

Российский физиологический журнал им. И.М. Сеченова, 2019, T. 105, № 12, стр. 1514-1525
Ингибирование гиперпродукции оксида азота в условиях прогрессивно нарастающей гипоксии на фоне действия ИЛ-1β снижает выживаемость крыс после острой гипоксии
Ж. А. Донина 1, *, Е. В. Баранова 1, Н. П. Александрова 1
1 Институт физиологии им. И.П. Павлова РАН
Санкт-Петербург, Россия
* E-mail: zdonina@mail.ru
Поступила в редакцию 02.07.2019
После доработки 07.08.2019
Принята к публикации 07.08.2019
Аннотация
На наркотизированных крысах линии Вистар оценивали роль оксида азота (NO) в реализации компенсаторных реакций организма на нарастающую острую нормобарическую гипоксию при повышенном уровне провоспалительного цитокина интерлейкина-1бета (ИЛ-1β). Прогрессивное нарастание гипоксии от нормоксии до апноэ создавали использованием экспериментальной модели “возвратного дыхания”. Было установлено, что блокада NO неселективным ингибитором NO-синтазы L-NAME угнетает компенсаторное увеличение легочной вентиляции на нарастающую гипоксическую гипоксию в большей степени, чем при дискретном повышении уровня ИЛ-1β, что могло явиться следствием снижения активности периферической хеморецепции. Полученное в настоящем исследовании снижение артериального давления при действии ИЛ-1β в нормоксических условиях и его повышение до контрольных значений при ингибировании NO-синтазы сопровождаются изменениями показателей внешнего дыхания и насыщения артериальной крови кислородом, что свидетельствует о роли NO в формировании интегративных реакций дыхательной и сердечно-сосудистой систем при патологии и экстремальных состояниях. У крыс с блокадой NO уровень сатурации при острой гипоксии не снижался, а превышал значения как в контроле, так и при действии ИЛ-1β. Такая реакция могла явиться следствием гипоксического гипометаболизма (снижение интенсивности обмена), который характеризуется уменьшением скорости потребления кислорода в результате резкого падения напряжения О2 в артериальной крови. Также установлено, что ингибирование гиперпродукции NO в условиях прогрессивно нарастающей гипоксии на фоне действия ИЛ-1β уменьшает спонтанное восстановление инспираторной активности и процент выживаемости крыс в постгипоксический период.Таким образом, анализ результатов свидетельствует об участии оксида азота в механизмах влияния провоспалительного цитокина ИЛ-1β на резистентность организма крыс к нарастающей острой нормобарической гипоксии.
В большинстве патологических процессов к наиболее тяжелой органной дисфункции относится острая дыхательная недостаточность, степень которой усугубляется стремительно развивающейся гипоксией. Изменения паттерна дыхания, нарушение механизмов регуляции дыхания и кровообращения, оксигенации тканей и органов, возникновение эпизодов апноэ являются частой причиной критических состояний, требующих проведения интенсивной терапии [1].
В патогенезе и тяжести протекания патологического состояния особое значение имеют многочисленные воспалительные медиаторы, среди которых важная роль принадлежит провоспалительным цитокинам и, в частности, интерлейкину-1бета (ИЛ-1β). При участии цитокинов происходит формирование иммунного ответа, интегрирование различных элементов иммунитета, развивается системная реакция острой фазы воспаления и запускается каскадная экспрессия цитокинов [2, 3]. В литературе имеется большое количество фактов о роли цитокинов в регуляции различных физиологических функций, в ответной реакции организма на экстремальные факторы, в том числе и на гипоксическое воздействие. Установлено, что в гипоксических условиях уровень цитокинов многократно увеличивается [4].
Считается, что ИЛ-1β является основным медиатором, обеспечивающим связь между воспалительной реакцией, снижением функциональных резервов кардиореспираторной системы и гипоксемией. В настоящее время показано, что повышенный уровень ИЛ-1β в плазме крови нарушает регуляцию дыхания, снижает устойчивость к острой гипоксии, возможность спонтанного возобновления дыхания после гипоксического апноэ и способствует развитию циркуляторного коллапса [5–7]. В то же время, в соответствии с современными данными, влияние ИЛ-1β на центральные механизмы регуляции кардиореспираторной системы осуществляется не в результате его прямого влияния на нейроны дыхательного и вазомоторного центров, а опосредовано действием вторичных мессенджеров, роль которых могут выполнять простагландины и оксид азота (NO), выделяемые эпендимными, периваскулярными клетками и клетками эндотелия церебральных сосудов при активации цитокиновых рецепторов [2, 3, 6]. Важно отметить, что оксид азота в физиологических условиях способствует нормальной деятельности сердечно-сосудистой системы, играет активную роль в развитии патофизиологических процессов, иммунных реакциях и является существенной частью неспецифической резистентности организма [8].
В организме человека и животных оксид азота образуется в результате реакции окисления аминокислоты L-аргинина, которая катализируется ферментом NO-синтазой (NOS) [9–11]. Различают три изоформы NOS: конститутивные – нейрональная (nNOS) – экспрессируется в основном в периферических и центральных нейронах и эндотелиальная (eNOS), которая локализуется в эндотелиальных и некоторых других клетках. К числу факторов, повышающих активность конститутивных изоформ NOS, относится концентрация кислорода. Индуцибельная изоформа (iNOS) – экспрессируется при воспалении, ее активность регулируется Ca+2-зависимым связыванием кальмодулина и может в значительной степени превышать активность конститутивных изоформ NOS. Показано, что iNOS в клетках индуцируется при воспалительных процессах, сопровождающихся развитием окислительного стресса и образованием активных форм кислорода (АФК) [2, 8].
Эффекты NO находятся в зависимости от его концентрации, в незначительных количествах он выполняет регуляторные функции, наиболее важной из которых является регуляция работы сердечно-сосудистой системы, в высоких дозах он может оказывать токсическое действие на клеточный и генетический аппарат [12]. В настоящее время известно, что образование NO в клетках модулируется гипоксией. Глубокая степень гипоксии (менее 0.5%) способна ограничивать продукцию NO на 60–80% в результате ингибирования всех изоформ NOS, менее тяжелая степень гипоксии сопровождается умеренным угнетением синтеза NO [13]. Все изоформы NOS присутствуют в респираторной системе и принимают участие в патогенезе различных бронхолегочных заболеваний [11].
Учитывая, что NO играет существенную роль в адаптации к гипоксии, можно предположить, что при повышении уровня провоспалительных цитокинов, усиленный синтез NO может инициировать изменение механизмов регуляции деятельности кардиореспираторной системы.
Целью работы явилось исследование роли NO-зависимых механизмов в формировании резистентности организма крыс к острой гипоксии на фоне повышенного системного уровня ИЛ-1β.
МЕТОДЫ ИССЛЕДОВАНИЯ
Работа проведена на животных из биоколлекции “Коллекция лабораторных млекопитающих разной таксономической принадлежности” Института физиологии им. И.П. Павлова РАН, поддержанной программой биоресурсных коллекций ФАНО России. Исследования проводились в соответствии с регламентом, установленным МЗСР РФ № 708н от 23.08.10 “Правила лабораторной практики” и Директивой 2010/63/EU Европейского парламента и Совета Европейского Союза по охране животных, используемых в научных целях.
Опыты поставлены на 24 наркотизированных (уретан, 1000 мг/кг) и трахеостомированных крысах линии Вистар массой 250–300 г. Проведено 3 серии экспериментов по 8 крыс в каждой. 1-я-контрольная (интактные крысы), 2-я и 3-я – экспериментальные, в которых при нарастающей гипоксии исследовали влияние на физиологические резервы кардиореспираторной системы ИЛ-1β и ИЛ-1β на фоне действия L-NАME – неселективного ингибитора синтазы оксида азота. Препараты вводили в левую бедренную вену.
Каждому животному ИЛ-1β вводился в количестве 500 нг, растворенных в 1 мл физиологического раствора, L-NАME – в количестве 3 мг растворенных в 1 мл физиологического раствора за 10 мин до введения ИЛ-1β. В контрольной серии экспериментов животным внутривенно вводился 1 мл физиологического раствора. Через 60 мин после инъекций препаратов проводили тестирование крыс нарастающей гипоксией от нормоксии до полной остановки дыхания. Контрольная серия проводилась по вышеописанной схеме. После окончания эксперимента проводили эвтаназию животных передозировкой уретана.
Для изучения динамических показателей, характеризующих состояние животных в условиях развития различной тяжести гипоксии, использовали метод возвратного дыхания (ререспирация), описанный ранее [7]. При возвратном дыхании животное производило вдох из мешка, заполненного атмосферным воздухом. Выдыхаемый газ поступал обратно в мешок, при этом избыток СО2, выделяемый организмом, поглощался натронной известью. В результате концентрация СО2 во вдыхаемом воздухе поддерживалась на уровне нормокапнии, а содержание О2 уменьшалось по мере его потребления животным в процессе дыхания от нормоксии до наступления апноэ. После остановки дыхания крыс отключали от гипоксического воздействия. По мере прогрессивного нарастания гипоксии фиксировали продолжительность дыхания до остановки дыхательных движений (время жизни, ВЖ), длительность гипоксического апноэ до спонтанного появления первого вдоха, фракционное содержание О2 во вдыхаемой газовой смеси (FIO2), насыщение артериальной крови кислородом (SpO2%) и процент выживаемости в постгипоксическом периоде. Поскольку концентрация О2 и время жизни до наступления апноэ имели существенные различия в разных экспериментальных группах, сравнительную оценку исследуемых параметров в обеих группах проводили на уровне острой гипоксии (10% O2).
В ходе эксперимента проводилась синхронная регистрация параметров внешнего дыхания и сердечно-сосудистой системы. Методом пневмотахографии регистрировали: объемную скорость инспираторного потока (Vi), дыхательный объем (ДО), частоту дыхания (ЧД), рассчитывали минутный объем дыхания (МОД). Внутригрудное (внутриплевральное) давление (ВГД) измерялось по величине инспираторных колебаний пищеводного давления, которые регистрировались баллонографическим методом. Переменные величины давлений преобразовывались в электрический сигнал при помощи электроманометра ПДП-1000. Систолическое и диастолическое давление (АДс, АДд) регистрировали в общей сонной артерии прямой катетеризацией сосудов, среднее АД (САД) рассчитывали по формуле: САД = Pд + (Рс – Рд)/2. Частоту сердечных сокращений (ЧСС) подсчитывали по электрокардиограмме, зарегистрированной с помощью игольчатых электродов при биполярном отведении. Непрерывный мониторинг SpO2% осуществляли методом пульсоксиметрии, используя ветеринарный пульсоксиметр типа UT (Zoomed, Россия). Перечисленные показатели регистрировали каждую минуту в течение всего периода гипоксического воздействия. Концентрацию кислорода во вдыхаемой газовой смеси (FIO2) измеряли анализатором кислорода ПГК-06 (“Инсовт”, Санкт-Петербург), диоксида углерода (FIСO2) многокомпонентным газоанализатором МАГ-6П (“Эксис”, Москва). Все перечисленные параметры для последующего анализа вводили в память персонального компьютера IBMPС с помощью аппаратно-программного комплекса “Biograph” (ГУАП, Санкт-Петербург).
Проверка выборки на нормальность распределения и статистическая обработка данных производилась компьютерными средствами с использованием программы Microsoft Excel. Вычислялась средняя величина и ошибка средней регистрируемых показателей. Достоверность различий оценивали с помощью t-критерия по Стьюденту. Различия считали достоверными при p < 0.05.
РЕЗУЛЬТАТЫ ИССЛЕДОВАНИЯ
В нормоксических условиях в экспериментальных группах с дискретным введением ИЛ-1β и ИЛ-1β + L-NAME наблюдались изменения показателей кардиореспираторной системы по сравнению с контрольными животными, которым вводился физиологический раствор (табл. 1). Однако если в серии с введением ИЛ-1β МОД увеличился на 180 ± 19% по сравнению с контрольными величинами, то в серии с введением ИЛ-1β на фоне действия L-NAME МОД возрастал только на 97 ± 9% (p < 0.05). Увеличение МОД происходило за счет роста дыхательного объема. Кроме того, было установлено снижение среднего АД на 41 ± 5% (p < 0.05) у крыс с повышенным системным уровнем ИЛ-1β, тогда как на фоне предварительного введения L-NAME ИЛ-1β не вызывал снижения АД. После введения ИЛ-1β в обеих экспериментальных группах также отмечалось уменьшение SpO2, причем в группе с ингибированием NO-синтазы это снижение было более значительным.
Таблица 1.
Исходные значения показателей внешнего дыхания и сердечно-сосудистой системы при введении NaCl, ИЛ-1β, ИЛ-1β + L-NAME при нормоксии (n = 8)
| Показатели | Контроль (NaCl) |
ИЛ-1β | ИЛ-1β + L-NAME |
|---|---|---|---|
| Дыхательный объем, мл | 0.95 ± 0.21 | 2.30 ± 0.10* | 1.30 ± 0.20* |
| Частота дыхания, мин–1 | 80 ± 4 | 93 ± 2 | 121 ± 28*# |
| Минутный объем дыхания, мл/мин | 76 ± 3 | 214 ± 6* | 150 ± 10*# |
| Внутригрудное давление, см вод. ст. | 0.50 ± 0.05 | 0.50 ± 0.10 | 1.00 ± 0.10*# |
| Насыщение артериальной крови кислородом, % | 96 ± 2 | 88 ± 1 | 82 ± 7* |
| Среднее артериальное давление, мм рт. ст. | 115 ± 6 | 68 ± 10* | 124 ± 19# |
| Частота сердечных сокращений, мин–1 | 320 ± 13 | 313 ± 15 | 237 ± 31*# |
По мере нарастания гипоксии, как в контрольной, так и в экспериментальных группах наблюдалось увеличение легочной вентиляции. Сравнительная оценка МОД на уровне 10% О2 выявила наибольший прирост легочной вентиляции в контрольной группе крыс – МОД увеличивался на 146 ± 12% (p < 0.05), у крыс с повышенным системным уровнем ИЛ-1β достоверный прирост МОД в ответ на гипоксическую стимуляцию отсутствовал. При блокаде NOS на фоне повышенного уровня ИЛ-1β рефлекторного повышения вентиляторной реакции на гипоксию не происходило, а наблюдалось снижение МОД по сравнению с нормоксическим периодом (рис. 1, МОД).
Рис. 1.
Ингибирование активности NOS сопровождается снижением прироста легочной вентиляции в условиях нарастающей гипоксии. По оси абсцисс – параметры дыхания: дыхательный объем (ДО), частота дыхания (ЧД), минутный объем дыхания (МОД) и внутригрудное давление (ВГД) на уровне максимальной гипоксической стимуляции (10% О2) в экспериментах с введением NaCl (контроль), ИЛ-1β и L-NAME. Данные представлены как M ± m в % от нормоксии (21% О2), принятой за 100%. По оси ординат – изменения показателей в % от уровня нормоксии. Примечание. *p < 0.05 по сравнению с контролем (NaCl).

Угнетение вентиляторной реакции на гипоксический стимул в экспериментальных группах животных происходило как в результате уменьшения прироста ДО, так и ЧД. При этом снижение прироста ДО было выражено в значительно большей степени. Увеличение ДО на гипоксический стимул у контрольных животных составил 140 ± 11% (p < 0.05), у крыс с повышенным уровнем ИЛ-1β – 35 ± 4% (p < 0.05), а на фоне ингибирования NOS (ИЛ-1β + L-NAME) – 23 ± 5% (р > 0.05) (рис. 1, ДО). ЧД в контрольной группе снижалась на 15 ± 3%, а в экспериментальных на 21 ± 3% (р > 0.05) и 34 ± 6% (р < 0.05) по сравнению с нормоксическим периодом (рис. 1, ЧД).
При действии ИЛ-1β в условиях нарастающей гипоксии снижался и прирост внутригрудного давления. Так, если в контроле на уровне 10% О2 ВГД увеличивалось на 220 ± 17% (p < 0.05), то в группах с повышенным уровнем ИЛ-1β и блокадой оксида азота этот прирост был менее выражен и составлял не более 18–30% (р > 0.05).
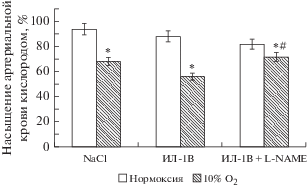
Гипоксическое воздействие также сопровождалось уменьшением насыщения артериальной крови кислородом. Причем на уровне 10% О2 в контрольной группе SpO2 снижалось до 70 ± 4%, с дискретным введением ИЛ-1β до 56 ± 3%, а в серии ИЛ-1β + L-NAME SpO2 соответствовало контрольным значениям и составляло 72 ± 2% (рис. 2).
Рис. 2.
Ингибирование активности NOS не снижает насыщение артериальной крови кислородом (SpO2%) при нарастающей гипоксии. На рисунке представлены изменения SpO2% в абсолютных единицах на уровне максимальной гипоксической стимуляции (10% О2) в экспериментах с введением NaCl (контроль), ИЛ-1β и L-NAME. Данные представлены как M ± m в % от нормоксии (21% О2), принятой за 100%. Примечание *p < 0.05 по сравнению с нормоксией (21% О2), # – p < 0.05 по сравнению с ИЛ-1β.
Повышение уровня ИЛ-1β в условиях нормоксии сопровождалось выраженным снижением среднего артериального давления до 68 мм рт. ст., т.е. на 41 ± 3% (p < 0.05), тогда как влияние ИЛ-1β на фоне ингибирования NO-синтазы не вызывало падения артериального давления (табл. 1, рис. 3). При действии гипоксического стимула (10% О2) во всех группах животных САД снижалось относительно нормоксического периода. При этом у контрольной группы и у животных, которым ИЛ-1β вводился на фоне действия L-NAME, степень снижения САД была практически одинаковой и составила 12 ± 1% и 13 ± 2% (р > 0.05 соответственно), тогда как в группе с повышенным уровнем ИЛ-1β артериальное давление снижалось на 20 ± 3% по сравнению с нормоксическим периодом (рис. 3). Однако достоверных различий между группами не установлено.
Рис. 3.
Ингибирование активности NOS не вызывает падения среднего артериального давления (САД) при нарастающей гипоксии. На рисунке представлены изменения САД в абсолютных единицах при нормоксии и максимальной гипоксической стимуляции (10% О2) в экспериментах с введением NaCl (контроль), ИЛ-1β и L-NAME. Данные представлены как M ± m в % от нормоксии (21% О2), принятой за 100%. *p < 0.05 по сравнению с контролем; # – p < 0.05 по сравнению с ИЛ-1β.

Дальнейшее прогрессивное нарастание гипоксии (FIO2 в дыхательной смеси менее 10% О2) у всех исследованных групп сопровождалось дестабилизацией показателей сердечно-сосудистой системы, нарушением дыхательного ритма и остановкой дыхания (апноэ). Однако концентрация О2, при которой происходила остановка дыхания была различной в контрольной и экспериментальных группах. В контрольной серии апноэ зафиксировано на уровне FIO2 3–4%, а в обеих экспериментальных группах при FIO2 7–8%. Длительность апноэ также отличалась у всех исследованных групп: в контроле она составляла 44.2 ± 3 с, в группе с ИЛ-1β – 31.2 ± 4 с, при действии ИЛ-1β на фоне L-NAME – 18.0 ± 8 с.
В постгипоксическом периоде стойкое самопроизвольное восстановление ритмичного дыхания у животных контрольной серии происходило в 100% случаев, у крыс с повышенным уровнем ИЛ-1β возобновление дыхания наблюдалось в 50%, а у животных группы ИЛ-1β+L-NAME спонтанное восстановление инспираторной активности происходило только в 25% случаев.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Повышение системного уровня основного провоспалительного цитокина ИЛ-1β оказывает активирующее влияние на систему дыхания, вызывая увеличение вентиляции легких за счет роста дыхательного объема и частоты дыхания. Этот факт подтверждается известными данными литературы [14], а также результатами наших предыдущих исследований, выполненных в других экспериментальных условиях [15, 16]. Кроме того, одновременно наблюдается снижение артериального давления, частоты сердечных сокращений и сатурации крови кислородом.
При системном введении ИЛ-1β на фоне действия L-NAME, неселективного ингибитора NO-синтазной активности, влияние ИЛ-1β на дыхательный объем и вентиляцию легких ослабевает. Прирост ДО при действии ИЛ-1β по сравнению с контрольными значениями снижается, что вызывает и соответствующее снижение прироста МОД. Этот факт указывает на то, что NO-ергические механизмы могут участвовать в реализации респираторных эффектов ИЛ-1β. Хотя гораздо более значительную роль, в соответствии с полученными нами данными, они играют в обеспечении влияний ИЛ-1β на параметры сердечно-сосудистой системы. Так, в наших экспериментах ингибирование NO-синтазной активности полностью устраняло влияние ИЛ-1β на величину среднего артериального давления. Это позволяет утверждать, что снижение АД при повышении системного уровня ИЛ-1β вызвано увеличением синтеза NO, которое происходит при взаимодействии ИЛ-1β с соответствующими рецепторами, локализованными на клетках сосудистого эндотелия.
Дыхание гипоксической газовой смесью в контрольной группе животных вызывало увеличение инспираторных колебаний ВГД, увеличение ДО и соответствующий рост МОД. Достоверных изменений в частоте дыхания при этом не наблюдалось. Увеличение системного уровня ИЛ-1β приводило к ослаблению вентиляторного ответа на гипоксию. Наблюдалось резкое снижение прироста инспираторных колебаний ВГД, достоверное уменьшение прироста ДО, а также снижение частоты дыхания. В результате не наблюдалось и увеличения вентиляции легких – основного компонента компенсаторной реакции на действие гипоксии. В наших предыдущих исследованиях было показано, что повышение содержания ИЛ-1β как в циркулирующей крови, так и в цереброспинальной жидкости ослабляет вентиляторный ответ на изменение газового состава крови [16, 17]. Вероятно это свойство ИЛ-1β как одного из провоспалительных цитокинов и является причиной срыва компенсаторной реакции на действие острой гипоксии, который был выявлен в данном исследовании.
Учитывая, что увеличение легочной вентиляции при гипоксии осуществляется рефлекторным путем через хеморецепторы каротидных и аортальных зон, угнетение вентиляторной реакции на гипоксическую стимуляцию на фоне действия ИЛ-1β в первую очередь могло быть связано с изменением активности периферических хеморецепторов. Основные провоспалительные цитокины, такие как ИЛ-1β, ФНО-α (фактор некроза опухоли), ИЛ-6 экспрессируются на сенсорных клетках каротидного тела и, с одной стороны, стимулируют активность синусного нерва [18], но в то же время снижают чувствительность гломусных клеток каротидного тела к дефициту кислорода [19]. Эти данные объясняют полученные нами результаты. Можно было бы также предположить, что одним из механизмов срыва компенсаторной реакции на гипоксию является увеличение синтеза NO, обусловленное повышенным уровнем ИЛ-1β. Установлено, что оксид азота может оказывать влияние на активность периферических хеморецепторов в результате непосредственного воздействия на гломусные клетки. В экспериментах in vitro было показано, что введение донора NO в препарат каротидного тела уменьшает его хемочувствительность к снижению концентрации кислорода [20]. Другим возможным механизмом, посредством которого NO может оказывать свое действие на каротидное тело, является увеличение внутриклеточного уровня циклического гуанозинмонофосфата (cGMP), действующего как вторичный мессенджер с соответствующим изменением функции кальциевых каналов. Это может привести к снижению выброса нейротрансмиттеров (ацетилхолин, аденозин нуклеотидов, допамин) и дальнейшему подавлению сенсорных разрядов синусного нерва [20, 21].
В работах ряда авторов также есть сведения о том, что оксид азота может изменять вентиляторную реакцию на гипоксию через центральные механизмы регуляции дыхания. Однако в этом случае гипоксический ответ не ослабевает, а усиливается. NO, продуцируемый нейрональной синтазой (nNOS), повышает активность диафрагмального нерва при гипоксии, соответственно системное введение ингибитора NOS угнетает вентиляторную реакцию на гипоксию [22, 23]. Эксперименты с введением донора NO непосредственно в область солитарного тракта также обнаружили увеличение импульсации инспираторных нейронов и соответственно вентиляции легких [24]. Таким образом, известные на сегодняшний день данные достаточно противоречивы и свидетельствуют о разнонаправленном влиянии молекул NO на центральные и периферические механизмы реализации гипоксического ответа.
В проведенном нами исследовании было установлено, что блокада активности NOS неселективным ингибитором L-NAME на фоне действия ИЛ-1β не восстанавливает вентиляторный ответ на гипоксию. Влияние ИЛ-1β на изменение дыхательных параметров, вызванное гипоксическим воздействием, не зависело от активности NOS. Степень снижения прироста МОД, ДО и инспираторных колебаний внутригрудного давления на гипоксию при повышенном уровне ИЛ-1β не изменялась при ингибировании активности NOS. Это свидетельствует о том, что срыв компенсаторной реакции на гипоксию, вызванный повышением системного уровня ИЛ-1β, не был связан с усилением синтеза NO. По всей вероятности, NO-синтазные пути участвуют в реализации респираторных влияний ИЛ-1β преимущественно в условиях нормоксии. Однако следует учитывать то, что мы использовали неселективный блокатор NO-синтазы L-NAME. Дальнейшее изучение роли NO-синтазных механизмов в осуществлении респираторных эффектов ИЛ-1β с использованием селективных ингибиторов нейрональной, индуцибельной и эндотелиальной NOS позволит уточнить степень участия разных изоформ NOS в реализации эффектов провоспалительных цитокинов. Кроме того, угнетение легочной вентиляции при гипоксии после введения ИЛ-1β могло быть связано с влиянием не только NO, но и других физиологически активных веществ, синтез которых усиливается при воспалительных реакциях, например, простагландинов. Активация этих медиаторов угнетает дыхание и изменяет паттерн дыхания при гипоксии [23, 25].
Хорошо известно, что NO является мощным эндогенным вазодилататором [26, 27]. В настоящем исследовании сдвиги артериального давления при действии ИЛ-1β и блокаде NOS сопровождались изменениями показателей внешнего дыхания и насыщения артериальной крови кислородом, что свидетельствует о роли NO в формировании интегративных реакций дыхательной и сердечно-сосудистой систем при патологии и экстремальных состояниях.
Кроме того, полученные нами данные указывают на то, что повышение системного уровня ИЛ-1β усиливает действие гипоксии на насыщение артериальной крови кислородом. В гипоксических условиях в группе животных с повышенным уровнем ИЛ-1β наблюдалось более значительное снижение SpO2 по сравнению с контрольной группой. Обращает на себя внимание факт, что при ингибировании NOS уровень сатурации при острой гипоксии не снижался, а превышал значения как в контроле, так и при действии ИЛ-1β. Такая реакция могла явиться следствием гипоксического гипометаболизма (снижение интенсивности обмена), который характеризуется уменьшением скорости потребления кислорода в результате резкого падения напряжения О2 в артериальной крови. Следовательно, можно предположить, что в этих условиях происходит переключение аэробного энергообразования в анаэробное, что является одной из разновидностей реакций внешнего дыхания на острое гипоксическое воздействие [28, 29]. При анализе устойчивости к гипоксии следует отметить, что методические условия проведенных нами опытов – прогрессивно нарастающая острая гипоксия, могут вносить определенные различия с результатами других исследователей, полученными при стабильном предъявлении гипоксического воздействия. Кроме того, выбор адекватного показателя устойчивости к острой гипоксии является довольно проблематичным. В экспериментальной практике для определения гипоксической устойчивости у бодрствующих животных используется высотный порог (время потери позы при минимальной концентрации кислорода) и время восстановления позы в постгипоксический период [30 ] . У наркотизированных животных в остром эксперименте для сравнительной оценки устойчивости к гипоксии мы использовали спонтанное возобновление ритмичного дыхания после апноэ в постгипоксическом периоде (выживаемость). Выявленное в настоящем исследовании снижение процента выживаемости после острой гипоксии у животных с повышенным уровнем ИЛ-1β полностью подтверждают ранее полученные данные [7]. В качестве основных причин, формирующих снижение резистентности к острой гипоксии, нами рассматривалось, в первую очередь, снижение сатурации, гипотония и изменение функционального состояния вазомоторного центра, вызванное угнетающим влиянием гипоксии на нейроны мозгового ствола.
В настоящем исследовании установлено, что ингибирование гиперпродукции NO неселективным блокатором NOS L-NAME в условиях прогрессивно нарастающей гипоксии на фоне действия ИЛ-1β снижает выживаемость крыс после острой гипоксии.
Таким образом, анализ результатов исследования указывает на участие NO в механизмах влияния провоспалительного цитокина ИЛ-1β на резистентность организма крыс к нарастающей острой нормобарической гипоксии
Список литературы
Matuschak G.M., Lechner A.J. Acute lung injury and the acute respiratory distress syndrome: pathophysiology and treatment. Mol. Med. 107(4): 252–258. 2010.
Симбирцев А.С. Цитокины: классификация и биологические функции. Цитокины и воспаление. 3(2): 16–23. 2004. [Simbircev A.S. Cytokines: classification and biological functions. Cytokines and inflammation. 3(2): 16–23. 2004. (In Russ.)].
Мюльберг А.А., Гришина Т.В. Цитокины как медиаторы нейроимунных взаимодействий. Успехи физиол. наук. 37(1): 18–27. 2006. [Myul’berg A.A., Grishina T.V. Cytokines as mediators of neuroimmune interactions. Successes fiziol. sciences. 37(1): 18–24. 2006. (In Russ.)].
Лукьянова Л.Д., Кирова Ю.И., Сукоян Г.В. Сигнальные механизмы адаптации к гипоксии и их роль в системной регуляции. Биол. мембраны: Журн. мембр. клеточн. биол. 29(4): 238–252. 2012. [Luk’yanova L.D., Kirova Yu.I., Sukoyan G.V. Signaling mechanisms of adaptation to hypoxia and its role in systemic regulation. Biol. Membr. J. Membr. Cell Biol. 29(4): 238–252. 2012. (In Russ.)].
Hofstetter A.O., Herlenius E. Interleukin-1β depresses hypoxic gasping and autoresuscitation in neonatal DBA/1lacJ mice. Respir. Physiol. Neurobiol. 146(2–3): 135–146. 2005.
Eltzschig H.K., Carmeliet P. Hypoxia and inflammation. N. Engl. J. Med. 364(7): 656–665. 2011.
Донина Ж.А., Баранова Е.В., Александрова Н.П. Влияние провоспалительного цитокина интерлейкина 1-β на резистентность организма к острой гипоксии. Рос. физиол. журн. им. И.М. Сеченова. 102(11): 1333–1342. 2016. [Donina Zh.A., Baranova E.V., Aleksandrova N.P. The effect of pro-inflammatory cytokine interleukin 1-β on the body’s resistance to acute hypoxia. Russ. J. Physiol. 102(11): 1333–1342. 2016 (In Russ.)].
Малышев И.Ю., Манухина Е.Б. Стресс, адаптация и оксид азота. Биохимия. 63(7): 992–1006. 1998. [Malyshev I.Yu., Manuhina E.B. Stress, adaptation and nitrous oxide. Biochemistry. 63(7): 992–1006. 1998. (In Russ.)].
Jeffrey Man H., Tsui A., Marsden P. Nitric oxide and hypoxia signaling. Vitam. Horm. 96: 161–192. 2014.
Манухина Е.Б., Дауни Г.Ф., Маллет Р.Т., Малышев И.Ю., Ванин А.Ф. Депо оксида азота (NO) и его адаптивная роль в сердечно-сосудистой системе. Патогенез. 10(2): 19–27. 2012. [Manuhina E.B., Dauni G.F., Mallet R.T., Malyshev I.Yu., Vanin A.F. Depot nitric oxide (NO) and its adaptive role in the cardiovascular system. Pathogenesis. 10(2): 19–27. 2012. (In Russ.)].
Antosova M., Mokra D., Pepucha L., Plevkova J., Buday T., Sterusky M., Bencova A. Physiology of Nitric Oxide in the Respiratory System. Physiol. Res. 66(2): 159–172. 2017.
Moncada S., Higgs E. The discovery of nitric oxide and its role in vascular biology. Br. J. Pharmacol. 147(1): 193–201. 2006.
Le Cras T., McMurtry I. Nitric oxide production in the hypoxic lung. Am. J. Physiol. Lung Cell Mol. Physiol. 280(4): 575–582. 2001.
Graff G.R., Gozal D. Cardiorespiratory responses to interleukin-1 beta in adult rats: role of nitric oxide, eicosanoids and glucocorticoids. Arch. Physiol. Biochem. 107(2): 97–122. 1999.
Aleksandrova N.P., Danilova G.A., Aleksandrov V.G. Interleukin-1beta suppresses the ventilatory hypoxic response in rats via prostaglandin-dependent pathways. Can. J. Physiol. Pharmacol. 95(6): 681–685. 2017.
Aleksandrova N.P., Danilova G.A., Aleksandrov V.G. Cyclooxygenase pathway in modulation of the ventilatory response to hypercapnia by interleukin-1β in rats. Resp. Physiol. Neurobiol. (209): 85–90. 2015.
Fernandez R., Gonzalez S., Rey S., Cortes P.P., Maisey K.R., Reyes E.P., Larrain C., Zapata P. Lipopolysacharide-induced carotid body inflammation in cats: functional manifestation, histopathology and involvement of tumor necrosis factor-α. Exp. Physiol. 93(7): 892–907. 2008.
Gauda E., Shirahata M., Mason A., Pichard L., Kostuk E., Chavez-Valdez R. Inflammation in the carotid body during development and its contribution to apnea of prematurity. Respir. Physiol. Neurobiol. 185(1): 120–131. 2013.
Prabhakar N.R. NO and CO as second messengers in oxygen sensing in the carotid body. Respir. Physiol. 115(2): 161–168. 1999.
Iturriaga R., Alcayaga J. Neurotransmission in the carotid body: transmitters and modulators between glomus cells and petrosal ganglion nerve terminals. Brain Res. Rev. 47(1–3): 46–53. 2004.
Haxhiu M.A.,Changch., Dreshai I., Erokwu B., Prabhakar N.R., Cherniack N.S. Nitric oxide and ventilatory response to hypoxia. Resp. physiol. 101(3): 257–266. 1995.
Gozal D. Sleep, sleep disorders and inflammation in children. Sleep Med. 10(1): 12–16. 2009.
Ogawa H., Mizusawa A., Kikuchi Y., Hida W., Miki H., Shirato K. Nitric oxide as a retrograde messenger in the nucleus tractus solitarii of rats during hypoxia. J. Physiol. (Lond.) 486: 495–504. 1995.
Olsson A., Kayhan G., Lagercrantz H., Herlenius E. IL-1 beta depresses respiration and anoxic survival via a prostaglandin-dependent pathway in neonatal rats. Pediatr. Res. 54(3): 326–331. 2003.
Emery C., Teng G., Liu X., Barer G. Vasoreactions to acute hypoxia, whole lungs and isolated vessels compared modulation by NO. Respir. Physiol. Neurobiol. 134: 115–129. 2003.
Godo S., Shimokawa H. Divergent roles of endothelial nitric oxide synthases system in maintaining cardiovascular homeostasis. Radic. Biol. Med. 109: 4–10. 2017.
Бурых Э.А. Компенсаторные и адаптивные перестройки в системе дыхания у человека при остром гипоксическом воздействии. Физиология человека. 35(3): 82–93. 2009. [Buryh E.A. Compensatory and adaptive adjustment in the respiratory system in humans with acute hypoxic exposure. Human physiology. 35(3): 82–93. 2009. (In Russ.)].
Гришин О.В., Басалаева С.В., Устюжанинова Н.В., Уманцева Н.Д., Гладырь С.Н. Реакции внешнего дыхания и интенсивность энергетического обмена у неадаптированных к гипоксии людей в условиях нарастающей гипоксии. Бюлл. физиол. патол. дыхания. 51: 8–14. 2014. [Grishin O.V., Basalaeva S.V., Ustyuzhaninova N.V., Umanceva N.D., Gladyr’ S.N. Lung respiration reaction and energy metabolism intensity in not adapted to hypoxia people exposed to increased hypoxia. Bull. Physiol. Pathol. Respirat. 51: 8–14. 2014. (In Russ.)].
Шустов Е.Б., Капанадзе Г.Д., Станкова Н.В., Ревякин А.О., Матвеенко Е.Л., Ким А.Е., Шуленин Н.С. Гипоксия физической нагрузки у спортсменов и лабораторных животных. Биомедицина. 4: 4–16. 2014. [Shustov E.B., Kapanadze G.D., Stankova N.V., Revyakin A.O., Matveenko E.L., Kim A.E., Shulenin N.S. Hypoxia physical activity in athletes and laboratory animals. Biomedicine. 4: 4–16. 2014. (In Russ.)].
Дополнительные материалы отсутствуют.
Инструменты
Российский физиологический журнал им. И.М. Сеченова