

Российский физиологический журнал им. И.М. Сеченова, 2019, T. 105, № 2, стр. 165-177
Агонист ядерных ретиноидных X рецепторов бексаротен улучшает когнитивные функции у крыс, перенесших пренатальную гипоксию
Н. М. Дубровская 1, 2, Д. С. Васильев 1, 2, Н. Н. Наливаева 1, Н. Л. Туманова 1, О. С. Алексеева 1, И. А. Журавин 1, 2, *
1 Институт эволюционной физиологии и биохимии им. И.М. Сеченова РАН
Санкт-Петербург, Россия
2 анкт-Петербургский государственный педиатрический медицинский университет
Минздрава РФ
Санкт-Петербург, Россия
* E-mail: i-zhur@mail.ru
Поступила в редакцию 11.11.2018
После доработки 12.12.2018
Принята к публикации 14.12.2018
Аннотация
В работе проверена гипотеза о том, что агонист ядерных ретиноидных X рецепторов бексаротен может компенсировать нарушение обучения и памяти, возникающее после пренатальной гипоксии. Для этого в парадигме “распознавание нового объекта” в постнатальный период развития на Р27 и Р50 было протестировано поведение крыс линии Вистар, перенесших острую пренатальную гипоксию на Е14 (3 ч, О2 7%) и однократное введение бексаротена (4 мкл, 10–2 М) в желудочки мозга на Р20. Для сравнения анализировалось поведение крыс соответствующего возраста из групп интактного контроля и перенесших пренатальную гипоксию, но не получавших бексаротен. Также проведено иммуногистохимическое исследование влияния бексаротена на число нервных клеток и синаптоподин-позитивных лабильных дендритных шипиков в поле СА1 гиппокампа, снижающихся после пренатальной гипоксии. Показано, что введение бексаротена увеличивает число лабильных шипиков в гиппокампе при неизменном количестве нервных клеток поля СА1, повышает экспрессию транспортного белка транстиретина в сосудистом сплетении желудочков мозга и улучшает память крыс, перенесших пренатальную гипоксию.
Развитие когнитивного дефицита и деменции сопровождает большое число нейродегенеративных заболеваний, включая болезнь Альцгеймера, и поиск соединений, способных замедлить или восстановить нарушение памяти, является одной из основных задач современной нейрофизиологии и медицины. В наших исследованиях было показано, что пренатальная гипоксия в период интенсивного формирования нервной ткани у крыс может служить удобной моделью нейродегенеративных заболеваний, сопровождающихся когнитивными дисфункциями и снижением синаптической пластичности вследствие уменьшения числа синаптоподин-позитивных лабильных дендритных шипиков в теменной коре и гиппокампе [1–3]. После пренатальной гипоксии в этих структурах мозга также наблюдается снижение содержания и активности основного амилоид-деградирующего фермента неприлизина, который регулирует уровень амилоидного пептида (Аβ) в головном мозге и препятствует накоплению его токсических форм, приводящих к развитию болезни Альцгеймера у человека [4]. Показано также, что подавление активности неприлизина путем введения его ингибиторов в кору головного мозга крыс приводит к нарушению памяти [5, 6]. Несмотря на то, что Aβ имеет прямую связь с патогенезом болезни Альцгеймера, этот пептид присутствует и у здоровых индивидуумов и выполняет важные физиологические функции [7]. Помимо амилоид-деградирующих ферментов содержание Aβ в головном мозге также регулируется посредством его выведения из ткани мозга с помощью ряда транспортных белков [8, 9]. К их числу относятся переносчик холестерина – аполипопротеин-E (ApoE) и транстиретин (TTР) [10, 11]. В настоящее время активация экспрессии амилоид-деградирующих ферментов и транспортных белков рассматривается как одна из терапевтических стратегий для предотвращения развития болезни Альцгеймера [9].
К числу соединений, способных регулировать экспрессию амилоид-деградирующих и транспортных белков, относится селективный агонист ядерных ретиноидных Х рецепторов (RXR) бексаротен [12], известный как противоопухолевый препарат таргретин (США). Известно, что посредством активации RXR облегчается транскрипция AроE, который необходим для удаления Aβ [8, 13]. Более того, на клетках нейробластомы было показано, что бексаротен повышает экспрессию неприлизина и инсулин-деградирующего фермента [12]. Принимая во внимание, что механизм регуляции ТТР схож с таковым для неприлизина [14], есть основания предполагать, что его экспрессия также может регулироваться бексаротеном. Также есть свидетельства того, что ТТР способен связываться с RXR [15], однако данные о влиянии бексаротена на уровень экспрессии этого транспортного белка отсутствуют. Особый интерес представляют данные о способности ТТР регулировать процессы памяти стареющих животных посредством активации ретиноидных сигнальных путей в гиппокампе [16].
Исследования на трансгенных мышах, используемых в качестве модели болезни Альцгеймера, показали, что введение бексаротена быстро уменьшает количество амилоидных Aβ депозитов в головном мозге и снижает когнитивный дефицит за счет активации экспрессии ApoE [17]. Описана способность бексаротена улучшать память у трансгенных мышей, используемых в качестве модели болезни Альцгеймера, и снижать содержание растворимого Аβ без изменения числа сенильных бляшек [18]. В модельных экспериментах также была подтверждена способность бексаротена препятствовать фибриллогенезу Аβ [19].
Исходя из вышеизложенного, целью данного исследования был анализ влияния бексаротена на когнитивные функции и морфофункциональное состояние нервной ткани гиппокампа у крыс, перенесших пренатальную гипоксию, а также на уровень экспрессии TTР в структурах мозга.
МЕТОДЫ ИССЛЕДОВАНИЯ
Животные. Исследование проводили на самцах крыс линии Вистар контрольной (интактный контроль) и двух экспериментальных групп – после пренатальной гипоксии и после пренатальной гипоксии с введением бексаротена. Все опыты осуществлялись в соответствии с протоколом обращения с лабораторными животными, утвержденным биоэтическим комитетом ИЭФБ РАН и основанном на директиве Европейского Сообщества по гуманному обращению с экспериментальными животными (European Communities Council Directive #86/609 for the Care of Laboratory Animals).
Модель пренатальной нормобарической гипоксии. Самок крыс линии Вистар на 14-е сутки беременности (Е14, рис. 1) подвергали действию нормобарической гипоксии в специальной камере емкостью 100 л, содержащей системы терморегуляции, вентиляции, газового анализа и адсорбции выдыхаемого СО2. В ходе эксперимента содержание кислорода в камере снижали с 20.7 до 7.0% в течение 10 мин и поддерживали на этом уровне в течение 3 ч. Концентрация углекислоты в камере не превышала 0.2%, а температура поддерживалась на уровне 22°С. В камеру одновременно помещали не более 10 крыс. Контрольные самки содержались при нормальной концентрации кислорода. На 20-й день беременности (за 1 сутки до родов) самок рассаживали по отдельным клеткам. На 2-е сутки после рождения в каждом выводке оставляли по 8 крысят. При расчете возраста крысенка нулевым считали день его появления на свет.
Введение агониста RXR бексаротена. На 20-е сутки после рождения (Р20, рис. 1) часть самцов из потомства, перенесшего пренатальную гипоксию, наркотизировали золетилом (100 мг/кг массы тела, i.p.) и в теменной кости черепа делали отверстие для инъекций по координатам: OD = –0.8; L = 1.5 мм [20]. В стереотаксическом приборе СТМ-3 (Россия) в систему желудочков мозга (на глубину H = 2.5 мм [20]) однократно с помощью шприца Гамильтона вводили раствор бексаротена в физиологическом растворе (10–2 М, 4 мкл, в течение 10 мин). Бексаротен (Sigma, США) растворяли согласно протоколу производителя непосредственно перед инъекцией в диметилсульфоксиде (DMSO), добавляя стерильный физиологический раствор до конечной концентрации DMSO 1%. Оптимальную концентрацию бексаротена подбирали эмпирически, согласно представленным изготовителем данным о дозозависимости его действия.
Исследование поведения животных. Эксперименты проводили на крысах двух возрастных категорий – молодые (через 27 сут после рождения, Р27, рис. 1) и взрослые (50 сут после рождения, Р50, рис. 1), которые были разделены на следующие группы: 1 – контрольные – не подвергавшиеся пренатальной гипоксии и введению бексаротена (группа “интактный контроль”, К, n = 35); 2 – перенесшие пренатальную гипоксию без введения бексаротена (Г, n = 32); 3 – перенесшие пренатальную гипоксию с введением бексаротена на Р20 (Г + Bex, n = 15).
Тестирование памяти проводили в модифицированном тесте “Распознавание новых объектов” – novel object recognition test [21]. В начале тестирования животных в течение 5 мин адаптировали к условиям эксперимента, для чего крысу помещали на специальную площадку 100 × 100 см, ограниченную непрозрачными стенками высотой 20 см. Через 2 ч после адаптации в течение 5 мин на экспериментальной площадке проводили тренировочное предъявление животному двух предметов (№ 1 и 2). При последующем тестировании объект № 2 заменяли новым предметом, а объект № 1 оставляли неизменным, но меняли место его расположения. Память оценивали через 60 мин после тренировки, для чего животному предъявляли пару объектов, состоящую из известного (№ 1) и нового (№ 3) предметов. Время обследования каждого предмета, т.е. время, проведенное в непосредственном тактильном или обонятельном контакте животного с объектом, регистрировали в секундах и выражали в процентах. За 100% принимали суммарное время обследования обоих предметов в течение 5 мин тестирования. Все предметы были сделаны из толстого стекла, и после каждого предъявления их и экспериментальную площадку протирали 50%-ным раствором этилового спирта. Одинаковая схема экспериментов была использована для тестирования всех групп животных.
Гистологическое исследование ткани гиппокампа. Через 3 суток после введения бексаротена (рис. 1) животных группы Г + Bex (n = 5), а также К (n = 12) и Г (n = 10) групп соответствующего возраста декапитировали, извлекали мозг, ткань одного из полушарий фиксировали в 10%-ном растворе нейтрального формалина на фосфатном буфере (рН 7.4; длительность фиксации 7 сут) и изготавливали фронтальные срезы дорзального гиппокампа толщиной 20 мкм (3.3 мм от линии Bregma по [20]) на криостате Leica CM 1510S (Leica Microsystems, Германия), которые затем окрашивали крезиловым фиолетовым по Нисслю. Световую микроскопию проводили на микроскопе ImagerA (Zeiss, Германия). Количественные характеристики структуры нервной ткани оценивали с использованием программы анализа изображений “ВидеоТест-Мастер-Морфология” (ООО “ВидеоТест”, СПб, Россия). Анализировали состояние клеток (изменение тургора и распределение вещества Ниссля) и плотность их расположения на участке пирамидного слоя поля СА1 дорзального гиппокампа площадью 3000 мкм2 для каждого животного. Методика количественного анализа описана в [1].
Иммуногистохимическое исследование распределения актин-ассоциированного белка шипикового аппарата синаптоподина в stratum radiatum-moleculare поля СА1. Через 3 или 30 суток после введения бексаротена (рис. 1) животных группы Г + Bex (по n = 6 для каждой возрастной группы, Р23 и Р50), а также животных К и Г групп (по n = 8 для каждой возрастной группы, Р23 и Р50) декапитировали, извлекали мозг, одно из полушарий фиксировали в 4%-ном растворе параформальдегида на фосфатном буфере (рН 7,4; длительность фиксации 24 ч) и изготавливали фронтальные срезы дорзального гиппокампа толщиной 20 мкм (3.3 мм от линии Bregma по [20]) на криостате Leica CM 1510S (Leica Microsystems, Германия). Для анализа распределения белка синаптоподина использовали кроличьи моноклональные антитела к этому белку (S9567, SIGMA, в разведении 1 : 1000) с последующей их визуализацией с помощью FITC-конъюгированных моноклональных вторичных антител против IgG кролика (SIGMA, разведение 1 : 500). Иммунофлуоресцентное исследование выполняли на микроскопе Leica DMR, оборудованном конфокальным сканером Leica TCS SL (Leica Microsystems, Германия). Количество синаптоподин-позитивных дендритных шипиков рассчитывали на участке нейропиля stratum radiatum-moleculare поля СА1 гиппокампа площадью 10 000 мкм2 и вычисляли среднее значение по 10 срезам для каждого животного.
Анализ содержания мРНК TTР. Для исследования использовали ткань дорсального гиппокампа на уровне Bregma –3.50 мм и ткань сосудистого сплетения боковых желудочков мозга крыс, перенесших пренатальную гипоксию, через 24 ч после введения бексаротена (когда его эффект на экспрессию нейрональных генов наиболее ожидаем) (Г + Bex, n = 6), а также контрольных крыс соответствующего возраста (К, n = 8) и крыс, перенесших пренатальную гипоксию, но не получавших инъекцию бексаротена (Г, n = 8).
Выделение тотальной РНК производилось с использованием TRI Reagent (Molecular Research Center Inc., США) согласно инструкции производителя. Обратная транскрипция образцов РНК проводилась с использованием обратной транскриптазы вируса лейкемии мышей (M-MLV), затем полученная кДНК использовалась для ПЦР в реальном времени. Праймеры синтезировались фирмой “Бигль”, Санкт-Петербург. В качестве гена “домашнего хозяйства” использовали циклофилин А (CycA). Для праймеров выбраны следующие последовательности: CycA – прямой 5'- AGGATTCATGTGCCAGGGTG -3', обратный 5'- CTCAGTCTTGGCAGTGCAGA-3'); TTР - прямой 5'-GGCTCACCACAGATGAGAAGTTC-3', обратный 5'-ACAAATGGGAGCTACTGCTTTGGC-3'. Амплификация проводилась на приборе BIO-RAD Thermo Cycler C1000 touch с верхним блоком CFX96. Определение содержания мРНК (кДНК) производили относительно контрольной пробы с использованием 2ΔΔCt метода [22].
Статистическую обработку полученных данных проводили с помощью пакета программ SPSS 17.0 (SPSS Inc., Chicago, Il, США), используя одновыборочный t-критерий для поведенческой серии экспериментов, однофакторный ANOVA с апостериорным критерием Даннета (one-way ANOVA, Dunnet post-hoc) и Холма–Сидака (one-way ANOVA, Holm-Sidak post-hoc) для анализа числа дендритных шипиков и количества клеток в гиппокампе. При сравнении содержания мРНК TTР у контрольных и экспериментальных животных использовали непараметрический критерий Манна–Уитни (Mann–Witney U-test). Значимыми считались различия при р < 0.05.
РЕЗУЛЬТАТЫ ИССЛЕДОВАНИЯ
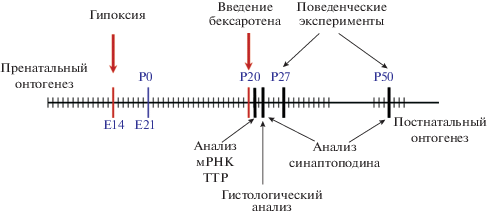
Оценка памяти животных. В норме интактные крысы (как молодые, так и взрослые) через 60 мин после тренировочного предъявления первой пары предметов и замены одного из них на новый уделяют больше внимания исследованию нового объекта. Так, в наших экспериментах время исследования нового объекта составляло 57.6 + 2.9% у молодых и 57.6 + 2.3% у взрослых контрольных крыс и достоверно отличалось (tк = 2,61 и tк = 3.22 соответственно) от 50%-го уровня, указывающего на отсутствие предпочтения времени обследования одного из пары предъявляемых объектов (рис. 2). Животные соответствующего возраста, перенесшие пренатальную гипоксию, уделяли одинаковое время исследованию знакомого и нового объектов, что свидетельствует о нарушении памяти (рис. 2). Введение бексаротена крысам, перенесшим пренатальную гипоксию, приводило к увеличению времени обследования новых объектов как через неделю (до 62.6 + 5.0%), так и через месяц после инъекций (до 60.3 + 3.4%). Это время достоверно отличалось от 50%-го уровня (tГ + Bex = 2.51, tГ + Bex = 3.06 соответственно) и не отличалось от контроля (рис. 2), что указывает на восстановление памяти у крыс, перенесших пренатальную гипоксию, после введения бексаротена.
Рис. 2.
Влияние пренатальной гипоксии и инъекции бексаротена на память у молодых (Р27) и взрослых (Р50) крыс. Анализ памяти у крыс в тесте распознавания нового объекта в группах: К – интактный контроль (n = 35); Г – крысы, перенесшие пренатальную гипоксию на Е14 (n = 32); Г + Bex (n = 15) – крысы, перенесшие пренатальную гипоксию на Е14 и получившие на Р20 инъекцию бексаротена. По оси ординат – среднее значение (± ошибка среднего) времени распознавания нового объекта, выраженное в процентах от суммарного времени исследования известного и нового объектов, принятого за 100%. * – р < 0.05, ** – р < 0.01 (одновыборочный t-критерий) – статистически достоверные различия между временем распознавания нового объекта и 50%-ным уровнем обследования, обозначенным горизонтальной линией и свидетельствующим об отсутствии предпочтения между новым и известным объектами.
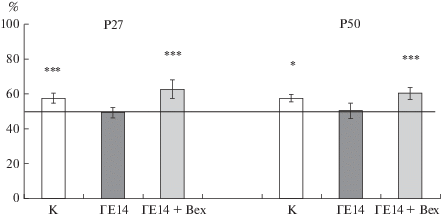
Состояние клеток поля СА1 гиппокампа. Светооптическое исследование препаратов нервной ткани головного мозга крыс, перенесших пренатальную гипоксию, показало, что у части пирамидных нейронов поля СА1 гиппокампа имело место набухание клеточных тел и появление неокрашенных участков цитоплазмы (рис. 3А, 3Б). На 3‑и сутки после введения бексаротена у крыс, перенесших пренатальную гипоксию, также наблюдались такие измененные нейроны (рис. 3В). Морфометрическое исследование на Р23 пирамидного слоя поля СА1 гиппокампа выявило снижение количества клеток у крыс, перенесших пренатальную гипоксию, которое составляло 80.0 ± 1.0% от уровня контроля. У крыс, которым вводили бексаротен, количество клеток также было снижено и составляло 81.2 ± 3.5% относительно контроля (рис. 3Г). Таким образом, введение бексаротена не приводило к изменению числа пирамидных нейронов поля СА1 гиппокампа у животных, перенесших пренатальную гипоксию (p = 0.72).
Рис. 3.
Исследование структурных изменений в поле СА1 гиппокампа у контрольных и экспериментальных крыс. А–В – микрофотографии ткани поля СА1 гиппокампа на Р23: А – интактного контроля (К); Б – перенесших пренатальную гипоксию на Е14 (Г); В – перенесших пренатальную гипоксию через 3-е суток после введения бексаротена на Р20 (Г + Bex). Окраска по Нисслю, масштаб 15 мкм. Стрелками отмечены изменённые клетки; Г – cреднее количество клеток пирамидного слоя поля СА1 гиппокампа крыс: интактный контроль (К, n = 12); перенесшие пренатальную гипоксию (Г, n = 10); перенесшие пренатальную гипоксию и введение бексаротена на Р20 (Г + Bex, n = 5). Данные представлены в виде среднего ± SEM. * – статистически значимое отличие от контроля (one-way ANOVA, Dunnet post-hoc, p < 0.01). Нет различий между Г и Г + Bex (p = 0.72).
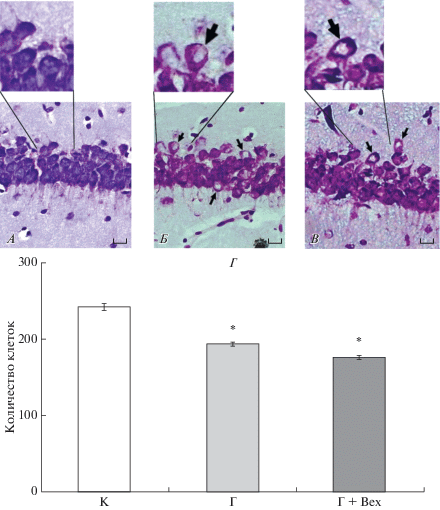
Анализ числа синаптоподин-позитивных дендритных шипиков в stratum radiatum-moleculare поля СА1. В ходе иммуногистохимического исследования распределения актин-ассоциированного белка шипикового аппарата синаптоподина было выявлено, что число лабильных синаптоподин-позитивных шипиков в stratum radiatum-moleculare поля СА1 гиппокампа молодых крыс (Р23), перенесших пренатальную гипоксию, составляло 77.3 ± 3.6% от уровня, наблюдаемого у контрольных животных. У взрослых крыс (Р50), перенесших пренатальную гипоксию, различия между контрольными и экспериментальными животными были еще больше: среднее число синаптоподин-позитивных шипиков у “гипоксических” крыс составляло 67.4 ± 3.8% от уровня контроля (one-way ANOVA, Dunnet post-hoc, FDunnet = 4,91; p < 0.01; рис. 4Б). Через три дня после введения бексаротена крысам, перенесшим пренатальную гипоксию, число лабильных дендритных шипиков статистически не отличалось от их количества в группе “гипоксических” животных того же возраста, не получавших данный препарат (one-way ANOVA, Dunnet post-hoc, FDunnet = 1.27; p = 0.84; рис. 4Б). Однако через месяц после инъекции бексаротена у крыс, перенесших пренатальную гипоксию, наблюдалось достоверное увеличение числа лабильных дендритных шипиков на 27.4 ± 6.7% относительно животных, не получавших этот препарат (one-way ANOVA, Holm-Sidak post-hoc, FHolm-Sidak = 3.63; p = 0.047).
Рис. 4.
Распределение белка синаптоподина в нейропиле stratum radiatum-moleculare поля СА1 гиппокампа. А – иммуногистохимическое окрашивание белка шипикового аппарата синаптоподина (зеленый цвет) в stratum radiatum-moleculare поля СА1 гиппокампа взрослых крыс. К – интактный контроль; Г – животные, перенесшие пренатальную гипоксию на Е14; Г + Вех – перенесшие пренатальную гипоксию животные через месяц после введения бексаротена. Масштаб 10 мкм. Места окрашивания соответствуют синаптоподин-позитивным дендритным шипикам на апикальных дендритах проекционных нейронов stratum pyramidale. Б – Результаты количественного анализа. Число шипиков подсчитывали на площади 10000 мкм2 и вычисляли среднее значение по 10 срезам для каждого животного в группах: К – интактный контроль (по n = 8 для каждого возраста); Г – животные, перенесшие пренатальную гипоксию на Е14 (по n = 8 для каждого возраста); Г + Вех – перенесшие пренатальную гипоксию животные через 3 дня и месяц после введения бексаротена (по n = 6 для каждого возраста). Возраст крыс указан над графиками. Данные представлены в виде среднего ± SEM. * – статистически значимое отличие от контроля (one-way ANOVA, Dunnet post-hoc, p < 0.01), # – статистически значимое отличие между группами Г и Г + Bex (one-way ANOVA, Holm-Sidak post-hoc, p = 0.047).
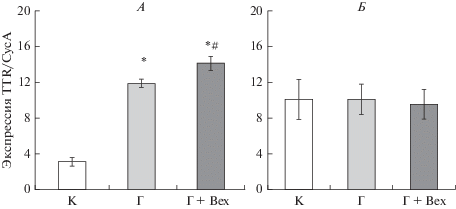
Анализ содержания мРНК TTР в гиппокампе и сосудистом сплетении мозга крыс. Содержание мРНК TTР в гиппокампе мозга крыс, перенесших пренатальную гипоксию, не отличалось от контрольных величин (рис. 5Б), тогда как в сосудистом сплетении оно было в 3.9 раз выше по сравнению с контрольными животными (рис. 5А). Через 24 ч после введения бексаротена уровень экспрессии мРНК TTР в сосудистом сплетении повышался на 18% (рис. 5А), в то время как в гиппокампе он оставался неизменным (рис. 5Б).
Рис. 5.
Изменение содержания мРНК ТТР в сосудистом сплетении и гиппокампе мозга крыс. Метод ртПЦР. По оси абсцисс - группы крыс на Р21: интактный контроль (К, n = 8), перенесшие пренатальную гипоксию (Г, n = 8), перенесшие пренатальную гипоксию и введение бексаротена на Р20 (Г + Bex, n = 6). По оси ординат – отношение содержания мРНК ТТР/CycA для сосудистого сплетения (А) и гиппокампа (Б). Данные представлены в виде среднего ± SEM. * – статистически значимое отличие от контроля (Mann–Witney U-test, p < 0.01), # – статистически значимое отличие между группами Г и Г + Bex (Mann–Witney U-test, p < 0.05).
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Несмотря на то, что в течение последних 30 лет ведется интенсивный поиск терапевтических подходов к лечению болезни Альцгеймера, все еще нет эффективных средств для борьбы с данным заболеванием [23]. Одной из причин неудачных попыток создания эффективных лекарственных препаратов является многофакторный патогенез болезни Альцгеймера, который включает в себя как генетические компоненты (при семейных формах заболевания), так и нарушения васкулярной системы мозга, приводящие к ишемии и инсультам, а также нейровоспаление и диабет [24]. Более того, различные патологии эмбрионального развития, включая пренатальную гипоксию, также приводят к нарушению развития мозга, нейродегенерации и раннему когнитивному дефициту [25]. В связи с этим большое значение имеет создание моделей ранних когнитивных нарушений, которые позволяют не только изучать молекулярные механизмы гибели нервных клеток, приводящей к деменции, но также осуществлять тестирование терапевтической эффективности различных соединений. Разработанная нами модель пренатальной гипоксии у крыс является удобным инструментом для таких исследований [3]. С ее использованием была показана эффективность применения вальпроата натрия и ингибиторов каспаз как для улучшения когнитивных функций животных, так и для повышения активности амилоид-деградирующего фермента неприлизина, уровень экспрессии которого существенно снижается в коре и гиппокампе не только с возрастом, но и после пренатальной гипоксии и ишемии мозга [6, 26–28].
К числу терапевтических средств, предложенных недавно для снижения уровня Аβ и устранения когнитивного дефицита при болезни Альцгеймера, относится агонист ядерных X ретиноидных рецепторов (RXR) бексаротен [17, 29, 30], хотя эффекты его применения не всегда воспроизводимы [31]. На молекулярном уровне бексаротен, помимо стимуляции ядерных Х ретиноидных рецепторов, также способен блокировать кальций-проницаемые ионные каналы, образуемые Аβ, благодаря его способности связываться с Аβ [32]. Защитный эффект бексаротена против повреждающего действия Аβ также может происходить через инсулин-опосредованные сигнальные пути [33]. Как было показано нами на клетках нейробластомы SH-SY5Y, помимо неприлизина бексаротен также может повышать экспрессию инсулин-деградирующего фермента, способного катаболизировать Аβ [12]. В этом случае эффект бексаротена осуществляется, вероятнее всего, через сигнальные системы, приводящие к снижению связывания и активности деацетилазы гистонов HDAC1 в районе промоторов этих генов. С помощью анализа экспрессии генов в ткани мозга мышей, используемых в качестве модели болезни Альцгеймера, также было показано, что бексаротен способен повышать экспрессию АроЕ и целого ряда других нейрональных генов, продукты которых вовлечены в клиренс Аβ, включая ТТР [29].
В данной работе на крысах, перенесших пренатальную гипоксию и характеризующихся ранним когнитивным дефицитом, однократная инъекция бексаротена в желудочки мозга в раннем онтогенезе (на Р20) приводила к улучшению памяти в тесте “Распознавание новых объектов”. Данный эффект наблюдался при тестировании крыс как через неделю, так и через месяц после инъекции бексаротена, что свидетельствует о длительном характере изменений, вызванных этим соединением.
Для выяснения механизмов влияния бексаротена на улучшение памяти у крыс, перенесших пренатальную гипоксию, нами был проведен морфологический и морфометрический анализ нервной ткани гиппокампа. Через месяц после введения бексаротена развивающимся крысам, перенесшим пренатальную гипоксию, в гиппокампе было выявлено увеличение числа синаптоподин-позитивных лабильных дендритных шипиков при неизменном количестве нейронов. Это может свидетельствовать о повышении нейрональной пластичности в данной структуре мозга [1, 3], лежащей в основе улучшения памяти у взрослых животных. Полученные данные согласуются с имеющимися представлениями о том, что бексаротен, связываясь с RXR, оказывает влияние на сигнальные пути, регулирующие нейрогенез, развитие нейрональных отростков и их структуру [34]. Этими же авторами было показано, что введение бексаротена мышам, используемым в качестве модели болезни Альцгеймера, и экспрессирующим аполипопротеины человека AроE3 и AроE4, приводит к увеличению числа стволовых клеток в зубчатой извилине гиппокампа. На нашей модели бексаротен не оказывал существенного влияния на число клеток пирамидного слоя поля СА1. Следует, однако, иметь в виду, что в исследованиях, проводимых другими авторами, использовались протоколы более длительного введения бексаротена. Мы допускаем, что и на нашей модели более длительное введение бексаротена может приводить к изменению клеточного состава гиппокампа.
Поскольку экспрессия генов NEP и TTR в нейрональных клетках регулируется сходным образом [14], а бексаротен способен повышать экспрессию неприлизина и инсулин-деградирущего фермента, также представляло интерес проведение сравнительного анализа влияния этого препарата на уровень содержания мРНК ТТР. Согласно нашим данным, у нормально развивающихся крыс наиболее высокое содержание ТТР наблюдается в сосудистом сплетении, в то время как в теменной коре и гиппокампе уровень его экспрессии относительно низкий. Это согласуется с представлениями о том, что в сосудистом сплетении ТТР составляет около 12% от уровня всех белков и его содержание на порядок выше, чем в печени [35]. Вновь синтезируемый в сосудистом сплетении ТТР секретируется в желудочки мозга и транспортируется к разным участкам мозга. Поскольку ТТР является основным переносчиком тиреоидных гормонов, в частности Т4, повышение его экспрессии после пренатальной гипоксии как на уровне мРНК, показанное в данной работе, так и на уровне белка, описанное нами ранее [36], говорит об острой необходимости в увеличении его содержания в патологически развивающейся ткани мозга. Вероятно, это связано с повышением апоптотических процессов в формирующейся ткани мозга, вызванных избыточной активацией каспаз после пренатальной гипоксии [27] и необходимостью как выведения из ткани мозга большего количества переработанных пептидов, так и транспорта регуляторных молекул для восстановления поврежденной нервной ткани. На это указывает способность ТТР усиливать процессы регенерации нервов [37]. С другой стороны, в ходе старения и при ранних когнитивных нарушениях, вызванных накоплением Аβ, ТТР может регулировать поддержание нормальных когнитивных функций посредством активации ретиноидных сигнальных путей в гиппокампе [16]. Важно отметить, что эффект бексаротена на повышение экспрессии TTР в сосудистом сплетении мозга крыс, перенесших пренатальную гипоксию, показан нами впервые. Это открывает возможность дальнейших исследований способности бексаротена предотвращать нарушения памяти в процессе старения и при развитии деменции, а также для выяснения молекулярных механизмов, регулирующих эти процессы.
Список литературы
Zhuravin I.A., Tumanova N.L., Vasil’ev D.S. Structural changes of the hippocampus nervous tissue in rat ontogenesis after prenatal hypoxia. J. Evol. Biochem. Physiol. 45 (1): 156–158. 2009. doi 10.1134/S0022093009010165
Dubrovskaya N.M., Zhuravin I.A. Ontogenetic characteristics of behavior in rats subjected to hypoxia on day 14 or day 18 of embryogenesis. Neurosci. Behav. Physiol. 40(2): 231–238. 2010. doi 10.1007/s11055-009-9235-2
Zhuravin I.A., Dubrovskaya N.M., Tumanova N.L., Vasilev D.S., Nalivaeva N.N. Ontogenetic and phylogenetic approaches for studying the mechanisms of cognitive dysfunctions. Evolutionary physiology and biochemistry: advances and perspectives. Intech: rijeka, croatia. Chapter 15: 205–224. 2018. doi 10.5772/intechopen.73666.10.5772/intechopen.73666
Nalivaeva N.N., Belyaev N.D., Zhuravin I.A., Turner A.J. The Alzheimer’s amyloid-degrading peptidase, neprilysin: can we control it? Int. J. Alzheimers Dis. 2012: 383 796. 2012. doi 10.1155/2012/383796
Dubrovskaya N.M., Nalivaeva N.N., Vasilev D.S., Bagrova D.I., Zhuravin I.A. Mechanisms of short-term working memory deficit. In: Short-term memory: new research. Nova science publishers, inc., n.y. chapter 6: 155–173. 2012. www.novapublishers.com.
Nalivaeva N.N., Belyaev N.D., Lewis D.I., Pickles A.R., Makova N.Z., Bagrova D.I., Dubrovskaya N.M., Plesneva S.A., Zhuravin I.A., Turner A.J. Effect of sodium valproate administration on brain neprilysin expression and memory in rats. J. Mol. Neurosci. 46: 569–577. 2012. doi 10.1007/s12031-011-9644-x
Gosztyla M.L., Brothers H.M., Robinson S.R. Alzheimer’s amyloid-β is an antimicrobial peptide: a review of the evidence. J. Alzheimers Dis. 62: 1495–1506. 2018. doi 10.3233/JAD-171133
Bell R.D., Sagare A.P., Friedman A.E., Bedi G.S., Holtzman D.M., Deane R., Zlokovic B.V. Transport pathways for clearance of human Alzheimer’s amyloid beta-peptide and apolipoproteins E and J in the mouse central nervous system. J. Cereb. Blood. Flow. Metab. 27: 909–918. 2007. doi 10.1038/sj.jcbfm.9600419
Nalivaeva N.N., Belyaev N.D., Kerridge C., Turner A.J. Amyloid-clearing proteins and their epigenetic regulation as a therapeutic target in Alzheimer’s disease. Front. Aging Neurosci. 6: 235. 2014. doi 10.3389/fnagi.2014.00235
Li X., Buxbaum J.N. Transthyretin and the brain re-visited: Is neuronal synthesis of transthyretin protective in Alzheimer’s disease? Mol. Neurodegener. 6: 79. 2011. doi 10.1186/1750-1326-6-79
Wildsmith K.R., Holley M., Savage J.C., Skerrett R., Landreth G.E. Evidence for impaired amyloid β clearance in Alzheimer’s disease. Alzheimers Res. Ther. 5: 33–38. 2013. doi 10.1186/alzrt187
Nalivaeva N.N., Belyaev N.D., Turner A.J. New insights into epigenetic and pharmacological regulation of amyloid-degrading enzymes. Neurochem. Res. 41(3): 620–630. 2016. doi 10.1007/s11064-015-1703-1
Tokuda T., Calero M., Matsubara E., Vidal R., Kumar A., Permanne B., Zlokovic B., Smith J.D., LaDu M.J., Rostagno A., Frangione B., Ghiso J. Lipidation of apolipoprotein E influences its isoform-specific interaction with Alzheimer’s amyloid beta peptides. Biochem. J. 348: 359–365. 2000. doi 3480359.10.1042/0264-6021
Kerridge C., Belyaev N.D., Nalivaeva N.N., Turner A.J. The Aβ-clearance protein transthyretin, like neprilysin, is epigenetically regulated by the amyloid precursor protein intracellular domain. J. Neurochem. 130(3): 419–431. 2014. doi 10.1111/jnc.12680
Escher N., Kob R., Kaatz M., Melle C., Hipler C., von Eggeling F. Confirmation of the biological significance of transthyretin as a biomarker for cutaneous T-cell lymphoma by its protein interaction partners. Mol. Med. Rep. 4: 157–161. 2011. doi 10.3892/mmr.2010.393
Brouillette J., Quirion R. Transthyretin: A key gene involved in the maintenance of memory capacities during aging. Neurobiol. Aging. 29(11): 1721–1732. 2008. doi 10.1016/j.neurobiolaging.2007.04.007
Cramer P., Cirroto J., Wesson D., Lee C. Y., Karlo J., Zinn A., Casali B., Restivo J., Goebel W., James M., Brunden K., Wilson D., Landreth G.E. ApoE-directed therapeutics rapidly clear β-amyloid and reverse deficits in AD mouse models. Science. 23(335): 1503–1506. 2012.
Fitz N.F., Cronican A.A., Lefterov I., Koldamova R. Comment on “ApoE-directed therapeutics rapidly clear beta-amyloid and reverse deficits in AD mouse models. Sci. Tech. Comments. 340(6135): 924-с. doi 2013.10.1126/science.1235809
Huy P.D.Q., Thai N.Q., Bednarikova Z., Phuc L.H., Linh H.Q., Gazova Z., Li M.S. Bexarotene does not clear amyloid β plaques but delays fibril growth: molecular mechanisms. ACS Chem. Neurosci. 8: 1960–1969. 2017. doi 10.1021/acschemneuro.7b00107
Paxinos G., Watson C. The rat brain in stereotaxic coordinates. San Diego. Acad. Press. 2005.
Mello P.B., Benetti F., Cammarota M., Izquierdo I. Effects of acute and chronic physical exercise and stress on different types of memory in rats. Ann. Acad. Bras. Cienc. 80: 301–309. 2008. doi 10.1016/j.ijdevneu.2008.09.200
Livak K.J., Schmittgen T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C (T)) method. Methods. 25(4): 402–408. 2001. doi 10.1006/meth.2001.1262
Karran E., de Strooper B. The amyloid cascade hypothesis: are we poised for success or failure? J. Neurochem. 139 (Suppl 2): 237–252. 2016. doi 10.1111/jnc.13632
Villegas-Llerena C., Phillips A., Garcia-Reitboeck P., Hardy J., Pocock J. M. Microglial genes regulating neuroinflammation in the progression of Alzheimer’s disease. Curr. Opin. Neurobiol. 36: 74–81. 2016. doi 10.1016/j.conb.2015.10.004
Nalivaeva N.N., Turner A.J., Zhuravin I.A. Role of prenatal hypoxia in brain development, cognitive functions and neurodegeneration. Front Neurosci. 12: 825. 2018. doi 10.3389/fnins.2018.00825
Zhuravin I.A., Dubrovskaya N.M., Vasilev D.S., Tumanova N.L., Nalivaeva N.N. Epigenetic and pharmacological regulation of the amyloid-degrading enzyme neprilysin results in modulation of cognitive functions in mammals. Dokl. Biol. Sci. 438: 145–8. 2011. doi 10.1134/S001249661103015X
Vasilev D.S., Dubrovskaya N.M., Nalivaeva N.N., Zhuravin I.A. Regulation of Caspase-3 content and activity in rat cortex in norm and after prenatal hypoxia. Neurochem. J. 10(2) 144–150. 2016. doi 10.1134/S1819712416020100
Dubrovskaya N.M., Tikhonravov D.L., Alekseeva O.S., Zhuravin I.A. Recovery of learning and memory impaired by prenatal hypoxic stress in rats after injection of caspase-3 inhibitor during early ontogenesis. J. Evol. Biochem. Physiol. 53(1): 66–68. 2017. doi 10.1134/S0022093017010082
Lefterov I., Schug J., Mounier A., Nam K.N., Fitz N.F., Koldamova R. RNA-sequencing reveals transcriptional up-regulation of Trem2 in response to bexarotene treatment. Neurobiol. Dis. 82: 132–140. 2015. doi 10.1016/j.nbd.2015.05.019
Mariani M.M., Malm T., Lamb R., Jay TR., Neilson L., Casali B., Medarametla L., Landreth G.E. Neuronally-directed effects of RXR activation in a mouse model of Alzheimer’s disease. Sci. Rep. 7: 42270. 2017. doi 10.1038/srep42270
LaClair K.D., Manaye K.F., Lee D.L., Allard J.S., Savonenko A.V., Troncoso J.C., Wong Ph. C. Treatment with bexarotene, a compound that increases apolipoprotein-E, provides no cognitive benefit in mutant APP/PS1 mice. Mol. Neurodegener. 8: 18. 2013. http: //www.molecularneurodegeneration.com/content/8/1/18.
Fantini J., Di Scala C., Yahi N., Troadec J. D., Sadelli K., Chahinian H., Garmy N. Bexarotene blocks calcium-permeable ion channels formed by neurotoxic Alzheimer’s β-amyloid peptides. ACS Chem. Neurosci. 5(3): 216–224. 2014. doi 10.1021/cn400183w
Dai W., Yang J., Chen T., Yang Z. Protective effects of bexarotene against amyloid-β25-35-induced dysfunction in hippocampal neurons through the insulin signaling pathway. Neurodegener. Dis. 14(2): 77–84. 2014. doi 10.1159/000358397
Mounier A., Georgiev D., Nam K.N., Fitz N.F., Castranio E.L., Wolfe C.M., Cronican A.A., Schug J., Lefterov I., Koldamova R. Bexarotene-activated retinoid X receptors regulate neuronal differentiation and dendritic complexity. J. Neurosci. 35: 11862–11876. 2015. doi 10.1523/JNEUROSCI.1001-15.2015
Schreiber G. The evolution of transthyretin synthesis in the choroid plexus. Clin. Chem. Lab. Med. 40(12): 1200–1210. 2002. doi 10.1515/CCLM.2002.210
Васильев Д.С., Туманова Н.Л., Дубровская Н.М., Алексеева О.С., Наливаева Н.Н., Журавин И.А. Пренатальная гипоксия вызывает усиление выработки белка транстиретин в сосудистом сплетении крыс в первый месяц после рождения. Рос. физиол. журн. им И.М. Сеченова. 104(6): 635–639. 2018. doi: 10.7868/S0869813918060035 [Vasilev D.S., Tumanova NL, Dubrovskaya N.M., Alekseeva O.S., Nalivaeva N.N., Zhuravin. I.A. Prenatal hypoxia causes an increase in transthyretin protein production in the vascular plexus of rats in the first month after birth. Russ. J. Physiol. 104 (6): 635–639. 2018. (In Russ.)]
Fleming C.E., Saraiva M.J., Sousa M.M. Transthyretin enhances nerve regeneration. J. Neurochem. 103(2): 831–839. 2007. doi 10.1111/j.1471-4159.2007.04828.x
Дополнительные материалы отсутствуют.
Инструменты
Российский физиологический журнал им. И.М. Сеченова