

Российский физиологический журнал им. И.М. Сеченова, 2021, T. 107, № 4-5, стр. 629-640
Роль рианодиновых и IP3-рецепторов в генерации кальциевых ответов, вызываемых трициклическими антидепрессантами в нейронах неокортекса крысы
С. И. Бойков 1, Д. А. Сибаров 1, *, Т. В. Карелина 1, Н. Н. Шестакова 1, С. М. Антонов 1
1 Институт эволюционной физиологии и биохимии им. И.М. Сеченова РАН
Санкт-Петербург, Россия
* E-mail: dsibarov@gmail.com
Поступила в редакцию 15.01.2021
После доработки 31.01.2021
Принята к публикации 31.01.2021
Аннотация
Трициклические антидепрессанты, в частности, амитриптилин (ATL) и дезипрамин (DES), применяются в настоящее время для терапии депрессий и лечения хронических болей различного происхождения, важную роль в которых играют дисфункции NMDA-рецепторов. Известно влияние терапевтических концентраций ATL на кальций-зависимую десенситизацию NMDA-рецепторов, управляемую уровнем свободного кальция в цитоплазме. К тому же в кардиомиоцитах ATL и DES могут вызывать выброс кальция в цитоплазму из внутриклеточных депо за счет открывания каналов инозитол-3-фосфатных рецепторов (IP3R) и/или рианодиновых рецепторов (RyR). Данный аспект действия этих препаратов на нейроны остается плохо исследованным. На нейронах неокортекса крыс в первичной культуре ткани мы изучили зависимость кальциевого ответа на DES и ATL от активации IP3R и RyR эндоплазматического ретикулума и митохондрий. Кратковременные (30 с) парные (с интервалом 5 мин) аппликации 200 мкМ DES или 200 мкМ ATL вызывают в нейронах коры кальциевые ответы, не различающиеся по величине. Использование антагонистов RyR и IP3R показало, что ответы на ATL блокируются антагонистом IP3R – 2-APB (100 мкМ), а ответы на DES блокируются рианодином – антагонистом RyR (100 нМ). Поскольку в нейронах RyR и IP3R распределены негомогенно, можно предполагать, что DES и ATL стимулируют высвобождение разных пулов депонированного кальция, сосредоточенных либо в разных сегментах ретикулума, либо в ретикулуме и митохондриях. Кроме того, ATL и DES, будучи каналоблокаторами NMDA-рецепторов, ингибировали вход кальция извне клетки через активированные NMDA-рецепторы. Учитывая высокие концентрации DES и ATL (более 100 мкМ), необходимые для стимуляции выброса депонированного кальция в нейронах, представляется маловероятным, что подобные эффекты проявляются при их терапевтическом действии. Тем не менее, обнаруженная специфичность DES и ATL в отношении RyR и IP3R соответственно может использоваться в качестве инструмента в экспериментальных целях.
Трициклические антидепрессанты (TCA) применяются в настоящее время как для терапии депрессий, так и для лечения хронических болей различного происхождения (невралгии, нейропатические боли, фибромиалгии и диабетической нейропатии [1–3]). Амитриптилин (ATL) принадлежит к TCA и в терапевтических концентрациях (до 3 мкМ) имеет широкий спектр эффектов на ионные каналы и транспортеры, действуя, например, как ингибитор транспортера серотонина [4], рецептора серотонина 5-HT2C [5], рецептора гистамина H1 [6] и другие. Известно также, что ATL в концентрациях >10 мкМ способен блокировать каналы NMDA-рецепторов [7–9]. К тому же недавно продемонстрировано кальций-зависимое ингибирование активности NMDA-рецепторов амитриптилином, которое не связано с каналоблокадой, но при физиологических концентрациях внеклеточного кальция может иметь терапевтическое значение при лечении нейропатической боли и проявляться в терапевтических дозах ATL (<1 мкМ) [9]. Предполагается, что ATL способен взаимодействовать с натрий-кальциевым обменником [10, 11], ингибирование активности которого провоцирует, подобно действию низких доз этанола [12], кальций-зависимую десенситизацию NMDA-рецепторов за счет накопления кальция внутри нейронов [13, 14]. Дезипрамин (DES), сходный по химической структуре с ATL, преимущественно ингибирует захват норадреналина [15], однако также блокирует каналы NMDA-рецепторов [8]. Следует отметить, что ATL при блокировании каналов NMDA-рецепторов может проходить через каналы внутрь клетки, хотя вероятность проникновения имеет очень низкое значение (<0.05) [9].
Таким образом, можно предполагать, что ТСА, в частности ATL, влияют на кальций-зависимую десенситизацию рецепторов, которая, в свою очередь, модулируется уровнем свободного кальция в примембранной области цитоплазмы [16]. Известны два источника повышения концентрации свободного кальция в нейронах. Кальций может поступать в цитоплазму извне через кальций-проницаемые ионные каналы, а также освобождаться из внутриклеточных депо (эндоплазматического или саркоплазматического ретикулумов, митохондрий, аппарата Гольджи). Последнее происходит за счет открывания каналов инозитол-3-фосфатных рецепторов (IP3R) и/или рианодиновых рецепторов (RyR). Интересно, что ATL уже в концентрации 10 мкМ может ослаблять сократимость сердца, нарушая прохождение кальциевых волн, а в более высоких концентрациях вызывает максимальный выброс Сa2+ из саркоплазматического ретикулума [17]. Многие TCA, в частности, амитриптилин, имипрамин и дезипрамин, вызывают временное повышение уровня свободного внутриклеточного кальция ([Ca2+]i) в культуре нейронов неокортекса в концентрации от 100 мкМ до 1 мМ [18]. Все три TCA увеличивают содержание инозитол-1,4,5-трифосфата (IP3) в клетках, т.е. мобилизуют Ca2+ из IP3-чувствительных депо, причем, независимо от цАМФ [18]. В кардиомиоцитах ATL активирует RyR2 в концентрации 0.5–3 мкМ, вызывая выброс кальция из саркоплазматического ретикулума [19], а также ингибирование захвата Ca2+ в депо [17].
Несмотря на то, что влияние TCA на высвобождение кальция из внутриклеточных депо в кардиомиоцитах ранее было изучено, подобный аспект действия этих препаратов на клетки, являющиеся основными мишенями ТСА в ЦНС, – нейроны, остается плохо исследованным. Очевидно, что нейроны значительно отличаются от кардиомиоцитов по многим биохимическим параметрам, включая экспрессию различных подтипов IP3R и RyR [20]. Хотя эффекты ATL на IP3R описаны на многих типах клеток, действие ATL на RyR в нейронах ранее не изучалось. Остаются неизвестными также эффекты DES на RyR. В настоящей работе, проведенной на нейронах коры большого мозга крыс, мы изучили кальциевые ответы нейронов на DES и ATL и их зависимость от активации IP3R и RyR эндоплазматического ретикулума и митохондрий.
МЕТОДЫ ИССЛЕДОВАНИЯ
Крыс Вистар (всего 20 беременных самок) подвергали эвтаназии путем 1-минутной ингаляции углекислого газа. Эмбрионы использовали для приготовления первичных культур кортикальных нейронов с использованием стандартных процедур, описанных ранее [21, 22]. Процедуры соответствовали требованиям по работе с лабораторными животными Federation of European Laboratory Animal Science Associations и протоколом, утвержденным этическим комитетом Института эволюционной физиологии и биохимии им. И.М. Сеченова РАН, находящимся в соответствии с национальными и международными требованиями. Выращивание культуры нейронов в среде Neurobasal с добавкой B-27 (Gibco-Invitrogen, Великобритания) осуществляли на покровных стеклах, покрытых поли-D-лизином. Культуры использовали для экспериментов через 10–16 дней инкубации in vitro.
В опытах использовали внеклеточный перфузионный раствор следующего состава (концентрации указаны в мМ): 144 NaCl; 2.8 KCl; 10 HEPES, при pH 7.2–7.4 и температуре 22–24°C. Бескальциевый раствор применяли, чтобы исключить влияние входа кальция в цитоплазму из внеклеточной среды. В экспериментах с использованием NMDA во внеклеточный раствор добавляли 1 мМ CaCl2. Непосредственно перед экспериментом в цитоплазму нейронов загружали флуоресцентный кальций-чувствительный краситель Fluo-8. Для этого нейроны инкубировали в растворе, содержащем 2 мкМ ацетоксиметилового эфира Fluo-8 (Fluo-8 AM), при комнатной температуре в течение 60 мин. Клетки отмывали от красителя путем 20-минутной инкубации в перфузионном растворе, затем покровные стекла переносили в инвертированный микроскоп Leica TCS SP5 MP (Leica Microsystems, GmbH, Wetzlar, Германия). Ванночку для визуализации постоянно перфузировали при скорости потока 1.2 мл/мин тем же раствором. Быструю смену раствора в области наблюдения осуществляли с помощью системы быстрой локальной перфузии (ALA Science, BPS-8). Флуоресценцию Fluo-8 возбуждали лазером 488 нм и регистрировали в спектральном диапазоне 510–560 нм с интервалом дискретизации ~2 с (кадр 1024 × 1024 пикселей, объектив 20×).
Для индукции внутриклеточных кальциевых ответов использовали 30-секундные аппликации DES или ATL отдельно или в сочетании с ингибиторами IP3-зависимого (2-аминоэтоксидифенил-борат, 2-APB, 100 мкМ) или рианодин-зависимого (рианодин, 100 нМ) выхода кальция из внутриклеточных депо. Выбор в пользу 2-APB для ингибирования IP3R в наших экспериментах относительно других ингибиторов, в частности, гепарина и кофеина [23], обусловлен мембранной проницаемостью 2-APB [24], действием на все подтипы IP3R и отсутствием эффектов на RyR [25], что присуще гепарину и кофеину [26, 27]. Кроме того, эффект 2-APB на депо-зависимый вход кальция в клетку [28–30] не проявляется в бескальциевом внеклеточном растворе.
Для анализа кальциевых ответов оценивали изменения интенсивности свечения тел отдельных нейронов. Для каждой клетки оценивали выход кальция в цитоплазму из внутриклеточных депо как интеграл превышения свечения над базовой линией (F) за время аппликации фармакологических агентов. Для статистического анализа за один опыт (n = 1) принято среднее значение кальций-индуцированного свечения для всех клеток с одного стекла с культурой нейронов. Для сравнения групп, состоящих из нескольких опытов, применяли парный критерий Стьюдента. Достоверность различий обозначали звездочками, соответствующими различным уровням доверительной вероятности: p < 0.05 (*), p < 0.01 (**) и p < 0.001 (***).
РЕЗУЛЬТАТЫ ИССЛЕДОВАНИЯ
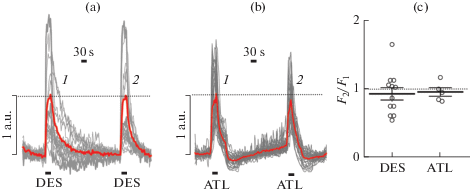
В каждом опыте использовали две 30-секундные аппликации ATL или DES, разделенные интервалом не менее 5 мин. Оказалось, что времени в 5 мин после первой аппликации достаточно для возвращения уровня [Ca2+]i к базовым значениям (рис. 1a, b). Кроме того, амплитуда кальциевого ответа при повторной аппликации достоверно не отличалась от таковой при первой аппликации (рис. 1c) как в случае DES (p = 0.46; n = 11), так и в случае ATL (p = 0.44; n = 5). Оба TCA вызывали кальциевые ответы, превышающие базовый уровень кальция в 3.8 ± 0.3 раза (n = 26) для DES и в 3.9 ± 0.6 раза (n = 21) для ATL. Таким образом, эффект аппликаций ATL или DES был обратим и в условиях использованного протокола не вызывал “истощения” запасов депонированного кальция. В связи с этим в следующей серии опытов во время второй аппликации мы комбинировали ATL или DES с фармакологическими блокаторами IP3-рецепторов или рианодиновых рецепторов как главных триггеров выхода кальция из внутриклеточных депо.
Рис. 1.
Кальциевые ответы нейронов на повторяющиеся 30-секундные аппликации 200 мкМ дезипрамина (DES) или 200 мкМ амитриптилина (ATL) в бескальциевом растворе. (a) Примеры кальциевых ответов отдельных нейронов на две аппликации DES с интервалом в 5 мин, полученные в одном опыте. Жирная линия соответствует усредненному флуоресцентному сигналу от тел нейронов в пределах поля зрения. (b) Опыт аналогичный (A), но при аппликациях ATL. (c) Усредненная величина второго кальциевого ответа на DES и ATL (F2) нормализованая на величину сигнала при первой аппликации (F1). Кружки соответствуют отдельным опытам. Показано среднее значение и стандартная ошибка среднего.
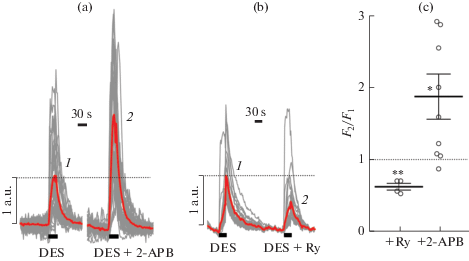
Кальциевый ответ нейронов на DES достоверно не подавлялся антагонистом IP3-рецепторов 2-APB (рис. 2a). Более того, 2-APB парадоксальным образом усиливал ответ нейронов на DES (p = 0.025; n = 9) (рис. 2c). Ингибирование рианодиновых рецепторов (RyR), напротив, достоверно подавляло кальциевые ответы на DES (p = 0.004; n = 4) (рис. 2b, c). Таким образом, кальциевые ответы на DES преимущественно зависят от активации RyR, но не IP3-рецепторов.
Рис. 2.
Модуляция кальциевых ответов нейронов на аппликации 200 мкМ дезипрамина (DES) антагонистами IP3-рецепторов (2-APB, 100 мкМ) и рианодиновых рецепторов (Ry, 100 нМ) в бескальциевом растворе. (a) Пример отдельного опыта, показывающего кальциевые ответы отдельных нейронов на DES и DES в присутствии 2-APB. Жирная линия соответствует усредненному флуоресцентному сигналу от тел нейронов в пределах поля зрения. (b) Пример отдельного опыта, показывающего кальциевые ответы отдельных нейронов на DES и DES в присутствии рианодина (Ry). Жирная линия соответствует усредненному флуоресцентному сигналу от тел нейронов в пределах поля зрения. (c) Средние по нескольким опытам значения флуоресцентных ответов нейронов на DES в присутствии антагонистов RyR и IP3R блокаторов (F2), нормализованные относительно флуоресцентных ответов в отсутствие блокаторов (F1, контроль). Кружки соответствуют отдельным опытам. Показано среднее значение и стандартная ошибка среднего. *, ** – значения достоверно отличаются от контроля.
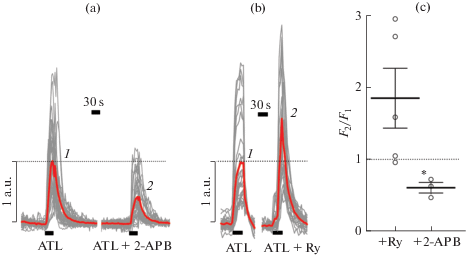
Кальциевый ответ нейронов на ATL подавлялся в присутствии 2-APB, но не рианодина (рис. 3a, b). Достоверное ингибирование кальциевых ответов в присутствии 2-APB (p = 0.033; n = 4) (рис. 3c) указывает на IP3-зависимый характер действия ATL. Несмотря на тенденцию к усилению ответов на ATL рианодином, эффект последнего был недостоверен (p = 0.11; n = 5).
Рис. 3.
Модуляция кальциевых ответов нейронов на аппликации 200 мкМ амитриптилина (ATL) антагонистами IP3-рецепторов (2-APB, 100 мкМ) и рианодиновых рецепторов (Ry, 100 нМ) в бескальциевом растворе. (a) Пример отдельного опыта, показывающего кальциевые ответы отдельных нейронов на ATL и ATL в присутствии 2-APB. Жирная линия соответствует усредненному флуоресцентному сигналу от тел нейронов в пределах поля зрения. (b) Пример отдельного опыта, показывающего кальциевые ответы отдельных нейронов на ATL и ATL в присутствии рианодина (Ry). Жирная линия соответствует усредненному флуоресцентному сигналу от тел нейронов в пределах поля зрения. (c) Средние по нескольким опытам значения флуоресцентных ответов нейронов на ATL в присутствии антагонистов RyR и IP3R блокаторов (F2), нормализованные относительно флуоресцентных ответов в отсутствие блокаторов (F1, контроль). Кружки соответствуют отдельным опытам. Показано среднее значение и стандартная ошибка среднего. * – значение достоверно отличается от контроля.
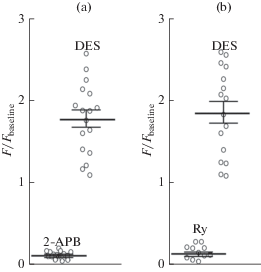
Учитывая обнаруженное в некоторых опытах потенцирование при использовании 2-APB или рианодина кальциевых ответов, были проведены дополнительные опыты, в которых проверяли возможность индуцирования кальциевых ответов нейронов аппликациями 2-APB или рианодина (рис. 4). Нами не выявлено достоверно детектируемых ответов нейронов ни на 30-секундные аппликации 100 мкМ 2-APB (рис. 4a), ни на аппликации 100 нМ рианодина (рис. 4b). При этом DES вызывал отчетливые кальциевые ответы в тех же нейронах. Таким образом, наблюдаемое нами потенцирование кальциевых ответов на DES в присутствии 2-APB или ответов на ATL в присутствии рианодина не связано с индукцией дополнительного кальциевого выброса 2-APB или рианодином.
Рис. 4.
Сравнение амплитуд кальциевых ответов на 30-секундные аппликации 200 мкМ дезипрамина (DES) и 100 мкМ (2-APB) (a) или 100 нМ рианодина (Ry) (b). Fbaseline соответствует яркости свечения нейронов в отсутствие фармакологических агентов. Кружки соответствуют отдельным опытам. Показано среднее значение и стандартная ошибка среднего.
Ввиду того, что DES и ATL являются ингибиторами NMDA-рецепторов [7–9], мы оценили влияние этих веществ на вход кальция в нейроны при активации NMDA-рецепторов 100 мкМ NMDA (+30 мкМ глицина в качестве коагониста) (рис. 5a, b). Опыты проводили во внеклеточном растворе, содержащем 1 мМ Ca2+. Как DES, так и ATL вызывали достоверное подавление входа кальция в нейроны при активации NMDA-рецепторов (рис. 5c), однако, даже сверхвысокие концентрации в 200 мкМ обоих TCA не вызывали полного блокирования входа кальция при действии NMDA. Этот результат был неожиданным, поскольку в электрофизиологических экспериментах такие высокие концентрации ATL и DES почти полностью блокировали токи через NMDA-рецепторы [7, 8]. Возможным объяснением неполного подавления кальциевого сигнала мог быть ранее показанный дополнительный выброс кальция из внутриклеточных депо при действии ATL или DES, проникающих в клетки через каналы NMDA-рецепторов.
Рис. 5.
Ингибирование дезипрамином (DES) и амитриптилином (ATL) входа кальция в нейроны, вызываемого 100 мкМ NMDA (+ глицин 30 мкМ) в присутствии 1 мМ кальция во внеклеточном растворе. Флуоресцентный ответ нейронов на NMDA в отсутствие TCA принят как контрольный (Fcontrol). (a) Пример отдельного опыта, показывающего кальциевые ответы отдельных нейронов на аппликации NMDA отдельно и в комбинации с различными концентрациями дезипрамина (DES). (b) Опыт аналогичный (A), но при аппликациях амитриптилина (ATL). (c) Средние значения величины кальциевых ответов нейронов в присутствии различных концентраций DES или ATL, нормированные относительно контроля. Звездочками показано достоверное подавление TCA входа кальция в цитоплазму через активированные NMDA-рецепторы относительно контроля. (d) Сравнение амплитуд кальциевых ответов на аппликации NMDA и 200 мкМ амитриптилина (ATL). Кружки соответствуют отдельным клеткам. Показано среднее значение и стандартная ошибка среднего. Fbaseline – флуоресценция без NMDA и ATL.
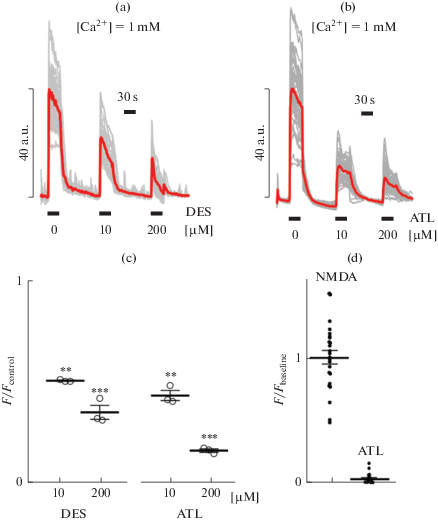
Соотношение кальциевых ответов нейронов на выход депонированного кальция и вход кальция из внеклеточной среды при активации NMDA-рецепторов исследовали в следующей серии экспериментов. Опыты проводили в базовом растворе, содержащем 1 мМ кальция. Амплитуда кальциевых ответов на аппликацию 200 мкМ ATL была в среднем в 40 раз меньше, чем при аппликации 100 мкМ NMDA (+30 мкМ глицина в качестве коагониста) (рис. 5d). Т.е. даже сверхвысокие концентрации ATL не вызывали в нейронах кальциевых ответов, сопоставимых по амплитуде с таковыми при активации NMDA-рецепторов. Тем не менее, неполное блокирование DES и ATL кальциевых ответов, активированных NMDA, по-видимому, может определяться совокупностью факторов, в частности, проникновением блокирующих каналы NMDA-рецепторов молекул в клетку и высвобождением депонированного в эндоплазматическом ретикулуме и митохондриях кальция.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Нами показано, что кратковременные аппликации как ATL, так и DES вызывают в нейронах коры кальциевые ответы, не различающиеся по величине. Однако использование антагонистов RyR и IP3R выявило, что ответы на ATL блокируются антагонистом IP3R – 2-APB (100 мкМ), а ответы на DES блокируются рианодином – антагонистом RyR при применении в концентрации 100 нМ.
Механизм действия ATL на IP3R слабо исследован, однако есть данные о том, что ATL прямо или косвенно стимулирует базовую активность фосфолипазы-C в мембране кортикальных нейронов и вызывает продукцию IP3, что стимулирует IP3-зависимый выход кальция из депо [31]. Кроме того, ATL и некоторые другие TCA связываются с сигма1-рецепторами, которые модулируют IP3-зависимый кальциевый сигналинг [32, 33]. Механизм действия DES на RyR в настоящее время неизвестен.
Распределение RyR и IP3R в пределах внутриклеточных органелл существенно различается [34]. Например, в клетках Пуркинье мозжечка [35], нейронах CA1 гиппокампа [36] RyR и IP3R преимущественно локализованы в разных частях клеток и управляют функционально отдельными пулами депонированного кальция [37]. Различные эффекты антагонистов RyR и IP3R на кальциевые ответы нейронов, вызванные ATL и DES, позволяют предполагать, что эти TCA, возможно, проявляют селективность в отношении различных внутриклеточных кальциевых депо. Поскольку ATL и DES вызывали кальциевые ответы нейронов в отсутствие активации кальций-проницаемых каналов, проникновение их в цитоплазму происходит, вероятно, не через поры ионных каналов, а напрямую через плазматическую мембрану [38].
Эффект потенцирования 2-APB выброса кальция при аппликации DES, вероятно, связан с неспецифическим подавлением захвата кальция митохондриями [28], за счет ингибирования 2-APB кальциевой АТФ-азы (SERCA) [28, 29]. Поэтому в отсутствие индуцированного кальциевого выхода из депо 2-APB не вызывал кальциевых ответов в наших экспериментах. 2-APB не действует на RyR [25], активируемые DES, поэтому при действии DES ингибирование 2-APB захвата кальция усиливает накопление последнего в цитоплазме. Однако кальциевые ответы на ATL IP3-зависимы. Поэтому при подавлении 2-APB вызываемого ATL IP3-зависимого кальциевого выхода побочный эффект на SERCA не играет значимой роли.
В растворе с физиологически нормальным содержанием кальция (1 мМ) флуоресцентный сигнал от вызываемого ТСА выхода кальция из депо был в десятки раз слабее такового при входе кальция из внеклеточной среды в результате активации кальций-проницаемых каналов NMDA-рецепторов. Учитывая концентрации ТСА (более 30 мкМ) необходимые для стимуляции выброса депонированного кальция в нейронах, представляется маловероятным, что подобные эффекты проявляются при их терапевтическом действии. Высокие концентрации ТСА, необходимые для генерации кальциевых ответов, являются отличительной чертой именно нейронов, так как в кардиомиоцитах концентрация всего 10 мкМ ATL полностью блокировала кальциевые волны [17]. Тем не менее, обнаруженная специфичность DES и ATL в отношении RyR и IP3R соответственно может использоваться в качестве инструмента в экспериментальных целях.
ATL и DES, будучи каналоблокаторами NMDA-рецепторов [7–9], ожидаемо ингибировали вход кальция через активированные NMDA-рецепторы. Однако даже в сверхвысоких концентрациях (200 мкМ) оба ТСА не смогли полностью заблокировать кальциевые ответы на NMDA. Остаточный кальциевый сигнал, по-видимому, может определяться совокупностью факторов, к которым относится проникновение ATL через каналы NMDA-рецепторов [9], позволяющий сохранить ничтожно малый вход кальция из внеклеточного раствора, и освобождение депонированного кальция из внутриклеточных органелл. Возможно, имеются и другие составляющие остаточного кальциевого сигнала, понимание которых требует дальнейшего изучения.
Список литературы
Rico-Villademoros F., Slim M., Calandre E.P. (2015) Amitriptyline for the treatment of fibromyalgia: a comprehensive review. Expert. Rev. Neurother. 15(10): 1123–1150. https://doi.org/10.1586/14737175.2015.1091726
Obata H. (2017) Analgesic Mechanisms of antidepressants for neuropathic pain. Int. J. Mol. Sci. 18(11): 2483. https://doi.org/10.3390/ijms18112483
Russell J.W., Zilliox L.A. (2014) Diabetic neuropathies. Peripheral Nervous System Disorders. 1226–1240. https://doi.org/10.1212/01.CON.0000455884.29545.d2
Tatsumi M., Groshan K., Blakely R.D., Richelson E. (1997) Pharmacological profile of antidepressants and related compounds at human monoamine transporters. Eur. J. Pharmacol. 340(2–3): 249–258. https://doi.org/10.1016/s0014-2999(97)01393-9
Cusack B., Nelson A., Richelson E. (1994) Binding of antidepressants to human brain receptors: focus on newer generation compounds. Psychopharmacology. 114(4): 559–565. https://doi.org/10.1007/BF02244985
Appl H., Holzammer T., Dove S., Haen E., Strasser A., Seifert R. (2012) Interactions of recombinant human histamine H1, H2, H3, and H4 receptors with 34 antidepressants and antipsychotics. Naunyn Schmiedebergs Arch. Pharmacol. 385(2): 145–170. https://doi.org/10.1007/s00210-011-0704-0
Tohda M., Urushihara H., Nomura Y. (1995) Inhibitory effects of antidepressants on NMDA-induced currents in Xenopus oocytes injected with rat brain RNA. Neurochem. Internat. 26(1): 53–58. https://doi.org/10.1016/0197-0186(94)00101-y
Barygin O.I., Nagaeva E.I., Tikhonov D.B., Belinskaya D.A., Vanchakova N.P., Shestakova N.N. (2017) Inhibition of the NMDA and AMPA receptor channels by antidepressants and antipsychotics. Brain Res. 1660: 58–66. https://doi.org/10.1016/j.brainres.2017.01.028
Stepanenko Y.D., Boikov S.I., Sibarov D.A., Abushik P.A., Vanchakova N.P., Belinskaia D., Shestakova N.N., Antonov S.M. (2019) Dual action of amitriptyline on NMDA receptors: enhancement of Ca-dependent desensitization and trapping channel block. Sci. Rep. 9(1): 19454. https://doi.org/10.1038/s41598-019-56072-z
Lavoie P.A., Beauchamp G., Elie R. (1990) Tricyclic antidepressants inhibit voltage-dependent calcium channels and Na+–Ca2+ exchange in rat brain cortex synaptosomes. Can. J. Physiol. Pharmacol. 68: 1414–1418. https://doi.org/10.1139/y90-215
Belinskaia D.A., Belinskaia M.A., Barygin O.I., Vanchakova N.P., Shestakova N.N. (2019) Psychotropic drugs for the management of chronic pain and itch. Pharmaceuticals. 12(2): 99. https://doi.org/10.3390/ph12020099
Boikov S.I., Sibarov D.A., Antonov S.M. (2020) Ethanol inhibition of NMDA receptors in calcium-dependent and -independent modes. Biochem. Biophys. Res. Commun. 522(4): 1046–1051. https://doi.org/10.1016/j.bbrc.2019.12.007
Sibarov D.A., Abushik P.A., Poguzhelskaya E.E., Bolshakov K.V., Antonov S.M. (2015) Inhibition of plasma membrane Na/Ca-exchanger by KB-R7943 or lithium reveals its role in Ca-dependent N-methyl-D-aspartate receptor inactivation. J. Pharmacol. Exp. Ther. 355(3): 484–495. https://doi.org/10.1124/jpet.115.227173
Sibarov D.A., Poguzhelskaya E.E., Antonov S.M. (2018) Downregulation of calcium-dependent NMDA receptor desensitization by sodium-calcium exchangers: a role of membrane cholesterol. BMC Neurosci. 19(1): 73. https://doi.org/10.1186/s12868-018-0475-3
Janowsky D.S., Byerley B. (1984) Desipramine: an overview. J. Clin. Psychiatry. 45 (10 Pt 2): 3–9. PMID: 6384207
Sibarov D.A., Antonov S.M. (2018) Calcium-dependent desensitization of NMDA receptors. Biochemistry (Mosc.) 83(10): 1173–1183. https://doi.org/10.1134/S0006297918100036
Zima A.V., Qin J., Fill M., Blatter L.A. (2008) Tricyclic antidepressant amitriptyline alters sarcoplasmic reticulum calcium handling in ventricular myocytes. Am. J. Physiol. Heart Circ. Physiol. 295(5): H2008–H2016. https://doi.org/10.1152/ajpheart.00523.2008
Joshi P.G., Singh A., Ravichandra B. (1999) High concentrations of tricyclic antidepressants increase intracellular Ca2+ in cultured neural cells. Neurochem. Res. 24: 391–398. https://doi.org/10.1023/a:1020937717260
Chopra N., Laver D., Davies S.S., Knollmann B.C. (2008) Amitriptyline activates cardiac ryanodine channels and causes spontaneous sarcoplasmic reticulum calcium release. Mol. Pharmacol. 75 (1): 183–195. https://doi.org/10.1124/mol.108.051490
Furuichi T., Kohda K., Miyawaki A., Mikoshiba K. (1994) Intracellular channels. Curr. Opin. Neurobiol. 4: 294–303. https://doi.org/10.1016/0959-4388(94)90089-2
Mironova E.V., Evstratova A.A., Antonov S.M. (2007) A fluorescence vital assay for the recognition and quantification of excitotoxic cell death by necrosis and apoptosis using confocal microscopy on neurons in culture. J. Neurosci. Methods. 163: 1–8. https://doi.org/10.1016/j.jneumeth.2007.02.010
Han E.B., Stevens C.F. (2009) Development regulates a switch between post- and presynaptic strengthening in response to activity deprivation. Proc. Natl. Acad. Sci. USA. 106: 10817–10822. https://doi.org/10.1073/pnas.0903603106
Ehrlich B.E., Kaftan E., Bezprozvannaya S., Bezprozvanny I. (1994) The pharmacology of intracellular Ca2+-release channels. Trends Pharmacol. Sci. 15: 145–149. https://doi.org/10.1016/0165-6147(94)90074-4
Maruyama T., Kanaji T., Nakade S., Kanno T., Mikoshiba K. (1997) 2APB,2-aminoethoxydiphenyl borate, a membrane-penetrable modulator of Ins(1,4,5)P3-induced Ca2+ release. J. Biochem. 122: 498–505. https://doi.org/10.1093/oxfordjournals.jbchem.a021780
Saleem H., Tovey S.C., Molinski T.F., Taylor C.W. (2014) Interactions of antagonists with subtypes of inositol 1,4,5-trisphosphate (IP3) receptor. Br. J. Pharmacol. 171(13): 3298–3312. https://doi.org/10.1111/bph.12685
Palade P., Dettbam C., Brunder D., Stein P., Hals G. (1989) Pharmacology of calcium release from sarcoplasmic reticulum. J. Bioenerg. Biomembr. 21: 295–319. https://doi.org/10.1007/BF00812074
Bezprozvanny I., Ondrias K., Kaftan E., Stoyanovsky D.A., Ehrlich B.E. (1993) Activation of the calcium release channel (ryanodine receptor) by heparin and other polyanions. Molec. Biol. Cell. 4: 347–352. https://doi.org/10.1091/mbc.4.3.347
Missiaen L., Callewaert G., De Smedt H., Parys J.B. (2001) 2-Aminoethoxydiphenyl borate affects the inositol 1,4,5-trisphosphate receptor, the intracellular Ca2+ pump and the non-specific Ca2+ leak from the non-mitochondrial Ca2+ stores in permeabilized A7r5 cells. Cell. Calcium. 29 (2): 111–116. https://doi.org/10.1054/ceca.2000.0163
Bilmen J.G., Wootton L.L., Godfrey R.E., Smart O.S., Michelangeli F. (2002) Inhibition of SERCA Ca2+ pumps by 2-aminoethoxydiphenyl borate (2-APB). 2-APB reduces both Ca2+ binding and phosphoryl transfer from ATP, by interfering with the pathway leading to the Ca2+-binding sites. Eur. J. Biochem. 269: 3678–3687. https://doi.org/10.1046/j.1432-1033.2002.03060.x
Goto J., Suzuki A.Z., Ozaki S., Matsumoto N., Nakamura T., Ebisui E., Fleig A., Penner R., Mikoshiba K. (2010) Two novel 2-aminoethyl diphenylborinate (2-APB) analogues differentially activate and inhibit store-operated Ca2+ entry via STIM proteins. Cell. Calcium. 47: 1–10. https://doi.org/10.1016/j.ceca.2009.10.004
Shimizu M., Nishida A., Yamawaki S. (1993) Forskolin and phorbol myristate acetate inhibit intracellular Ca2+ mobilization induced by amitriptyline and bradykinin in rat frontocortical neurons. J. Neurochem. 61(5): 1748–1754. https://doi.org/10.1111/j.1471-4159.1993.tb09812.x
Hayashi T., Su T.P. (2007) Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca2+ signaling and cell survival. Cell. 131(3): 596–610. https://doi.org/10.1016/j.cell.2007.08.036
Ryskamp D.A., Korban S., Zhemkov V., Kraskovskaya N., Bezprozvanny I. (2019) Neuronal Sigma-1 receptors: signaling functions and protective roles in neurodegenerative diseases. Front. Neurosci. 13: 862. https://doi.org/10.3389/fnins.2019.00862
Verkhratsky A. (2005) Physiology and pathophysiology of the calcium store in the endoplasmic reticulum of neurons. Physiol. Rev. 85: 201–279. https://doi.org/10.1152/physrev.00004.2004
Walton P.D., Airey J.A., Sutko J.L., Beck C.F., Mignery G.A., Südhof T.C., Deerinck T.J., Ellisman M.H. (1991) Ryanodine and inositol trisphosphate receptors coexist in avian cerebellar Purkinje neurons. J. Cell. Biol. 113(5): 1145–1157. https://doi.org/10.1083/jcb.113.5.1145
Segal M., Vlachos A., Korkotian E. (2010) The spine apparatus, synaptopodin, and dendritic spine plasticity. Neuroscientist. 16: 125–131. https://doi.org/10.1177/1073858409355829
Chen-Engerer H.J., Hartmann J., Karl R.M., Yang J., Feske S., Konnerth A. (2019) Two types of functionally distinct Ca2+ stores in hippocampal neurons. Nat. Commun. 10 (1): 3223. https://doi.org/10.1038/s41467-019-11207-8
Fisar Z. (2005) Interactions between tricyclic antidepressants and phospholipid bilayer membranes. Gen. Physiol. Biophys. 24 (2): 161–80. PMID: 16118470
Дополнительные материалы отсутствуют.
Инструменты
Российский физиологический журнал им. И.М. Сеченова