

Расплавы, 2021, № 4, стр. 391-405
Молекулярные модели комплексов на основе ионов РЗМ во фторидных солевых расплавах
И. А. Бабина a, *, Б. С. Воронцов b, **, В. В. Москвин b, А. О. Бабин a
a Южно-Уральский государственный университет
(национальный исследовательский университет)
Челябинск, Россия
b Курганский государственный университет
Курган, Россия
* E-mail: babina_inga@mail.ru
** E-mail: fizika@kgsu.ru
Поступила в редакцию 02.03.2021
После доработки 31.03.2021
Принята к публикации 04.04.2021
Аннотация
Фторидные солевые расплавы применяются во многих металлургических процессах и технологиях. Известно, что даже очень небольшие добавки редкоземельных металлов (РЗМ) существенно изменяют их структурно-чувствительные свойства. Обсуждаются возможные механизмы такого влияния и различные модели участия ионов РЗМ в структурообразовании фторидных расплавов. Методами молекулярной механики и квантовой химии подтверждена возможность образования ионами РЗМ комплексов состава LnF6Me14 (Ln – La, Ce, Pr, Nd, Sm, Eu; Me – Li, Na, K, Cs) во фторидных солевых расплавах. Подобный состав в литературе постулирован на основе интерпретации спектральных данных и гипотезы о близости структур фторидных солей в жидком и твердом состояниях при высоких температурах. Приведены полученные в расчетах с силовым полем ММ+ данные о геометрическом строении молекулярных моделей подобных комплексов в области минимумов поверхности потенциальной энергии. Выявлено три различных по геометрическому строению типа структур. Показана возможность формирования самосогласованной электронной подсистемы в моделях с оптимизированной геометрией. Проанализированы особенности плотности электронных состояний в валентном и SPARKLE приближениях. Показано, что электронные уровни координируемых ионами РЗМ атомов щелочных металлов существенно влияют на плотность электронных состояний основы комплекса, что свидетельствует о перспективности полученных моделей для последующего исследования спектральных свойств. Предложенный подход, который начинается с построения молекулярных моделей комплексов на основе РЗМ и завершается расчетом различного рода спектральных характеристик, связанных с электронной структурой и геометрией этих комплексов, является альтернативным традиционному пути от свойств к структуре.
ВВЕДЕНИЕ
Систематические исследования спектральных характеристик расплавов хлоридов, фторидов, бромидов и нитридов щелочных металлов с малыми добавками оксидов редкоземельных металлов [1–4] позволили достаточно обосновано предположить образование в них комплексов на основе ионов РЗМ.
Более того, на основе анализа зависимостей параметров спектров от состава и термодинамических условий [5–7] предложен и состав этих комплексов, включая вторую координационную сферу. Предполагается, что в расплаве сохраняются фрагменты кристаллов со структурой эльпасолита [8]. Схематично пример такого фрагмента, образованного ионом церия, показан на рис. 1.
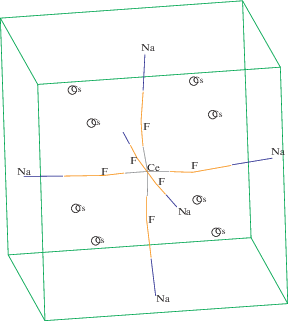
Следует отметить, что при таком подходе предполагаемые модели структуры являются чисто гипотетическими, не содержащими конкретных геометрических параметров.
Целью настоящей работы является разработка альтернативного подхода, который начинается с построения молекулярных моделей комплексов на основе РЗМ и завершается расчетом различного рода спектральных характеристик, связанных с электронной структурой и геометрией этих комплексов.
ТЕОРЕТИЧЕСКИЙ АНАЛИЗ
Сложность реализации поставленной задачи обусловлена спецификой элементов РЗМ. Имеющиеся стандартные комплексы квантово-химических программ [9–12] не позволяют полностью реализовать намеченную последовательность модельного эксперимента. В программах, реализующих неэмпирические расчеты и позволяющих рассчитывать электронные спектры в УФ и видимой областях, отсутствуют базисные наборы для атомов РЗМ. Полуэмпирические методы также не решают проблемы, т.к. только в одном методе РМ-7 программы MOPAC имеются наборы параметров для атомов РЗМ. Однако их применение позволяет лишь получить модель с оптимальными геометрическими параметрами, и не дает возможности, необходимого детального исследования электронной структуры.
МЕТОДИКА
В связи со сказанным, реализация намеченной программы разбита на этапы и начата с построения молекулярных моделей, нахождения для них равновесных конфигураций, соответствующих минимуму ППЭ (поверхность потенциальной энергии), и определения на этих моделях геометрических характеристик комплексов РЗМ различного состава.

Построение модели методом молекулярной механики включает следующие этапы:
1. Графическое представление гипотетической молекулярной структуры произвольной стехиометрии и с произвольными геометрическими параметрами – длинами связей, углами между связями и пространственными углами (пробная геометрия).
2. Запись информации об этой структуре в одном из форматов, используемых в квантовохимических программах.
3. С использованием какого-либо силового поля (мы брали стандартные силовые поля, хорошо зарекомендовавшие себя в практике молекулярной механики) проводится варьирование геометрических характеристик и взаимного расположение атомов в поисках устойчивой конфигурации. Такая конфигурация либо находится, и происходит оптимизация геометрии, либо нет, и тогда модель распадается на отдельные атомы или фрагменты.
Фрагменты структуры расплава, которыми являются построенные нами модели комплексов на основе РЗМ, дают определяемый их геометрией и электронной структурой локальный вклад в суммарную плотность состояний. Электронные переходы атомов РЗМ зависят от структуры ближнего порядка, что проявится в положении и форме спектральных линий. Кроме того, квантово-химический эксперимент позволяет учесть окружение локальной модели, например, вписав ее в континуум фторидной соли.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Модели молекулярной механики
Представленные ниже молекулярно-механические модели построены с использованием графического редактора программы HyperChem [10]. В расчетах были апробированы различные силовые поля. В итоге, наиболее систематично устойчивые модели, сходные по строению с фрагментом, показанным на рис. 1, удалось реализовать в рамках силового поля ММ+ [13]. Метод MM+ учитывает потенциальные поля, формируемые всеми атомами рассчитываемой системы и позволяет гибко модифицировать параметры расчета в зависимости от конкретной задачи, что делает его, с одной стороны, наиболее общим, а с другой – резко увеличивает необходимые ресурсы по сравнению с другими методами молекулярной механики.
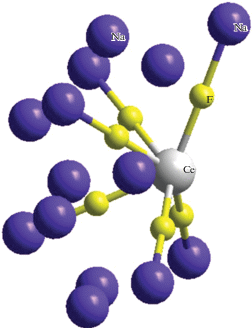
В зависимости от пространственного распределения атомов щелочных металлов по отношению к каркасу РЗМ–F6 модели можно разделить на три группы. Пример первого типа моделей показан на рис. 2. В этих моделях во второй координационной сфере наиболее удалены от центра атомы, лежащие на продолжении связей РЗМ-фтор. На рис. 2 это атомы Na, входящие в состав связей F–Na, атомы же цезия находятся ближе к центру комплекса.
Нами были апробированы сотни различных вариантов, прежде чем удалось получить первую удачную модель. Это рутинная и нетворческая работа, в связи с этим ее описание опущено и приведен только результат. Молекулярно-динамический расчет не проводился, так как была поставлена задача подтверждения или отрицания комплексов с такой стехиометрией, которая предложена при интерпретации спектральных данных в указанных нами статьях.
В моделях второго типа, пример для которых приведен на рис. 3, более удалены атомы, не входящие в состав направленных связей (помечены два таких атома K).
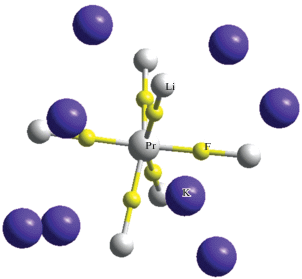
При этом каркас комплекса PrF6Li6 находится внутри сферы несвязанных атомов. Имеются также комплексы промежуточного характера (в них все атомы щелочных металлов примерно равноудалены от центра комплекса).
Например, в моделях комплексов состава MeF6Li14 все атомы лития примерно равноудалены от центрального атома РЗМ, т.е. эти модели являются промежуточными по отношению к комплексам первого и второго типов (рис. 4).

Геометрические характеристики построенных моделей приведены в табл. 1. Однако не для всех составов удается построить модели качественно сходные по строению со структурой на рис. 1. Так, например, для комплексов состава MРЗМF6Na14 атомы натрия координируются в одной полуплоскости так, как это показано на рис. 5.
Таблица 1.
Геометрические характеристики молекулярных моделей комплексов РЗМ, построенных методом молекулярной механики с силовым полем ММ+
| Состав комплекса | Длина связи ион РЗМ–F, Å | Длина
связи F–Me, Å |
Радиус координ. сферы R2, Å |
Радиус координ. сферы R3, Å | COSMO AREA, Å2 |
COSMO Volume, Å3 |
Тип модели (1 или 2) |
|---|---|---|---|---|---|---|---|
| CeF6Na6Cs8 | 2.37 | 3.07 | 4.6 | 3.5 | 581 | 1197 | 1 |
| CeF6Cs14 | 2.37 | 2.75 | 5.5 | 3.5 | 830 | 1870 | 1 |
| CeF6K14 | 2.37 | 2.75 | 5.1 | 6.1 | 902 | 1630 | 2 |
| CeF6Li14 | 2.37 | 1.95 | 4.3 | 4.3 | 434 | 663 | 1–2 |
| CeF6Li6Cs8 | 2.37 | 1.95 | 4.3 | 3.3 | 555 | 1129 | 1 |
| LaF6Na6Cs8 | 2.41 | 2.26 | 4.7 | 3.4 | 582 | 1199 | 1 |
| LaF6K6Cs8 | 2.37 | 2.75 | 5.1 | 3.3 | 582 | 1200 | 1 |
| LaF6Li14 | 2.41 | 1.95 | 4.4 | 4.3 | 436 | 667 | 1–2 |
| PrF6Cs14 | 2.37 | 3.07 | 5.5 | 3.4 | 830 | 1870 | 1 |
| PrF6Li14 | 2.37 | 1.95 | 4.3 | 4.3 | 434 | 663 | 1–2 |
| PrF6Na6Cs8 | 2.37 | 2.26 | 5.5 | 3.4 | 628 | 1278 | 1 |
| PrF6Li6Cs8 | 2.35 | 1.95 | 4.6 | 3.4 | 559 | 1139 | 1 |
| PrF6Li6K8 | 2.35 | 1.95 | 4.3 | 5.7 | 716 | 1384 | 2 |
| NdF6Na6Cs8 | 2.36 | 2.26 | 5.4 | 3.4 | 628 | 1278 | 1 |
| NdF6Na6K8 | 2.36 | 2.26 | 4.5 | 5.5 | 722 | 1464 | 2 |
| NdF6Cs14 | 2.36 | 3.07 | 5.6 | 3.3 | 826 | 1852 | 1 |
| NdF6K14 | 2.36 | 2.75 | 5.0 | 6.1 | 884 | 1919 | 2 |
| SmF6Na6Cs8 | 2.34 | 2.26 | 4.6 | 3.3 | 577 | 1189 | 1 |
| SmF6Cs14 | 2.34 | 3.07 | 5.4 | 3.3 | 825 | 1859 | 1 |
| SmF6K14 | 2.34 | 2.75 | 5.1 | 6.3 | 878 | 1907 | 2 |
| SmF6Na6K8 | 2.34 | 2.26 | 4.6 | 5.7 | 741 | 1466 | 2 |
| SmF6Li6K8 | 2.85 | 2.77 | 5.6 | 6.2 | 643 | 1545 | 2 |
| SmF6Li6Cs8 | 2.85 | 2.77 | 5.6 | 6.2 | 903 | 2029 | 2 |
| SmF6Li14 | 2.34 | 1.95 | 4.3 | 4.4 | 431 | 660 | 1–2 |
| EuF6Na6Cs8 | 2.57 | 2.26 | 4.8 | 3.3 | 589 | 1213 | 1 |
| EuF6K14 | 2.57 | 2.75 | 5.3 | 6.4 | 896 | 1952 | 2 |
| EuF6Li14 | 2.57 | 1.95 | 4.5 | 4.4 | 446 | 679 | 1–2 |
| EuF6Li6Cs8 | 3.08 | 2.77 | 5.8 | 6.3 | 743 | 1533 | 2 |
Следует отметить тот факт, что, несмотря на кажущуюся симметричность, все модели относятся к точечной группе С1.
В качестве приближенной оценки объема и площади поверхности построенных моделей использованы соответствующие величины, полученные при расчете в программе MOPAC-12 в модели COSMO (обычно используется при учете влияния растворителя [14]).
На следующем этапе моделирования в работе сделана попытка раскрытия механизма формирования электронной структуры построенных моделей комплексов РЗМ.
Расчеты в приближении “SPARKLE” для РЗМ и с параметрами PM7 для остальных атомов
В литературе достаточно подробно описана методика построения и “Sparkle” расчетов молекулярных комплексов лантанидов в составе органики [15]. В MOPAC-12 при выборе химического элемента нужного лантаноида в действительности загружается “overall neutral species”, который включает ион с зарядом +3 и три электрона, которые распределяются по орбиталям органической части комплекса. Внутренняя электронная структура иона при этом в расчетах не учитывается. Валентные орбитали атомов РЗМ при этом не включаются в базис для нахождения молекулярных орбиталей в приближении МО ЛКАО. Фактически данное приближение дает мало информации об электронной структуре. Однако в решаемой нами задаче оно дает ответ на вопрос о принципиальной возможности формирования самосогласованной электронной подсистемы в полученных моделях с сохранением их геометрии.
Основу всех построенных моделей составляют фрагменты XF6 (X = La, Ce, Pr). Расчеты показали, что для всех лантанидов они устойчивы по отношению к процедуре оптимизации, заложенной в указанном комплексе программ в рамках полуэмпирического метода NDDO с параметрами РМ-7 [16] и SPARKLE приближении для РЗМ. Качественно их структура практически одинакова. При этом, как и следовало ожидать, для данного приближения, наборы собственных значений энергий (энергетические диаграммы) для них практически одинаковы. На рис. 6 показана типичная молекулярная модель центрального фрагмента комплекса на основе иона лантана.
Рис. 6
Молекулярная модель центральной части комплекса на основе иона La (первая координационная сфера).

В табл. 2 приведены собственные значения энергий этой модели, а на рис. 7 – соответствующая им плотность электронных состояний для альфа-электронов.
Таблица 2.
Собственные значения энергии LaF6 в SPARKLE-приближении
| Альфа-собственные значения (Alpha eigenvalues) |
||||
|---|---|---|---|---|
| –46.48155 | –44.85311 | –43.52734 | –35.51889 | –35.50164 |
| –35.43231 | –21.43564 | –21.18031 | –19.91908 | –19.66081 |
| –18.32609 | –18.09443 | –16.51827 | –14.87024 | –14.65428 |
| –14.64370 | –14.58003 | –14.29066 | –14.27792 | –14.27438 |
| –14.26026 | –14.20851 | –14.19676 | –7.27713 | |
| Бета-собственные значения (Beta eigenvalues) |
||||
| –46.43191 | –43.57697 | –41.95144 | –35.51632 | –35.50098 |
| –35.43231 | –21.48428 | –21.22760 | –19.88959 | –19.87037 |
| –19.65954 | –19.61343 | –16.46607 | –14.65902 | –14.64590 |
| –14.58003 | –14.29206 | –14.28290 | –14.27827 | –14.26840 |
| –14.20851 | –14.19676 | –7.32936 | –5.65984 | |

Из сопоставления с энергетической диаграммой атомов фтора в параметризации PM-7 следует, что 45 электронов “каркаса” комплексов расположены на 24 МО – орбиталях, базисом для которых являются 2s и 2p атомные орбитали фтора. По три энергетических уровня, соответствующих 2s-орбиталям, расположены вблизи значений 45 и 35 эВ. Заселенные уровни, соответствующие 2p-орбиталям располагаются в области от –20 до –14 эВ.
Все рассматриваемые комплексы формируются в результате координации каркасом LaF6 из 14 атомов щелочных металлов. В результате модель дополняется 14 электронами, а в базис включаются 14s и 42p AO. Расчеты показали, что качественно изменение электронной структуры не зависит от индивидуальности щелочного металла. Три из дополнительных 14 электронов располагаются на вакантных уровнях каркаса. Оставшиеся электроны заселяют 6 новых энергетических уровней, расположенных в области отрицательных значений шириной порядка 4 эВ в высокоэнергетической части спектра. Вакантные уровни при этом расположены, как правило, в области положительных значений.
Интересен тот факт, что исчезает имевшее место в каркасе разбиение 6 уровней s-орбиталей на две группы. В комплексах они группируются в одной, достаточно узкой области. Область уровней p-орбиталей сдвигается вправо по оси энергий на величину ∆E, зависящую от природы щелочного металла.
В качестве примера на рис. 8 приведена, плотность электронных состояний для комплекса LaF6Li14.
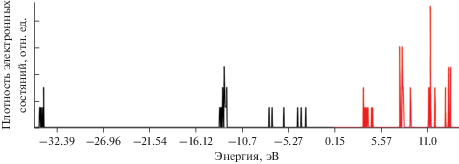
Видно, что уровни атомов лития образуют новую группу занятых и вакантных состояний в высокоэнергетической части спектра.
Подобна же плотность состояний для комплексов LaF6K14 и LaF6Cs14 после их оптимизации в данном приближении (рис. 9, 10).
Рис. 9.
Плотность электронных состояний: а) модели LaF6K14 в SPARKLE приближении, б) область плотности состояний, сформированная атомами калия.
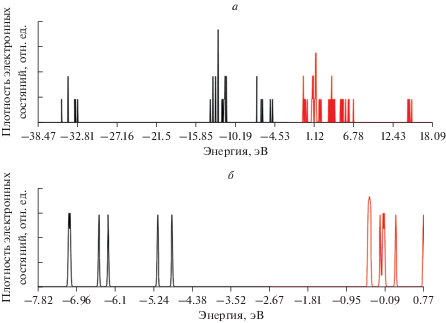
Рис. 10.
Область плотности электронных состояний модели LaF6Cs14 в SPARKLE приближении, сформированная атомами цезия.

Выборочные характеристики электронной структуры комплексов приведены в табл. 3.
Таблица 3.
Характеристики электронной структуры ряда комплексов в SPARKLE приближении
| Состав комплекса |
Энергия ВЗМО, эВ | Энергия НВМО, эВ | Область уровней s-орбиталей фтора, эВ | ∆E, эВ | Дип. момент, Дб |
|---|---|---|---|---|---|
| LaF6Li14 | –3.23/–3.93 | 3.48/–0.4 | –33.8…–34.3 | 2/1.5 | 1.42/24.5 |
| –34.1…–36.8 | |||||
| LaF6K14 | –4.83/–4.76 | –0.68/–0.47 | –32.7…–34.9 | 3/2 | 12.5/9.72 |
| –33.9…–35.7 | |||||
| LaF6Cs14 | –3.63/–3.67 | –0.21/–0.4 | –34.1…–36.8 | 2/1.5 | 21.9/15.4 |
| CeF6Li14 | –3.28 | 3.46 | –33.8…–34.4 | 2 | 2.04 |
| CeF6Cs14 | –3.61 | –0.22 | –31.7…–32.5 | 4 | 2.16 |
| SmF6Li14 | –3.4 | 3.48 | –34.0…–34.4 | 2 | 2.16 |
| EuF6Li14 | –3.33 | 3.36 | –33.7…–34.4 | 2 | 4.12 |
| PrF6Cs14 | –3.62 | –0.22 | –31.7…–32.4 | 3.7 | 21.9 |
| NdF6Li14 | –3.26 | 3.45 | –33.9…–34.5 | 2.1 | 2.16 |
Расчеты с параметрами РМ-7 для комплексов на основе лантана
Лантан – единственный элемент из РЗМ, для которого проведена параметризация методом PM-7 в валентном приближении [16].
В отличие от расчетов предыдущего раздела, здесь в базис для разложения МО включены 9 валентных АО лантана (одна 6s, три 6p и пять 5d). Из сопоставления плотности электронных состояний на рис. 7 и 11 и в табл. 2 и 3 видно, что уровни, внесенные атомом лантана в энергетический спектр каркаса LaF6, не заполнены и лежат в высокоэнергетической области.
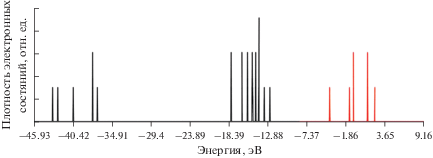
Для комплексов с атомами щелочных металлов интерпретация энергетического спектра усложняется тем, что в базис дополнительно включено по 4 валентных s и p АО от каждого атома. 56 энергетических уровней этих атомов перемешиваются с уровнями каркаса комплекса. В качестве примера на рис. 13 приведена плотность состояний для модели LaF6Li14 (рис. 12). Однозначно можно утверждать, что только шесть из них попадает в область отрицательных значений, и они заполнены.
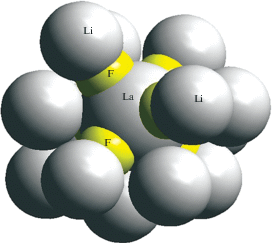
Рис. 13.
Плотность электронных состояний комплекса LaF6Li14: а – полный диапазон, б –высокоэнергетическая область.

Плотность электронных состояний для этого комплекса приведена на рис. 13.
Плотность состояний внутренних уровней также претерпевает изменения: средняя область несколько сужается и примерно на 2 эВ смещается в область более высоких энергий (вправо по оси энергий). Качественные особенности плотности состояний для других комплексов лантана аналогичны. Количественные данные для ряда из них приведены в табл. 4. Сравнение расчетных данных показывает, что отличия двух использованных приближений незначительны.
Таблица 4.
Собственные значения энергии модели LaF6 в валентном приближении с параметрами PM-7
| Альфа-собственные значения (alpha eigenvalues) |
||||
|---|---|---|---|---|
| –43.25658 | –2.48868 | –40.34189 | –37.56618 | –37.55844 |
| –36.90627 | –18.10436 | –18.06967 | –16.54633 | –16.51360 |
| –15.77500 | –15.76171 | –15.08670 | –15.08373 | –15.03018 |
| –14.61523 | –14.60942 | –14.22067 | –14.18237 | –14.16512 |
| –14.16327 | –13.37792 | –12.60725 | –4.17809 | –1.39117 |
| –0.85994 | –0.84965 | 1.14408 | 1.14869 | 2.12764 |
| 72.75209 | 73.91774 | 73.92318 | ||
| Бета-собственные значения (Вета eigenvalues) |
||||
| –43.24757 | –40.35081 | –39.58284 | –37.56615 | –37.55841 |
| –36.90624 | –18.11228 | –18.08020 | –17.33761 | –17.32831 |
| –16.53863 | –16.50313 | –15.08733 | –15.08610 | –15.03086 |
| –14.61528 | –14.60945 | –14.22066 | –14.18236 | –14.16515 |
| –14.16331 | –13.36641 | –4.18943 | –3.41860 | –1.39104 |
| –0.86011 | –0.84965 | 1.14425 | 1.14872 | 2.12770 |
| 72.75212 | 73.91777 | 73.92324 | ||
Основываясь на данных работы [17] предполагается на основе расширения базиса АО атомов РЗМ более адекватное описание электронной структуры, позволяющее перейти к расчету спектральных характеристик.
ЗАКЛЮЧЕНИЕ
Построены молекулярные модели комплексов, формирование которых постулируется при введении в солевые фторидные расплавы ионов редкоземельных элементов группы лантана. Методом молекулярной механики с силовым полем ММ+ определены их геометрические характеристики для состояний, соответствующих минимумам поверхности потенциальной энергии.
Установлено, что эти модели устойчивы по отношению к оптимизации, проведенной полуэмпирическим квантовохимическим методом PM7 в SPARKLE приближении и в валентном приближении с учетом s, p, d AO для атомов РЗМ.
Показано, что, несмотря на определяющий вклад в плотность состояний каркаса комплекса RF6 (R – ион редкоземельного металла – La, Ce, Pr, Nd, Sm), влияние атомов во второй координационной сфере на электронный спектр значимо и зависит от природы щелочного металла в ней.
Построенные модели перспективны для последующих расчетов спектральных характеристик.
Список литературы
Photiadis G.M., Voyiatzis G.A., Kipouros G.J., Papatheodorou G.N. Raman spectroscopic study of NdCl3–ACl (A = Li, Na, K, Cs and Ca) systems in the solid and liquid states // Oeye Symp. Conference. 1995. P. 313–324.
Photiadis G.M., Borresenf B., Papatheodorou G.N. Vibration modes and structures of lanthanide halide-alkali halide binary melts LnBr3–KBr (Ln = La, Nd, Gd) and NdCl3–ACl (A = Li, Na, K, Cs) // J. Chemical Society. Faraday trans. 1998. 94. № 17. P. 2605–2613.
Барбанель Ю.А., Котлин В.П., Клокман В.Р. Комплексообразование лантаноидов и актиноидов в галидных расплавах // Радиохимия. 1979. 5. С. 694–705.
Барбанель Ю.А., Лумпов А.А. Высокотемпературные спектры трихлоридов плутония и празеодима // Радиохимия. 1987. 6. С. 730–741.
Хохряков А.А., Пайвин А.С., Норицин С.И., Истомин С.А. Электронные спектры отражения Ce3+ в расплавах BeF2 и B2O3 // Физика и химия стекла. 2014. 40. С. 407–412.
Хохряков А.А., Вершинин А.О., Пайвин А.С., Лизин А.А. Электронные спектры ионов Nd(III) в расплавленных фторидах щелочных металлов // Расплавы. 2015. 4. С. 3–11.
Хохряков А.А., Вершинин А.О., Пайвин А.С., Лизин А.А. Электронные спектры растворов трифторида празеодима в расплавленных фторидах щелочных металлов // Журнал неорганической химии. 2015. 60. С. 1717–1772.
Барнабель Ю.А. Координационная химия f-элементов расплавов. М: энергоатомиздат. 1985.
Stwart J.J.P. MOPAC 2012 // Stewart Computational Chemistry, Colorado Springs, CO, USA. 2012. http://Open MOPAC.net.
HyperChem version 8.10. http://www.hyper.com/index.html
Alex A. Granovsky, Firefly version 8. http://classic.chem.msu.su/gran/firefly/index.html.
https://sourceforge.net/projects/mocalc2012/.
Хельтье Х.-Д., Зиппель В., Роньян Д., Фолькерс Г. Молекулярное моделирование: теория и практика. Изд-во “Лаборатория знаний”. 2015.
Klamt A., Schüürmann G. COSMO: a new approach to dielectric screening in solvents with explicit expressions for the screening energy and its gradient // J. Chemical Society: Perkin Transactions. 1993. 2. P. 799–805.
Filho M.A.M., Dutra J.D.L., Rocha G.B., Freire R.O., Simas A.M. Sparkle / RM1 Parameters for the Semiempirical Quantum Chemical Calculations of Lanthanide Complexes // The Royal Society of Chemistry. 2013. 3. P. 16 747–16 755.
Stewart J.J.P. Optimization of parameters for semiempirical methods VI: more modifications to the NDO approximations and re-optimization of parameters // J. Molecular Modeling. 2013. 19. № 1. P. 1–32.
Юрьева Э.И., Резницких О.Г., Бамбуров В.Г. Квантовохимические особенности межатомных La–O – взаимодействий в системах LnNbxOy (La = Ln, Ce, Pr, Nd, Sm, Eu) // ФТТ. 2010. 52. № 2. С. 215–220.
Дополнительные материалы отсутствуют.