

Расплавы, 2021, № 1, стр. 55-64
Анализ механизмов протонирования для моделей поверхностных комплексов N-(пропил)этилендиаминтриуксусной кислоты с кремнеземом
И. А. Бабина b, *, Б. С. Воронцов a, **, В. В. Москвин a, И. Н. Грехов b, А. О. Бабин b
a Курганский государственный университет
Курган, Россия
b Южно-Уральский государственный университет
(национальный исследовательский университет)
Челябинск, Россия
* E-mail: babina_inga@mail.ru
** E-mail: fizika@kgsu.ru
Поступила в редакцию 03.02.2020
После доработки 13.02.2020
Принята к публикации 25.03.2020
Аннотация
Ввиду использования сорбентов в различных областях науки, техники и в быту, представляет интерес разработка новых материалов с сорбционными свойствами. Для исследования свойств модифицированных сорбентов выбран аморфный кремнезем. Так как существуют проблемы строгого описания взаимодействий поверхности кремнеземов с привитыми структурами, было решено использовать модельный расчет энергий протонирования структуры с привитой N-(пропил)этилендиаминтриуксусной кислотой. Показано, что модельный эксперимент, основанный на полуэмпирических квантовохимических расчетах, свидетельствует о том, что протонирование кремнезема, модифицированного молекулами со слабоосновным азотом, сопровождается формированием различных по строению поверхностных комплексов, в том числе с образованием водородных связей.
ВВЕДЕНИЕ
Широкое применение сорбционных технологий очистки сред, выделения конечных продуктов требует создания все новых материалов на основе модифицированных сорбентов, в том числе и с привитыми группами [1]. Удобным объектом для исследования свойств модифицированных поверхностей является аморфный кремнезем, что обусловлено накопленными знаниями о составе поверхностных групп и их реакционной способности [2, 3]; возможностью получения прочных поверхностных структур. Известным и подтвержденным фактом является взаимодействие кислотных силанольных групп поверхности кремнеземов с основными атомами азота привитых групп.
ТЕОРЕТИЧЕСКИЙ АНАЛИЗ
Несмотря на большое количество экспериментального материала, вопрос о строгом описании этих взаимодействий и возможности проведения на его основе предрасчета сорбционных равновесий проработан недостаточно [4, 5]. Одним из путей решения проблемы является модельный расчет энергий протонирования привитых структур с кластерами кремнеземной поверхности. Первоначально целесообразно рассмотреть привитые молекулы со слабоосновным азотом. В качестве модели для рассмотрения в данной публикации выбрана структура с привитой N-(пропил)этилендиаминтриуксусной кислотой. Это распространенный комплексон, прививка которого на поверхность позволяет усилить ее сорбционную активность по отношению к ионам металлов. Имеющиеся экспериментальные данные говорят о слабых взаимодействиях подобных веществ с поверхностными силанольными группами.
МЕТОДИКА
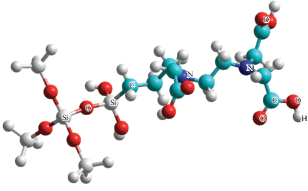
Модели строились с использованием графического редактора пакета HyperChem [6] поэтапно через ряд промежуточных стадий. При этом на каждом этапе число оптимизируемых параметров было относительно небольшим, что позволяло достаточно быстро реализовать процедуру оптимизации с максимальным градиентом 0.01 ккал/моль ∙ A. Первые три модели отличаются друг от друга размером фрагмента, представляющего поверхность кремнезема. На рис. 1 представлена такая модель с минимальным поверхностным кластером. Подробную информацию о представительных кластерах поверхности кремнезема можно найти, например, в работе [7], и опыте авторов по построению поверхностных комплексов SiO2 со сложными органическими молекулами в работе [8].
Рис. 1.
Поверхностный комплекс N-(пропил)этилендиаминтриуксусной кислоты с минимальным представительством кремнезема группировкой Si–(OSiH3)3–O–.
Протолитические свойства моделей были оценены по простейшей методике, основанной на расчете энергий протонирования и депротонирования. Эти энергии приравнивались к разности полных энергий нейтральной модели и заряженной модели с присоединенным (удаленным) ионом Н+ [9].
По результатам наших неэмпирических расчетов, приведенных в [9], энергия протонирования жестких моделей SiO2 по атому кислорода в составе поверхностных O–H-групп и по мостиковому кислороду достаточно близки и равны соответственно 882 и 869 кДж/моль.
Полуэмпирические расчеты дают существенно разнящиеся значения энергий протонирования. Одинаково то, что протонирование по мостиковому кислороду связано с меньшим энергетическим эффектом, как и в случае неэмпирических расчетов. Сопоставительные данные для модели на рис. 1 приведены в табл. 1.
Таблица 1.
Энергии протонирования поверхности кремнезема в кДж/моль по данным полуэмпирических расчетов
| Протонирование
по гидроксильной группе Si–OH–H+ |
Протонирование
по мостиковому кислороду Si–Oм–Si |
|
|---|---|---|
| Параметры PM3 Программа Hyper Chem |
946.3 | 928.1 |
| Параметры MNDO программа Hyper Chem |
760.6 | 739.7 |
| Параметры PM7 Программа MOPAC |
580.9 | 565.4 |
Из данных табл. 1 следует, что применение полуэмпирических методов неэффективно для установления абсолютных значений энергий протонирования. Имеет смысл только сопоставительный эксперимент в рамках одного метода.
Расчет электростатического потенциала для поверхностного комплекса, приведенного на рис. 1, показал наиболее вероятные центры протонной атаки (рис. 2).
Рис. 2.
Распределение потенциала для модели с максимальным представительством кремнезема; розовым цветом показаны области отрицательного потенциала.

В соответствии с этим распределением, центрами протонной атаки выбирались атомы кислорода, связанные двойной связью с атомами углерода (на рис. 1 и 2 внизу и вверху справа).
При расчете депротонированной модели удалялся один из атомов водорода, и заряд полагался равным –1. Помимо изменений полной энергии нами отслеживались также изменения в распределении зарядов (рассчитанных по Малликену [10]) для наиболее важных, с химической точки зрения, атомов. При необходимости дополнительно анализировалась информация о значениях энергии связывания и теплоты образования моделей.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Для квантовохимических расчетов было решено использовать три полуэмпирических метода, упомянутых в табл. 1.
1. Расчеты методом MNDO
В начале исследования нами был выбран полуэмпирический метод ПДДП (MNDO), результаты использования которого даны в табл. 2.
Таблица 2.
Энергии протонирования поверхности кремнезема в ккал/моль по данным полуэмпирических расчетов методом ПДДП (MNDO)
| Расчетные значения, ккал/моль | Исходная модель | После протонирования | После депротонирования |
|---|---|---|---|
| Полная энергия | Eполн = –151417.9 | –15 1570.4 | –15 1115.48 |
| Энергия связывания | Eсвяз = –5754.34 | –5632.31 | –5726.49 |
| Теплота образования | dH = –830.746 | –656.60 | –855.02 |
Таким образом, энергия протонирования равна –638.65 кДж/моль (понижение энергии модели), а изменение теплоты образования составляет +729.13 кДж/моль.
Энергия депротонирования +1905 кДж/моль (повышение полной энергии). Теплота образования при этом возрастает на 101.8 кДж/моль.
Анализ перераспределения зарядов на атомах, произошедшего в результате протонирования можно провести на основе информации, представленной на рис. 2. Очевидно, что перераспределение затрагивает достаточно большую часть молекулы, прилежащую к месту протонирования, и приводит к симметризации области протонирования с образованием двух одинаковых C–O–H связей.
Качественно сходны результаты для модели с увеличенной до кластера состава Si–O(SiH3)2–O–SiH2–OSiH3 кремнеземной части (табл. 3).
Таблица 3.
Энергии протонирования поверхности кремнезема в ккал/моль для кластера состава Si–O(SiH3)2–O–SiH2–OSiH3
| До протонирования | После протонирования | После депротонирования | |
|---|---|---|---|
| Полная энергия | Eполн = –161 593.5 | –16 1746.1 | –16 1290 |
| Энергия связывания | Eсвяз = –6139.64 | –6028.12 | –6111.6 |
| Теплота образования | dH = –943.84 | –770.21 | 967.96 |
Энергия протонирования –639.3 кДж/моль, изменение теплоты образования +725.63 кДж/моль.
Энергия депротонирования +1266.3 кДж/моль, увеличение теплоты образования 101.5 кДж/моль.
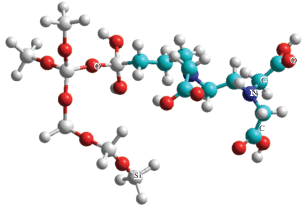
Модель с максимальным представительством кремнезема показана на рис. 4. Расчетные значения: Епр = 689.5 кДж/моль, изменение теплоты образования – +424.4 кДж/моль.
Рис. 3.
Заряды на атомах, рассчитанные по Малликену, в области протонирования: а) фрагмент модели до и б) после протонирования.
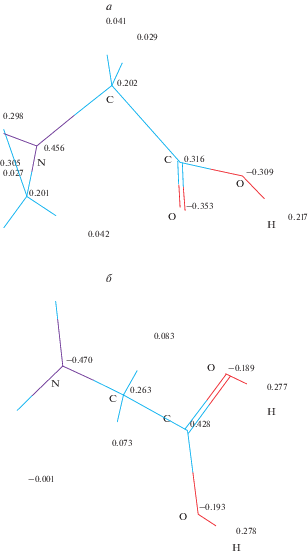
Рис. 4.
Модель поверхностного комплекса с наибольшим фрагментом, представляющим поверхность SiO2 (расчет MNDO Hyper Chem).
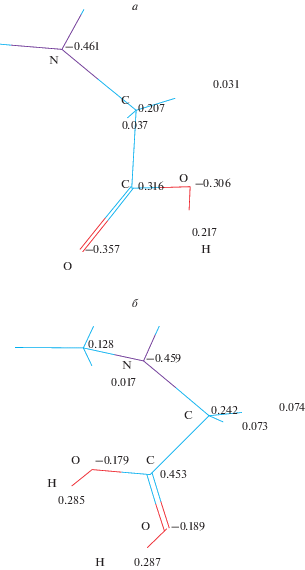
Перераспределение заряда при протонирование этой модели иллюстрируется на рис. 5.
Рис. 5.
Заряды на атомах для фрагмента модели с максимальным представительством кремнезема, рассчитанные методом ПДДП; а – до протонирования, б – после протонирования.
Таким образом, три модели с различным представительством кремнезема дают как одинаковые количественные оценки энергии протонирования, так и качественно одинаковое перераспределение заряда в этой реакции. Следовательно, можно считать представительность матрицы кремнезема в модели комплекса достаточной и не влияющей на оценку протолитических свойств модели в целом.
2. Расчеты методом РМ3
При расчетах методом РМ3 в качестве пробных взяты охарактеризованные выше модели оптимизированные методом МПДДП и далее проводилась их оптимизация по энергии уже с параметрами РМ3 [11].
В результате такой “дооптимизации” качественный вид нейтральной модели с максимальным участием SiO2 (рис. 4) не изменяется. Однако для протонированных моделей результаты отличаются. В зависимости от выбранного центра протонирования формируются различные модели. При этом возможно образование водородной связи привитой молекулы с поверхностью кремнезема. Этот результат не изменяется при достройке фрагмента, представляющего SiO2, до более реалистичного (рис. 6).
Рис. 6.
Максимальная по размеру модель в процессе протонирования сворачивается за счет образования водородных связей (расчет с параметрами РМ3).
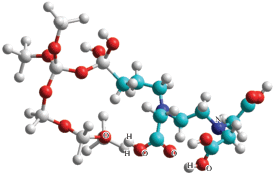
Для моделей без образования водородных связей энергия протонирования равна 691.13 кДж/моль и практически совпадает со значением, полученным методом MNDO.
Образование водородной связи приводит к существенному увеличению энергии протонирования (917.7 кДж/моль). На рис. 7 выделена область образования водородной связи.
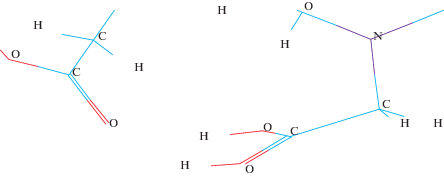
Расстояния: O1–H, равное 1.6 А и O2–H, равное 1.00 Å, характерны для водородной связи.
Далее был проведен модельный эксперимент обратного процесса депротонирования, у протонированной модели с водородными связями убран протон и вновь проведена оптимизация геометрии. В итоге была получена нейтральная по заряду модель, в которой, в отличие от приведенной на рис. 4, сохранилась водородная связь. Геометрия модели при этом близка к приведенной на рис. 6.
Энергетические же характеристики этих двух моделей: полная энергия, энергия связывания и теплота образования практически одинаковы.
3. Расчет с параметрами PM7
Развитие полуэмпирических методов расчета в квантовой химии продолжается до настоящего времени. Наиболее последовательно их применение проводится в программе MOPAC [12]. Последний набор параметров PM7 был получен в 2013 г. [13] и к настоящему времени прошел достаточно успешную апробацию (см., например, [14, 15]). В качестве исходной для расчетов, проведенных с использованием MOPAC и параметрами PM7, была взята непротонированная модель (без образования водородных связей), оптимизированная с параметрами PM3 (рис. 4). В независимости от выбора центра протонирования после оптимизации с параметрами PM7 образовывался поверхностный комплекс с водородной связью. Энергия протонирования при этом составляла примерно 650 кДж/моль, что хорошо согласуется с приведенными выше данными, полученными с другими наборами параметров.
ЗАКЛЮЧЕНИЕ
Модельный эксперимент, основанный на полуэмпирических квантовохимических расчетах, свидетельствует о том, что протонирование кремнезема, модифицированного молекулами со слабоосновным азотом, может сопровождаться формированием различных по строению поверхностных комплексов, в том числе с образованием водородных связей. Значения энергий протонирования при этом значимо изменяются, что может являться причиной различий в константах протонирования фиксируемым экспериментально.
Список литературы
Лисичкин В.Г. Химия привитых поверхностных соединений. М.: Физматлит, 2003.
Дункен Х., Лыгин В.И. Квантовая химия адсорбции на поверхности твердых тел. М.: Мир, 1988.
Лыгин В.И. Модели “жесткой” и “мягкой” поверхности. Конструирование микроструктуры поверхности кремнеземов // Российский химический журн. 2002. XLVI. № 3. С. 12–18.
Шаров А.В., Филистеев О.В. Протолитические равновесия на поверхности кремнеземов, содержащих аминогруппы B2O3–Na2O–La2O3 // Вестник Курганского госуниверситета. Серия: Естественные науки. 2011. 2. № 21. С. 103–110.
Попов И.С., Шаров А.В. Синтез и адсорбция хлорметилдиметилхлорсилана и дихлорметилдиметилхлорсилана на кремнеземной поверхности // Компьютерное моделирование физико-химических свойств стекол и расплавов. Труды XI Российского семинара. г. Курган. 2012. С. 73.
Hyper Chem version 8.10 URL: http://www.hyper.com/index.html.
Лыгин В.И. Молекулярные модели поверхностных структур кремнеземов // Журн. физической химии. 1997. 71. № 10. С. 1735–1742.
Воронцов Б.С., Шаров А.В. Визуализация процесса сборки на поверхности силикагеля и квантовохимическая оценка свойств поверхностного комплекса с имидоуксусной кислотой // Вестник Курганского университета. Серия: Естественные науки. 2015. 38. № 4. С. 80–83.
Воронцов Б.С., Шаров А.В. Квантовохимическая оценка энергий протонирования поверхности кремнеземов на наноразмерных моделях // Наука и мир. 2014. 12. № 8. С. 30–33.
Stwart J.J.P. Optimization of parameters for semiempirical methods. I. Method // J. Comp. Chem. 1989. 10. № 2. P. 205–220.
Степанов Н.Ф. Квантовая механика и квантовая химия. М.: Мир, 2001.
Stwart J.J.P. MOPAC 2012 // Stewart Computational Chemistry, Colorado Springs, CO, USA. 2012. http://openmopac.net.
Stwart J.J.P. Optimization of parameters for semiempirical methods VI: more modification to the NDDO approximations and re-optimization of parameters. // J. Molecular Modeling. 2013. 1. № 1. P. 1–32.
Каткова Е.В., Оферкин И.В., Сулимов В.Б. Применение квантово-химического полуэмпирического метода PM7 для разработки новых ингибиторов урокиназы // Вычислительные методы и программирование. 2014. 15. С. 258–273.
Воронцов Б.С., Москвин В.В. Кольцевые молекулярные фрагменты оксидов системы B2O3–Na2O–La2O3 по данным квантовохимического моделирования // Вестник Курганского государственного университета. Серия: Естественные науки. 2015. 38. № 4. С. 77–80.
Дополнительные материалы отсутствуют.