

Расплавы, 2021, № 3, стр. 294-302
Расчет диаграммы плавкости бинарной солевой смеси методом двухфазного молекулярно-динамического моделирования
А. С. Татаринов a, *, М. А. Кобелев a, Д. О. Закирьянов a, Н. К. Ткачёв a
a Институт высокотемпературной электрохимии УрО РАН
Екатеринбург, Россия
* E-mail: tolja170497@gmail.com
Поступила в редакцию 24.12.2020
После доработки 23.01.2021
Принята к публикации 25.01.2021
Аннотация
Предложена методика двухфазного молекулярно-динамического моделирования для бинарных солевых смесей. На примере фазовой диаграммы KF–KCl показано: нагревание двухфазной ячейки жидкость–кристалл со скоростью 2 К/нс позволяет наблюдать изменение концентрации расплава и уменьшение объема кристаллической фазы; и при дальнейшем повышении температуры, увидеть излом мольного объема на линии ликвидус. Скорость нагрева в 2 К/нс оказывается достаточной для количественного описания диаграммы плавкости. Важно, что методика расчета фазовой диаграммы опирается на модельные парные потенциалы Борн-Майеровского типа, параметры которых могут быть найдены с помощью методов квантовой химии.
ВВЕДЕНИЕ
Развитие теоретических подходов к описанию фазовых равновесий в многокомпонентных системах является актуальной задачей в силу того, что далеко не всегда имеются экспериментальные данные или они доступны только в ограниченной области концентраций и температур [1, 2]. В последние десятилетия широкое распространение получили программы на основе пакета CALPHAD, опирающиеся на термодинамические базы данных по ионным системам. Однако, достоверность данного эмпирического метода напрямую зависит от количества и качества имеющихся в базе экспериментальных данных, а также от зачастую неконтролируемых отклонений от модели субрегулярного раствора, лежащего в основе вычислений [3]. Немаловажно, что такое термодинамическое моделирование фазовых равновесий основывается на неявном предположении о существовании решения уравнений, описывающих условия равновесия.
В последние годы, благодаря существенному прогрессу методов компьютерного моделирования, где моделируются статистические ансамбли, возникает значительный интерес к прямому наблюдению и расчету фазовых равновесий посредством молекулярной динамики. Важно, что для построения фазовых диаграмм такими методами необходимы только парные или более сложные, многочастичные потенциалы, характеризующие микроскопические силы взаимодействия между атомами [4]. Однако, даже для простых моделей, взаимодействия между частицами в которых описываются парными потенциалами типа Леннард-Джонса, твердые сферы, прямоугольная яма [5–7], задача расчета фазовой диаграммы является весьма непростой. Это в полной мере касается и фазовых равновесий “жидкость–твердое”.
В методологическом отношении особенно интересны системы с кулоновским дальнодействием, для растворов и смесей которых, в общем случае не всегда удается свести задачу к субрегулярным растворам (или параметры модели неизбежно приобретают зависимость от плотности и концентрации) [8]. Если же говорить о полностью микроскопических подходах, то в этой области известны всего несколько работ [9, 10], где был проведен молекулярно-динамический расчет диаграмм плавкости для галогенидных смесей. Однако, свободная энергия в твердом состоянии рассчитывалась при этом отдельно, с использованием методов ab initio. Отдавая должное указанным работам, отметим, что в данном случае прямого моделирования равновесий “жидкость–твердое” не проводилось, и сами термодинамические модели жидкости и твердого тела были разные.
Таким образом, в настоящее время созрела необходимость в разработке прямых методов моделирования для расчета фазовых диаграмм плавкости в сложных по составу смесях галогенидов различных металлов, в которых скачкообразные изменения, например, объема или плотности свидетельствуют о границе сосуществования одно-, двух- или многофазных областей. При этом, особую роль, с нашей точки зрения, должны сыграть методы прямого наблюдения за процессами плавления или кристаллизации, при использовании единообразной модели взаимодействий между ионами и для твердой, и для жидкой фазы. Тогда сама точка фазового перехода определяется нюансами короткодействующего отталкивания и дальнодействия, как обычно, и ставится задача в статистической теории.
Ранее нами было показано, что, нагревая полностью кристаллический образец, можно рассчитать линию фазовых равновесий “твердое тело–жидкость” для бинарной смеси LiCl–KCl [11]. Однако, в области умеренных концентраций и особенно эвтектического состава точность такого моделирования невелика при типичных скоростях нагрева. Отметим, что в этом случае получается описать основные особенности линии ликвидуса, а именно, смещение эвтектической точки в сторону компонента с меньшей температурой плавления. Для дальнейшего улучшения количественного согласия предлагаемой методики расчета линий фазовых равновесий “твердое тело–расплав” с экспериментальными данными можно предложить следующие шаги. Первое, значительно увеличить размеры молекулярно-динамических ячеек. При этом среднеквадратичное отклонение энергии от среднего значения уменьшилось бы кратно, так как среднеквадратичные флуктуации пропорциональны корню квадратному из числа частиц. Второе, уменьшить скорость изменения температуры для более точной идентификации возникающих равновесий.
В качестве объекта исследования, в данной работе выбрана бинарная смесь KF–KCl, фазовая диаграмма которой хорошо изучена и относится к простому эвтектическому типу [12]. В последние годы большой практический интерес вызывает расплавленная смесь KF–KCl эвтектического состава в качестве реакционной среды для получения кремния [13] и титана [14]. В связи с этим актуализировались работы по изучению диаграмм плавкости и физико-химических свойств многокомпонентных смесей в которых, в качестве основы выступает бинарный расплав KF–KCl. К таким работам относится экспериментальное исследование фазовой диаграммы тройной смеси KF–KCl–KI методом кривых охлаждения [15], измерения электрической проводимости методом импедансной спектроскопии в смеси KF–KCl–K2SiF6 [16] при добавлении 4 мол. % SiO2 и ряд других работ данной группы исследователей.
Целью данной работы является разработка улучшенной методики построения фазовой диаграммы посредством моделирования двухфазной ячейки “жидкость–твердое”, в которой гомогенная фаза жидкого раствора моделируется предварительно, и затем приводится в контакт с твердым кристаллом. Для иллюстрации и проверки точности метода выбрана система эвтектического типа KF–KCl.
ДЕТАЛИ КОМПЬЮТЕРНОГО МОДЕЛИРОВАНИЯ МОДЕЛЬ ПАРНОГО ВЗАИМОДЕЙСТВИЯ
На данном этапе разработки проблемы описания температур фазовых равновесий в ионных системах, мы ограничимся моделью парных межионных взаимодействий Борна–Майера. Функция потенциальной энергии в зависимости от расстояния в этом случае Uij(r) содержит лишь два слагаемых:
(1)
${{U}_{{ij}}}\left( r \right) = \frac{{{{Z}_{i}}{{Z}_{j}}{{e}^{2}}}}{{\varepsilon \cdot r}} + A \cdot {\text{exp}}\left[ { - \frac{r}{\rho }} \right],$Таблица 1.
Параметры парного потенциала (1) полученные методом ab initio в сравнении с параметрами Фуми–Тоси (ФТ) [22] для солей KF и KCl
| K–K | K–F | F–F | ||||
|---|---|---|---|---|---|---|
| $A$, эВ | $\rho $, Å | $A$, эВ | $\rho $, Å | $A$, эВ | $\rho $, Å | |
| ab initio | 6546 | 0.26151 | 3295 | 0.25948 | 1062 | 0.27645 |
| ФТ (KF) | 1516 | 0.338 | 524 | 0.338 | 169 | 0.338 |
| K–K | K–Cl | Cl–Cl | ||||
| $A$, эВ | $\rho $, Å | $A$, эВ | $\rho $, Å | $A$, эВ | $\rho $, Å | |
| ab initio | 6546 | 0.26151 | 3533 | 0.3091 | 1380 | 0.33897 |
| ФТ (KCl) | 1556 | 0.337 | 1788 | 0.337 | 1925 | 0.337 |
В то же время для хлорида калия, обсуждаемые параметры имеют значения в пределах одного порядка. В случае фторидов лития, натрия и калия, параметры отталкивания Фуми–Тоси [19, 20] могут приводить к нефизическим наблюдениям при моделировании расплава. А именно, недостаточная величина отталкивательной части приводит к “слипанию” разноименных ионов в процессе моделирования и невозможности дальнейших вычислений. В работе [21] был проведен расчет характеристик плавления галогенидов щелочных металлов при использовании парных потенциалов Фуми–Тоси. Для фторидов натрия и калия наблюдались существенные расхождения, рассчитанных и экспериментальных температур плавления в сторону их уменьшения: для KF различие составляло 270 градусов, для NaF – более 650 градусов. В то же время, расчет характеристик плавления с помощью параметров, определяемых по методу ab initio показал [17], что для фторида калия разность между расчетной и экспериментальной величиной температуры плавления составляет 40 градусов.
По нашему мнению, для теоретических задач больше подходит квантово-химическое вычисление параметров, которые устраняют указанные расхождения. Нами показано ранее при расчетах температур плавления индивидуальных галогенидов лития, натрия и калия [17], что результаты молекулярно-динамического вычисления температуры плавления ближе всего к экспериментальным данным. Это позволяет ожидать и достаточной точности оценки температур ликвидуса для многокомпонентых систем.
МЕТОДИКА ДВУХФАЗНОГО МОДЕЛИРОВАНИЯ
Чтобы минимизировать эффект переоценки температуры ликвидуса, с которым мы столкнулись в [11], были предложены следующие методологические улучшения двухфазного способа моделирования. Было найдено, что на начальном этапе моделирования лучше стартовать с двухфазной ячейки “расплав–кристалл”. При этом возможны два варианта формирования исходной двухфазной ячейки. В первом случае (метод 1) расплав представляет собой гомогенный раствор компонентов примерно эквимолярного состава. Твердая фаза состоит из кристаллов одного компонента, например, $_{{{\text{расплав}}}}^{{\,\,\,\,\,\,\,\,\,\,\,{{N}_{1}}}}({\text{KF}} + {\text{KCl}})$ + $_{{{\text{кристалл}}}}^{{\,\,\,\,\,\,\,\,\,\,\,\,{{N}_{2}}}}{\text{KCl,}}$ где Ni – число ионов в i фазе. Изменяя число частиц твердой фазы N2, при фиксированной жидкофазной части ячейки (N1 = const), можно получить образец с требуемой концентрацией. Во втором случае (метод 2) расплав и твердая фаза состоят из различных чистых компонентов бинарной смеси (рис. 1), например, $_{{{\text{расплав}}}}^{{\,\,\,\,\,\,\,\,\,\,\,{{N}_{1}}}}{\text{KF}}$ + $_{{{\text{кристалл}}}}^{{\,\,\,\,\,\,\,\,\,\,\,\,\,{{N}_{2}}}}{\text{KCl}}{\text{.}}$ Температура ликвидуса определяется по скачку объема моделируемой ячейки, и подтверждается анализом ее мгновенных конфигураций.
Рис. 1.
Мгновенные конфигурации ячеек: a) стартовая конфигурация: сосуществование кристалла одного компонента и расплава другого компонента, б) постепенное плавление кристалла в процессе нагрева, в) полный переход системы в расплав.

Все ячейки представляют собой прямоугольный параллелепипед, содержащий от 5856 до 13 664 ионов в зависимости от концентрации компонентов смеси. На ячейки налагаются периодические граничные условия. Кулоновское взаимодействие рассчитывается по методу Эвальда. Параметр обрезания межионного взаимодействия выбирается как половина длины наименьшего ребра МД-ячейки. Геометрические размеры моделируемых ячеек составляют 40 Å × 40 Å × Lz, где параметр решетки вдоль координаты z принимает значения Lz = 85–220 Å, в зависимости от концентрации компонентов бинарной смеси. Нагревание образца состоит из последовательных этапов: моделирование NPT – ансамбля при постоянном внешнем давлении равном 1 бар и начальной температуре, равной экспериментальной эвтектической температуре Т0 = 880 К. В том случае, когда образец полностью плавился при температуре Т0, а это наблюдалось для бинарных смесей при концентрациях фторида калия x(KF) = 0.3–0.4 мольных долей, начальную температуру понижали на 100 градусов. На следующем этапе моделируется NPT – ансамбль с температурой Т1 увеличенной на 20 градусов при аналогичных значениях временных параметров и так, вплоть до полного плавления ячейки. Максимальная температура нагрева соответствовала температуре, при которой происходило полное плавление моделируемой ячейки.
Временнóй шаг моделирования для всех смесей выбирался равным Δt = 0.001 пс, а общее время моделирования при каждой температуре составляло 10 нс. Значение скорости 2 К/нс было подобрано в результате серии предварительных, молекулярно-динамических моделирований со скоростями нагрева 10, 5, 2 К/нс и, было показано, что значение скорости 2 К/нс оказалось удачным и, с точки зрения, времени моделирования, которое даже в этом случае составляло 150–200 ч. Контроль температуры и давления задается с помощью термостата и баростата Нозе–Гувера [23]. Параметр релаксации температуры равен 0.1 пс, аналогичный параметр для давления – 0.5 пс. Все расчеты проведены в пакете LAMMPS с использованием вычислительных ресурсов суперкомпьютера “Уран” СКЦ ИММ УрО РАН.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Расчет температур ликвидуса с помощью двух способов формирования исходных двухфазных ячеек показал, что результаты моделирования различаются на 10–20 градусов и находятся в пределах оцениваемой погрешности метода, которая составляет ΔT = ±30 K. Такой результат вполне объясним, учитывая, что наблюдаемый процесс плавления происходит по границе двух сосуществующих фаз “твердое тело–расплав”. Для сравнения, было проведено моделирование плавления полностью кристаллической исходной ячейки, содержащей x(KF) = 0.4 мольных долей. В результате для указанной концентрации расчетная температура ликвидуса составила Tликвидус = 940 K, что на 130 градусов выше результата, полученного с помощью двухфазной методики.
Было изучено влияние скорости нагрева на величину температуры ликвидуса. В табл. 2 приведены результаты моделирования по двухфазной методике при различных скоростях нагрева ячеек. Как видно из данных табл. 2, этот параметр оказывает существенное влияние на результаты моделирования. А именно, снижение темпа нагрева ячеек с 5 до 2 К/нс приводит к уменьшению рассчитанных температур ликвидуса в среднем на 55 градусов. Полагаем, что необходимый баланс между временем моделирования и получаемыми величинами температур ликвидуса соблюдается при значении скорости нагрева в 2 К/нс. Поэтому, приводимые далее результаты моделирования плавления двухфазных ячеек бинарной смеси KF–KCl, получены для этой скорости нагрева.
Таблица 2.
Рассчитанные температуры ликвидуса в зависимости от концентрации (x(KF) – мольная доля фторида калия) бинарной смеси KF–KCl при разной скорости нагрева
| x(KF) | 0.1 | 0.2 | 0.3 | 0.4 | 0.6 | 0.7 | 0.8 | 0.9 |
|---|---|---|---|---|---|---|---|---|
| ${{T}^{{{\text{ликв}}}}}$, °C; 2 К/нс | 657 | 627 | 567 | 537 | 697 | 722 | 787 | 817 |
| ${{T}^{{{\text{ликв}}}}}$, °C; 5 К/нс | 687 | 660 | 633 | 612 | 770 | 797 | 827 | 875 |
На рис. 2 представлены результаты расчета температур ликвидуса для бинарной смеси KF–KCl в сопоставлении с экспериментальными данными. Результаты моделирования для исходных ячеек, созданных по схеме $_{{{\text{расплав}}}}^{{\,\,\,\,\,\,\,\,\,\,\,{{N}_{1}}}}({\text{KF}} + {\text{KCl}})$ + $_{{{\text{кристалл}}}}^{{\,\,\,\,\,\,\,\,\,\,\,\,\,{{N}_{2}}}}{\text{KCl}}$ приведены с помощью черных квадратных маркеров (■). Погрешность метода определения температуры ликвидуса оцениваемая в ΔT = ±30 K, отмечена на рис. 2 через соответствующие метки погрешностей. Общее наблюдение из рис. 2 состоит в том, что с помощью предлагаемой методики удается с достаточной точностью воспроизвести основные ключевые особенности фазовой диаграммы эвтектического типа. А именно, понижение температуры плавления с уменьшением концентрации одного из компонентов, выпуклость кривой ликвидуса в области умеренных концентраций и смещение эвтектической точки в сторону компонента с меньшей температурой плавления.
Рис. 2.
(◼) Рассчитанные температуры ликвидуса методом двухфазного моделирования в сопоставлении с экспериментальными данными (пунктирная линия).
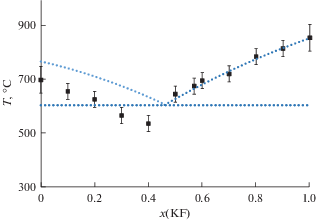
Открытым остается вопрос о расчете линии солидуса в рамках предлагаемой методологии. Так как моделировании начинается уже с двухфазной ячейки, это исключает изучение эвтектического равновесия с помощью нагревания. Определенные сложности возникают при моделировании исходных ячеек с понижением температуры. Как показали предварительные данные по охлаждению жидкофазных ячеек, состоящих из двух компонентов, кристаллизация наблюдается только в области концентраций близких к чистому соединению. Более того, получаемые температуры затвердевания существенно, на сотни градусов, ниже экспериментальных значений температуры солидуса. А в области концентраций x(KF) = 0.3–0.7 происходит стеклование ячеек, даже при скоростях охлаждения порядка 1 К/нс, что делает процесс моделирования чрезвычайно длительным.
Переходя к количественному сопоставлению рассчитанной линии ликвидуса (■) и экспериментальных данных, отметим хорошее согласие на ветви, описывающей равновесие “расплав–KFтв” (рис. 2). Результаты, полученные по этой же методике для второй ветви ликвидуса, а именно “расплав–KClтв”, демонстрируют заниженные по сравнению с экспериментом значения температур ликвидуса. В предыдущей работе [11], где изучалась линия ликвидуса для бинарной смеси LiCl–KCl, также наблюдалось несколько худшее согласие рассчитанных значений температур ликвидуса по сравнению с экспериментом на ветви фазовых равновесий “расплав–KClтв”. Можно предположить, что учет эффектов поляризации при описании парного взаимодействия, окажет определенное влияние на результаты моделирования, так как поляризуемость ионных пар увеличивается в ряду KF < LiCl < KCl. Рассмотрение этих эффектов выходит за рамки данной работы, но является перспективной задачей для дальнейших исследований.
ВЫВОДЫ
В заключении подчеркнем, что предлагаемая методика расчета линий фазовых равновесий “жидкость–твердое тело” не требует анализа термодинамических функций и опирается на прямое наблюдение эволюции двухфазной ячейки с температурой. При этом такой подход оказался достаточно продуктивным и является полностью неэмпирическим, что позволяет использовать его для изучения фазовых равновесий в ионных смесях, для которых отсутствуют необходимые термодинамические данные.
Работа выполнена при поддержке Российского фонда фундаментальных исследований (номер гранта 18-03-00606).
Список литературы
Jayaraman S., Thompson A.P., von Lilienfeld O.A. // Phys. Rev. E. 2011. 84. 030201. https://doi.org/10.1103/physreve.84.030201
Peng Q., Ding J., Wei X., Jiang G. // Thermochimica Acta. 2017. 654. P. 28–34. https://doi.org/10.1016/j.tca.2017.04.015
Jung I.H., van Ende M.A. // Metal Mater Trans B. 2020. 51B. P. 1851–1874. https://doi.org/10.1007/s11663-020-01908-7
Dorrell J., Partay L.B. // J. Phys. Chem. B. 2020. 124. P. 6015–6023. https://doi.org/10.1021/acs.jpcb.0c03882
Miller M.A. // J. Chem. Phys. 2004. 121. P. 535–545. https://doi.org/10.1063/1.1758693
Lamm M.H., Hall C.K. // AIChE. 2001. 47. № 7. P. 1664–1675. https://doi.org/10.1002/aic.690470718
Рыжов В.Н., Тареева Е.Е., Фомин Ю.Д., Циок Е.Н. // УФН. 2020. 190. С. 449–473. https://doi.org/10.3367/UFNr.2018.04.038417
Margheritis C., Flor G., Sinistri C.Z. // Z. Naturforsch. 1973. 28a. P. 1329–1334. https://doi.org/10.1515/zna-1973-0813
Beneš O., Zeller Ph., Salanne M., Konings R.J.M. // J. Chem. Phys. 2009. 120. 134716. https://doi.org/10.1063/1.3097550
Seo W.G., Matsuura H., Tsukihashi F. Calculation of phase diagrams for the FeCl2, PbCl2, and FeCl2 binary systems by using molecular dynamics simulations // Metall. Mater. Trans. B. 2006. 37. P. 239–251.
Kobelev M.A., Tatarinov A.S., Zakiryanov D.O., Tkachev N.K. // Phase Transitions. 2020. 93. P. 504–508. https://doi.org/10.1080/01411594.2020.1758318
Wang K., Robelin C., Jin L., Zeng X., Chartrand P. // J. Mol. Liquids. 2019. 292. P. 111384. https://doi.org/10.1016/j.molliq.2019.111384
Maeda K., Yasuda K., Nohira T., Hagiwara R., Homma T. // J. Electr. Soc. 2015. 162. P. 444–448. https://doi.org/10.1149/2.0441509jes
Norikawa Y., Yasuda K., Nohira T. // Electrochemistry. 2018. 86. P. 99–103. https://doi.org/10.5796/electrochemistry.17-00082
Khudorozhkova A.O., Isakov A.V., Redkin A.A., Zaikov Yu.P. // Russ. Metal. 2019. № 8. P. 830–834. https://doi.org/10.1134/s0036029519080081
Apisarov A.A., Redkin A.A., Zaikov Yu.P, Chemezov O.V., Isakov A.V. // J. Chem. Eng. Data. 2011. 56. P. 4733–4735. https://doi.org/10.1021/je200717n
Zakiryanov D., Kobelev M., Tkachev N. // Fluid Phase Equil. 2020. 506. P. 112369. https://doi.org/10.1016/j.fluid.2019.112369
Fumi F.G., Tosi M.P. Ionic sizes and born repulsive parameters in the NaCl-type alkali halides – I. The Huggins-Mayer and Pauling forms // J. Phys. Chem. Solids. 1964. № 25. P. 31–43.
Belonoshko A.B., Ahuja R., Johansson B. // Phys. Rev. B. 2000. 61. P. 11928–11935. https://doi.org/10.1103/PhysRevB.61.11928
Ribeiro M.C.C. // J. Phys. Chem. B. 2003. 107. P. 4392–4402. https://doi.org/10.1021/jp027261a
Aragones J.L., Sanz E., Valeriani C., Vega C. // J. Chem. Phys. 2012. 137. P. 104507. https://doi.org/10.1063/1.4745205
Sangster M.J.L., Dixon M. // Adv. Phys. 1976. 25. P. 247–342. https://doi.org/10.1080/00018737600101392
Nosé S. // Prog Theor Phys Supp. 1991. 103. P. 1–46. https://doi.org/10.1143/PTPS.103.1
Дополнительные материалы отсутствуют.