Попов В.О. // Прикл. биохимияи микробиология. 2014. Т. 50. № 4. С. 347–373.
Cabrera Z., Fernandez-Lorente G., Palomo J.M., Fernandez-Lafuente R., Guisan J. M. // J. Mol. Catal. B: Enzym. 2009. V. 57. P. 171–176.
Turati D.F.M., Morais W.G, Jr., Terrasan C.R.F., Moreno-Perez S., Pessela B.C., Fernandez-Lorente G., Guisan J.M., Carmona E. C. // Molecules. 2017. V. 22. P. 339–353.
Silveira E.A., Moreno-Perez S., Basso A., Serban S., Mamede R.P., Tardioli P.W., Farinas C.S. et al. // BMC Biotechnol. 2017. V. 17. P. 88–101.
Bohr S.S., Lund P.M., Kallenbach F.S., Pinholt Y., Thomsen J., Iversen L. et al. // Scientific Reports . 2019. V. 9. P. 16169–16180. doi.org/https://doi.org/10.1038/s41598-019-52539-1
Нуруллина П.В., Перминова Л.В., Коваленко Г.А. // Вестн. Моск. ун-та. сер. 2. Химия. 2020. Т. 61. № 2. С. 134–140. https://doi.org/10.3103/S0027131420020108
Беклемишев А.Б., Пыхтина М.Б., Перминова Л.В., Коваленко Г.А. // Биотехнология. 2021. Т. 37. № 5. С. 5–19. https://doi.org/10.21519/0234-2758-2021-37-5-5-62
Kovalenko G.A., Perminova L.V., Krasnikov D.V., Kuznetsov V.L. // J. Porous Mater. 2018. V. 25. P. 1017–1026. DOI: 10.1007/s10934-017-0512-0
Kovalenko G.A., Perminova L.V., Beklemishev A.B. // Reac. Kinet. Mech. Catal. 2019. V. 128. P. 479–491. https://doi.org/10.1007/s11144-019-01648-z
Kovalenko G.A., Perminova L.V., Pykhina M.B., Beklemishev A.B. // Biocatal. Agr. Biotechnol. 2021. V. 36. 102124. https://doi.org/10.1016/j.bcab.2021.102124
Kovalenko G.A., Perminova L.V., Pykhina M.B., Bek-lemishev A.B. // Catalysis Today. 2021. V. 379. P. 36–41. https://doi.org/10.1016/j.cattod.2020.11.018
Коваленко Г.А., Перминова Л.В., Шашков М.В., Беклемишев А.Б. // Кинетика и катализ. 2022. Т. 63. № 2. С. 212–222. https://doi.org/10.31857/S0453881122020046
Bearden J. // Biochim. Biophys. Acta. 1978. V. 533. P. 525–529.
Kuznetsov V.L., Suslyaev V.I., Dorofeev I.D., Kazakova V.F., Moseenkov S.I., Smirnova T.E., Krasnikov D.V. // Phys. Status Solidi B. 2015. V. 252. № 11. P. 2519–2523. https://doi.org/10.1002/pssb.201552254
Патент RU 2 577 273 C1. Бюл. № 7. 2016.
Kumar M., Mukherjee J., Sinha M., Kaur P., Sharma S., Gupta M.N., Singh T.P. // Sustain Chem Process. 2015. V. 3. 10 p. https://doi.org/10.1186/s40508-015-0042-5
Laane C., Boeren S., Vos K., Veeger C. // Biotechnol. Bioeng. 1987. V. 30. P. 81–87.
Коваленко Г.А., Перминова Л.В., Беклемишев А.Б., Яковлева Е.Ю., Пыхтина М.Б. // Катализ в промышленности. 2014. № 6. С. 71–79.
Palomo J.M., Ortiz C., Fuentes M., Fernandez-Lorente G., Guisan J.M., Fernandez-Lafuente R. // J. Chromatogr. A. 2004. V. 1038. P. 267–273. https://doi.org/10.1016/j.chroma.2004.03.058
Naik S., Basu A., Saikia R., Madan B., Paul P., Chatejree R., Brask J., Svendsen A. // J. Mol. Catal. B: Enzym. 2010. V. 65. P. 18–23. https://doi.org/10.1016/j.molcatb.2010.01.002
Fernandez-Lafuente R. // J. Mol. Catal. B: Enzym. 2010. V. 62. P. 197–212. https://doi.org/10.1016/j.molcatb.2009.11.010
Rodrigues R.C., Fernandez-Lafuente R. // J. Mol. Catal. B: Enzym. 2010. V. 66. P. 15–32. https://doi.org/10.1016/j.molcatb.2010.03.008
Дополнительные материалы отсутствуют.
Инструменты
Прикладная биохимия и микробиология