

Российский физиологический журнал им. И.М. Сеченова, 2021, T. 107, № 4-5, стр. 458-473
Холинергическая модуляция секреции ацетилхолина в нервно-мышечном соединении
Э. А. Бухараева 1, *, А. И. Скоринкин 1
1 Казанский институт биохимии и биофизики Федерального исследовательского центра “Казанский научный центр РАН”
Казань, Россия
* E-mail: elbukhara@gmail.com
Поступила в редакцию 18.01.2021
После доработки 29.01.2021
Принята к публикации 29.01.2021
Аннотация
Влияние холинергических соединений (активаторов и блокаторов никотиновых холинорецепторов) на секрецию ацетилхолина из двигательных нервных окончаний представляет интерес в связи с вопросом о наличии механизма обратной связи в нервно-мышечном синапсе. Предполагается, что на нервных окончаниях могут быть ауторецепторы к ацетилхолину, изменение активности которых влияет на выделение медиатора в ответ на нервный стимул. Однако многочисленные экспериментальные данные не дают однозначного представления о направленности и механизмах действия как эндогенного ацетилхолина, так и других холинергических соединений на вызванную квантовую секрецию медиатора в нервно-мышечном синапсе. Актуальность таких исследований обусловлена необходимостью расшифровки эффектов этих соединений, так как многие из них применяются в клинической практике. Обзор посвящен анализу результатов исследований, проведенных на классических для нейрофизиологии объектах – нервно-мышечных препаратах теплокровных животных с помощью радиоизотопного метода оценки количества секретируемого из нервных окончаний медиатора и электрофизиологического метода определения числа квантов, выделяющихся в ответ на нервный стимул. Сопоставлены многочисленные данные, полученные при использовании активаторов и блокаторов ионотропных никотиновых рецепторов, а также вероятные механизмы действия холинергических соединений, модулирующих секреторный процесс. Предложена схема регуляции квантовой секреции, учитывающая новые сведения о возможном участии Шванновской клетки и о пресинаптической гомеостатической пластичности.
Вопрос о влиянии ацетилхолина (АХ), основного медиатора нервно-мышечного синапса, на процесс собственного освобождения продолжает интересовать нейрофизиологов. Он затрагивает не только фундаментальные аспекты ауторегуляции секреторного процесса, но и важные особенности клинического применения миорелаксантов разного типа действия. В отечественной нейрофизиологии значимый вклад в изучение пресинаптического действия холинергических соединений и их эффектов на процессы секреции в периферических синапсах внесли исследования академика РАН Л.Г. Магазаника [1–3] и его ученика и последователя академика РАН Е.Е. Никольского [4, 5]. В нервно-мышечном соединении скелетной мускулатуры ведущую роль в обеспечении сократительной функции мышечного волокна играют ионотропные ацетилхолиновые рецепторы (АХР) никотинового типа. Несмотря на достаточно длительную историю исследований влияния активаторов и блокаторов АХР на процессы секреции АХ и немалое количество обзорных публикаций, до сих пор нет однозначного мнения о свойствах пресинаптических ионотропных АХР на двигательных нервных окончаниях; о том, какое действие они оказывают на процесс секреции АХ – облегчающее или тормозное; о механизмах, участвующих в реализации влияния агонистов и антагонистов АХР; о физиологической роли пресинаптических АХР. В связи с этим целью данного обзора является анализ сведений о влиянии холинергических соединений, активных в отношении никотиновых ионотропных АХР в синапсах теплокровных. Эти ограничения обусловлены тем, что в нервно-мышечном синапсе присутствуют также и мускариновые метаботропные АХР [6], вносящие вклад в модуляцию работы синапса, а регуляторные процессы в холинергических синапсах других видов животных (лягушки, змеи, жабы) имеют свои специфические особенности [7, 8].
МЕТОДЫ ИССЛЕДОВАНИЯ ВЛИЯНИЯ ХОЛИНЕРГИЧЕСКИХ СОЕДИНЕНИЙ НА СЕКРЕЦИЮ АЦЕТИЛХОЛИНА ИЗ ДВИГАТЕЛЬНЫХ НЕРВНЫХ ОКОНЧАНИЙ
Для изучения эффектов активаторов и блокаторов никотиновых АХР на процесс выделения АХ из нервных окончаний применяются, в основном, два метода: радиоизотопный анализ количества выделившегося при стимуляции двигательного нерва меченого [3H]АХ [9, 10] и электрофизиологический метод определения количества освободившихся в ответ на нервный стимул квантов АХ [11].
При использовании достаточно чувствительного радиоактивного метода нервно-мышечный препарат предварительно инкубируется меченным [3H] холином при стимуляции двигательного нерва. После инкубации в ответ на нервный стимул освобождается вновь синтезированный [3H]АХ. Оценивается соотношение количества [3H]АХ при стимуляции до введения исследуемого холинергического соединения и после его аппликации. Особенностью этого метода является то, что его можно использовать без применения других веществ, способных модифицировать синаптические процессы, таких как ингибиторы ацетилхолинэстеразы или блокаторы мышечных сокращений. Хотя и считается, что меченый [3H]АХ, выделяется только из нервной терминали при стимуляции нерва, нельзя полностью исключить его освобождение в неквантовой форме, а может быть и из других источников (мышечное волокно, Шванновская клетка).
Анализ количества освободившихся квантов АХ в ответ на нервный стимул (квантовый состав синаптического ответа) осуществляется при микроэлектродной регистрации потенциалов или токов концевой пластинки – участка постсинаптической мембраны мышечного волокна. Квантовый состав чаще всего определяется путем деления амплитуды вызванного нервным стимулом постсинаптического потенциала или тока на амплитуду миниатюрного потенциала или тока концевой пластинки. Ограничением этого метода является необходимость блокировать сокращения мышечного волокна в ответ на развитие потенциала действия. Все применяемые для этого способы: поперечное рассечение мышечных волокон, обработка нервно-мышечного препарата глицерином, супрамаксимальное растяжение мышцы – могут оказывать собственное влияние на работу нейросекреторного аппарата. В последние годы для блокады мышечных сокращений стали использовать µ-GIIIB конотоксин, который избирательно блокирует потенциал-зависимые натриевые каналы мембраны мышечного волокна и таким образом предотвращает его сокращение [12]. Однако сравнительно небольшое число исследований эффектов холинергических соединений в нервно-мышечных синапсах проведено с использованием этого метода.
ЭФФЕКТЫ АКТИВАТОРОВ НИКОТИНОВЫХ ХОЛИНОРЕЦЕПТОРОВ НА СЕКРЕЦИЮ АЦЕТИЛХОЛИНА
С помощью радиоизотопного метода определения эффектов агонистов никотиновых рецепторов на выделение меченого АХ в нервно-мышечном препарате крысы или мыши получено относительно немного данных. В качестве агонистов АХР использовали никотин, цитизин, диметилфенилпиперазин (ДФПП), холин, 2-(4-аминофенил)-этил-триметиламмонийиодид. Под действием всех исследованных активаторов никотиновых рецепторов наблюдалось увеличение количества освобожденного меченого [3H]АХ. ДФПП в диапазоне концентраций 1–30 мкМ повышал выделение [3H]АХ на 76–92%, и этот эффект устранялся при действии блокатора никотиновых АХР тубокурарина [13, 14]. Однако в присутствии блокатора ацетилхолинэстеразы неостигмина ДФПП существенно не влиял на вызванное высвобождение [3H]АХ [14]. Степень эффективности облегчающего действия агонистов на секрецию уменьшалась в следующем порядке: никотин > цитизин > > ДФМП > 2-(4-аминофенил)-этил-триметиламмонийиодид [14]. При этом пресинаптические эффекты никотина сильно зависели от времени воздействия: облегчение выявлялось после короткой 20-секундной аппликации, а уже после 3 мин действия наблюдалось снижение секреции АХ [15]. На основании полученных данных были сделаны выводы о том, что двигательные нервные окончания наделены пресинаптическими никотиновыми рецепторами. Эти ауторецепторы обеспечивают механизм положительной обратной связи, который может быть запущен ранее высвобожденным эндогенным АХ. Снижение секреции после длительной экспозиции агониста может быть связано с десенситизацией рецепторов высокими концентрациями агонистов (эндогенных или экзогенных). Вероятно, это один из механизмов, ограничивающих процесс фасилитации секреции. Было высказано предположение, что пресинаптические рецепторы, по-видимому, отличаются по своим фармакологическим свойствам от постсинаптических рецепторов [16].
Электрофизиологический анализ для определения количества квантов АХ, выделившихся в ответ на нервный стимул (квантовый состав) применялся чаще всего на нервно-мышечных препаратах диафрагмальной мышцы мыши или крысы. Использование ингибиторов ацетилхолинэстеразы эзерина и неостигмина для увеличения амплитуды регистрируемых постсинаптических сигналов – вызванных стимулом потенциалов концевой пластинки и спонтанных миниатюрных потенциалов концевой пластинки – приводило к снижению высвобождения АХ [17].
Цитизин при низкой (0.5 Гц) частоте стимуляции двигательного нерва диафрагмальной мышцы крысы вызывал кальций-зависимое снижение квантового состава постсинаптического ответа на 20% [18, 19]. Этот эффект наблюдался при концентрации ионов кальция 1.8 мМ, а при понижении или повышении содержания ионов кальция отсутствовал. Повышение частоты стимуляции нерва до 50 Гц приводило к устранению эффекта цитизина на количество освобождаемых квантов АХ. Эти наблюдения позволили авторам предположить, что цитизин активирует пресинаптические никотиновые ауторецепторы по механизму отрицательной обратной связи.
ДФПП в диапазоне концентраций от 1 до 4 мкМ не влиял на спонтанное квантовое высвобождение АХ, но увеличивал количество квантов АХ в ответ на нервный импульс во время стимуляции с частотой 50 Гц [20]. Индуцированное ДФПП увеличение вызванного освобождения АХ зависело от частоты стимуляции, отсутствовало при низкой частоте (0.5 Гц) и не зависело от содержания ионов кальция. Облегчение вызванной секреции АХ при 50 Гц устранялось антагонистом кальмодулина W7 (N-(6-аминогексил)-5-хлор-1-нафталинсульфонамида гидрохлорид). Был сделан вывод, что ДФПП вызывает изменения вызванного освобождения АХ из двигательных нервных окончаний крысы, которые согласуются с существованием пресинаптических стимулирующих никотиновых рецепторов. Полученные данные также указывают на роль кальмодулин-зависимых систем в стимулирующем секрецию эффекте [20].
В исследованиях Балезиной и ее учеников [21] на нервно-мышечном препарате диафрагмы мыши показано, что никотин в низкой концентрации (10 нМ) не влиял на освобождение квантов АХ в ответ на первые стимулы в пачке, но вызывал небольшое подавление секреции при ритмической стимуляции. Экзогенный холин также оказывал пресинаптическое ингибирующее действие на квантовый состав потенциалов концевой пластинки, вызванных одиночными и ритмическими стимулами. Данный эффект подавлялся антагонистами АХР, такими как метилликаконитин и α-кобратоксин [22], это позволило высказать предположение о том, что ингибирующее действие опосредуется активацией пресинаптических АХР нейронального типа.
Анализ результатов исследований действия агонистов никотиновых АХР показывает, что они могут вызывать как облегчающее, так и ингибирующее влияние на процесс выделения АХ. Это приводит к формированию представления о том, что, вероятно, могут существовать разные системы, активация которых никотиновыми агонистами, в том числе и эндогенным АХ, реализует положительную и отрицательную обратную связь в нервно-мышечном синапсе. Однако необходимо отметить, что если при определении количества выделившегося меченого [3H]АХ чаще наблюдалось начальное повышение уровня секреции под действием агонистов, то при анализе количества квантов АХ более выраженным было угнетение секреции медиатора при повышенной частоте стимуляции двигательного нерва.
ВЛИЯНИЕ БЛОКАТОРОВ НИКОТИНОВЫХ РЕЦЕПТОРОВ НА ОСВОБОЖДЕНИЕ АЦЕТИЛХОЛИНА
При изучении вопроса о существовании пресинаптических никотиновых АХР в нервно-мышечном синапсе, существенно больше исследований было проведено при использовании блокаторов этих рецепторов. Это вполне объяснимо, т.к. такие соединения, недеполяризующие и деполяризующие миорелаксанты, препятствующие возникновению мышечных сокращений, широко применяются в клинической практике. Ранее описанные два метода использовались для выяснения эффектов антагонистов никотиновых АХР на классических нервно-мышечных препаратах мышей и крыс.
Радиоизотопным методом было показано, что блокатор ганглионарных никотиновых рецепторов гексаметоний (0.001–1 мМ) и блокатор мышечных никотиновых АХР тубокурарин (1–10 мкМ) снижали на 52–60% освобождение [3H]АХ при стимуляции двигательного нерва с частотой 5 Гц [23]. Однако в присутствии ингибитора ацетилхолинэстеразы неостигмина (10 мкМ) тубокурарин существенно не влиял на вызванное освобождение [3H]АХ [13]. Были проверены эффекты нейротоксинов змеиного яда, блокирующих никотиновые АХР [23]. α-бунгаротоксин, α-кобратоксин и эрабутоксин-β подавляли вызванные нервным импульсом сокращения диафрагмы, но не изменяли интенсивность освобождения АХ. Эти данные позволили авторам сделать вывод, что пресинаптические никотиновые АХР отличаются от никотиновых рецепторов, локализованных на мышечной мембране и в вегетативных ганглиях, поскольку α-нейротоксины и κ-бунгаротоксин, устраняя мышечные сокращения, не блокировали пресинаптические никотиновые АХР двигательных нервов. Эффективность антагонистов АХР по уменьшению вызванной секреции [3H]АХ (при кратковременной высокочастотной стимуляции нерва) снижалась в ряду: µ-конотоксин MII > дигидро-β-эритроидин > гексаметоний > d-тубокурарин > панкуроний = пипекуроний > мекамиламин [24–26]. Необходимо отметить, что большинство исследований, где показано ингибирующее действие антагонистов никотиновых АХР на секрецию [3H]АХ, проводилось при высокочастотной стимуляции двигательного нерва от 5 до 50 Гц [27, 28], тогда как при низких частотах (менее 5 Гц) и в состоянии покоя такое влияние отсутствовало.
Деполяризующий миорелаксант сукцинилхолин, в отличие недеполяризующих холинолитиков, в низкой концентрации вызывал, напротив, усиление вызванного освобождения [3H]АХ. Однако в более высокой концентрации он также, как и тубокурарин, подавлял секрецию [29].
Данные, полученные при анализе эффектов антагонистов АХР на вызванную секрецию меченого [3H]АХ, указывают на то, что освобождаемый в синаптическую щель АХ при высокочастотной стимуляции нерва может достигать концентрации, достаточной для того, чтобы влиять на собственное выделение за счет действия на пресинаптические рецепторы. Увеличение высвобождения [3H]АХ при ингибировании ацетилхолинэстеразы сульфатом физостигмина, которое устранялось при действии тубокурарина, панкурония и пипекурония подтверждает это заключение. Снижение выделения [3H]АХ при действии антагонистов, специфически взаимодействующих с рецепторами, содержащими α2β3 субъединицы, свойственные нейрональным никотиновым рецепторам, указывает на их вероятное существование на нервных терминалях в синапсах скелетных мышц [26].
Много сведений о действии антагонистов никотиновых АХР в нервно-мышечных синапсах теплокровных животных было получено при использовании электрофизиологического метода анализа синаптической передачи. Мы не рассматриваем здесь данные о сопоставлении изменений силы сокращения мышечных волокон и амплитудных параметров постсинаптического ответа в ходе пачки ритмических стимулов под действием антагонистов. Эти многочисленные сведения требуют отдельного анализа, поскольку затрагивают не только влияние блокаторов на секрецию медиатора, но и постсинаптические механизмы, участвующие в реализации мышечных сокращений.
При физиологической внеклеточной концентрации ионов кальция (2 мМ) тубокурарин вызывал снижение квантового состава тока концевой пластинки на 30% при высоких частотах стимуляции двигательного нерва (50–150 Гц), которое не зависело от содержания ионов кальция. Напротив, при низких частотах стимуляции (0.5–1.0 Гц) антагонист увеличивал квантовый состав постсинаптического ответа примерно на 20%, и этот эффект устранялся при снижении концентрации ионов кальция [30]. Довольно большое число работ представило аналогичные данные об увеличении квантового состава под действием тубокурарина [19, 31–33]. Анализ показал, что изменения квантового состава под действием антагониста были вызваны влиянием на пул везикул, доступный для немедленного освобождения. Гексаметоний и метилликаконитин также при низких частотах стимуляции (0.5–2 Гц) увеличивали квантовый состав потенциалов концевой пластинки на 30–40%, а при высоких частотах (50–150 Гц) не оказывали никакого влияния. При этом эффект гексаметония наблюдался только при нормальной физиологической концентрации ионов кальция (2 мМ) и отсутствовал при ее снижении [34]. Аналогичный эффект увеличения секреции АХ наблюдался под действием гексаметония для первых нескольких стимулов, но не сохранялся во время серии последующих стимулов [35]. Было сделано предположение о том, что гексаметоний и метилликаконитин увеличивают вызванное освобождение АХ за счет блокирования никотиновых пресинаптических АХР, которые относятся к нейрональному типу и реализуют отрицательную обратную связь. Однако исследования, проведенные в условиях сниженной интенсивности вызванной секреции квантов АХ (при повышении содержания ионов магния в среде), показали, что тубокурарин и гексаметоний подавляют выделение нейромедиатора, предположительно, без участия рецепторных механизмов [36]. Интересно, что α-бунгаротоксин, который считается антагонистом никотиновых АХР преимущественно мышечного типа, также вызывал кратковременное повышение квантового состава первых постсинаптических ответов на высокочастотную стимуляцию нерва [37].
На основании полученных электрофизиологическими методами данных были сделаны выводы о том, что в нервно-мышечном соединении скелетных мышц млекопитающих существуют системы, посредством которых АХ может воздействовать на пресинаптические ауторецепторы либо усиливая, либо подавляя собственное вызванное освобождение [30, 38]. Действие антагонистов приводит к кальций-независимому снижению квантовой секреции АХ при высоких частотах стимуляции двигательного нерва и к кальций-зависимому увеличению освобождения при низкочастотной стимуляции. Таким образом, по аналогии с результатами, полученными при исследовании эффектов агонистов никотиновых АХР, данные о влиянии блокаторов показали наличие двух противоположно направленных эффектов на квантовую секрецию АХ из двигательных нервных окончаний. Эти результаты дали основания предполагать наличие, как минимум, двух систем, контролирующих квантовую секрецию АХ [30, 39]. Одна реализует отрицательную обратную связь, снижая выброс медиатора при ее активации и повышая при блокировании [32, 33, 35], другая, напротив, облегчает освобождение АХ в условиях активации эндогенным АХ или агонистами. Следовательно, блокируя эти рецепторы, антагонисты должны уменьшать секрецию АХ [15, 27, 40]. Таким образом, возникла гипотеза о том, что на двигательных нервных окончаниях существуют два разных типа никотиновых ауторецепторов для АХ. Один тип реализует механизм положительной обратной связи, повышающий уровень секреции, и отрицательной, которая ограничивает избыточное выделение медиатора [32, 33, 35].
ДОКАЗАТЕЛЬСТВА НАЛИЧИЯ НИКОТИНОВЫХ ХОЛИНОРЕЦЕПТОРОВ НА ДВИГАТЕЛЬНЫХ НЕРВНЫХ ОКОНЧАНИЯХ
Несмотря на множество экспериментальных данных об изменении секреции АХ под действием экзогенных агонистов и антагонистов никотиновых АХР, существование самой белковой молекулы рецептора на пресинаптической мембране двигательных нервных окончаний долгое время было предметом дискуссий. Jones и Salpeter [41] пытались выявить никотиновые АХР на двигательных нервных окончаниях в препаратах диафрагмы мыши с помощью электронной микроскопии и меченого [125I] α-бунгаротоксина после отделения терминалей от мышцы с помощью коллагеназы и протеазы. Анализ локализации радиоактивной метки показал, что α-бунгаротоксин не связывался с мембраной нервного окончания. На этом основании был сделан вывод об отсутствии никотиновых АХР на пресинаптической мембране. Действительно, из-за тесного контакта нервной терминали и постсинаптической мембраны, а также прилегающей к синапсу Шванновской клетки довольно трудно физически установить наличие пресинаптических АХР. Наличие субъединиц нейронального никотинового АХР было исследовано в криостатных срезах диафрагмы мыши методом непрямой иммунофлуоресценции [42]. Специфическое мечение моноклональными антителами (mAb) к β2- и α8-субъединицам нейронального АХР наблюдалось в области нервно-мышечного контакта, определяемого путем мечения флуоресцентным красителем конъюгированным с α-бунгаротоксином. На двигательном нерве, включая нервные окончания, была обнаружена иммунореактивность mAb к субъединице α3 нейронального АХР. Эти результаты подтвердили, что подтипы постсинаптических никотиновых АХР, распознаваемых антителом antit2 и/или mAb против α8, и пресинаптического АХР, распознаваемого mAb против α3, присутствуют в нервно-мышечном соединении в дополнение к классическим мышечным никотиновым АХР. На основании пресинаптических эффектов холина, имеющего более высокое сродство к нейрональным АХР, высказывалось предположение о существовании на моторных нервных окончаниях нейрональных АХР, содержащих α7 субъединицу [22]. Однако иммуногистохимический анализ с использованием специфической метки для α7 АХР не выявил ее связывания с пресинаптической мембраной нервного окончания в синапсах мыши, а показал их наличие на рядом расположенной Шванновской клетке [43].
Пресинаптический никотиновый АХР на двигательных нервных окончаниях в настоящее время идентифицируется как нейрональный, содержащий α3β2 субъединицы, который не ингибируется α-бунгаротоксином и предпочтительнее взаимодействует с недеполяризующими миорелаксантами и гексаметонием [42, 44]. Это заключение подтверждается данными о влиянии подтип-специфических агонистов на вызванную секрецию квантов АХ [26], а также способностью недеполяризующих миорелаксантов связываться с рецепторами α3β2 типа, экспрессируемыми в ооцитах Xenopus laevis, которым вводили информационную РНК, кодирующую эти рецепторные субъединицы [45].
МЕХАНИЗМЫ, УЧАСТВУЮЩИЕ В РЕАЛИЗАЦИИ ЭФФЕКТОВ ХОЛИНЕРГИЧЕСКИХ СОЕДИНЕНИЙ НА СЕКРЕЦИЮ АЦЕТИЛХОЛИНА
Проведенный анализ результатов исследований влияния агонистов и антагонистов никотиновых АХР на освобождение АХ из двигательных нервных окончаний в нервно-мышечных синапсах крысы и мыши показал, что холинергические соединения оказывают как облегчающее, так и угнетающее действие на секрецию медиатора, выраженность которого зависит от функционального состояния синапса (интенсивность ритмической активности, исходный уровень секреции).
Имеющиеся данные позволяют считать, что на окончаниях аксона, иннервирующих скелетную мышцу, имеются никотиновые рецепторы, субъединичный состав и свойства которых отличаются от таковых для никотиновых рецепторов постсинаптической мембраны мышечного волокна [26, 44, 46]. Однако остаются спорными заключения о роли ауторецепторов: служат ли они для ограничения секреции, формируя отрицательную обратную связь [31, 32], или усиливают освобождение по механизму положительной обратной связи [13, 47]. Кроме того, возникает вопрос: благодаря каким механизмам происходит изменение интенсивности секреторного процесса при активации пресинаптических АХР. Неоднозначность феноменологических данных приводит также к разнообразию моделей, пытающихся объяснить наблюдаемые эффекты.
Облегчающее действие агонистов на секрецию, т.е. положительную обратную связь, реализуемую пресинаптическими никотиновыми АХР связывают:
1) с повышением концентрации ионов калия в синаптической щели вследствие деполяризации концевой пластинки при активации агонистами постсинаптических АХР [48]. Однако это предположение было опровергнуто, поскольку при изменении внеклеточной концентрации ионов калия с 2.0 на 7.5 мМ не наблюдалось значимого изменения квантового состава постсинаптического ответа;
2) с повышением входа ионов кальция в аксоплазму. Считается, что нейрональные никотиновые рецепторы имеют более высокую относительную проницаемость для ионов кальция по сравнению с их аналогами мышечного типа [49]. Активация агонистом рецепторов, содержащих α3 субъединицу, приводит к возрастанию внутриклеточной концентрации кальция [50]. Кратковременный приток кальция через рецепторно-канальный комплекс может активировать внутриклеточные ферментные системы (например, протеинкиназу C, Ca2+-кальмодулин-зависимую киназу II), способствуя экзоцитозу за счет быстрого увеличения пула везикул, готового к освобождению [20].
В настоящее время прямых доказательств участия Ca2+-кальмодулин-зависимой киназы II (СаМКII) в облегчении секреции АХ недостаточно. Однако показано, что активация холинорецепторов потенциирует синаптическую передачу в нейрональной сети за счет повышения активности CaMKII [51]. К тому же, на нервно-мышечном синапсе лягушки было показано, что CaMKII играет важную роль в обеспечении краткосрочной синаптической пластичности, вероятно, влияя на скорость мобилизации синаптических везикул [52]. Было также показано, что в ответ на действие агонистов может происходить повышение внутриклеточного кальция за счет его выхода из клеточных депо [53]. Нельзя исключить, что кальций, входящий в аксоплазму при активации нейрональных АХР эндогенным АХ, может участвовать в регуляции низкопороговых кальциевых каналов L-типа. Активация этих каналов приводит к повышению уровня секреции при участии рианодиновых рецепторов [54];
3) с деполяризацией нервных окончаний входом ионов натрия, основываясь на данных о том, что на нервных окончаниях выявлены АХР α3β2 субъединичного состава, которые обеспечивают приток ионов натрия и, как следствие, увеличивается внутриклеточное содержание ионов кальция за счет активации потенциал-зависимых кальциевых каналов [53].
Представляется наиболее вероятным, что облегчающее секрецию АХ действие агонистов и эндогенного АХ может реализовываться благодаря мобилизации синаптических везикул и ускорению экзоцитоза вследствие повышения внутриклеточного кальция [39, 54]. Этот процесс может происходить как за счет усиления его входа из внеклеточной среды, так и при усилении выброса из внутриклеточных депо.
Более сложная ситуация складывается с гипотезами, объясняющими механизм отрицательной обратной связи или аутоингибирования, обеспечивающего снижение секреции АХ при действии эндогенного АХ и агонистов, а также ее повышение под влиянием антагонистов. Как было описано ранее, часть исследований показывает снижение секреции АХ под действием агонистов, причем даже после начального повышения с течением времени появляется падение уровня освободившегося медиатора. Другие демонстрируют кальций-зависимое повышение квантового состава под действием тубокурарина и блокаторов никотиновых рецепторов нейронального типа при низкочастотной стимуляции нерва.
Аутоингибирование, наблюдаемое преимущественно при низких частотах активации, зависит от концентрации внеклеточных ионов кальция и считается опосредованным никотиновым АХР нейронального подтипа [30, 34].
Одним из возможных механизмов реализации снижения секреции АХ под действием агонистов и/или эндогенного АХ предполагается изменение активности кальциевых и кальций-активируемых калиевых каналов. Блокатор кальций-активируемых калиевых каналов малой проводимости апамин и блокатор рианодиновых рецепторов снимали угнетающее секрецию АХ действие холина [22]. Был сделан вывод, что активация холином рецепторно-канального комплекса нейронального типа, включающего α7 субъединицу и обладающего относительно высокой способностью пропускать ионы кальция, запускают выброс кальция из рианодин-чувствительных депо, что, в свою очередь, приводит к активации кальций-зависимых калиевых каналов. По-видимому, последующий выход ионов калия из аксоплазмы вызывает усиление следовой гиперполяризации терминали и снижение эффективности потенциала действия. Однако доказательств такого развития событий пока нет. В работах группы Балезиной на основании наблюдений о том, что блокирование CaMKII устраняло угнетающее секрецию действие холина, был сделан вывод о том, что помимо кальций-зависимых калиевых каналов в реализации ингибирующего действия агониста может участвовать CaMKII [55, 56]. Таким образом, холинергическая модуляция секреции – аутоингибирование, опосредованное α7 АХР так же, как и аутофасилитация через активацию α3β2 АХР, могут быть связаны с изменением внутриклеточного кальциевого метаболизма при участии кальций-зависимых каналов и депонированного кальция [57].
Активация нейрональных АХР может опосредовать три типа цитоплазматических сигналов кальция: (1) прямой приток кальция через сами рецепторы; (2) непрямой приток кальция через потенциал-зависимые кальциевые каналы, которые активируются АХР-опосредованной деполяризацией; 3) кальций-индуцированное высвобождение кальция (CICR) (запускаемое первыми двумя источниками) из эндоплазматического ретикулума через рецепторы рианодина и рецепторы инозитол (1,4,5)-трифосфата, а также повышение активности фосфолипазы C [58, 59]. Все эти процессы могут быть включены в модуляцию синаптического процесса.
Prior и соавт. [19, 38] считают, что существуют два отдельных механизма, обеспечивающих аутофасилитацию и аутоингибирование секреции АХ в нервно-мышечном синапсе. По их мнению, аутофасилитация подтверждается данными о кальмодулин-зависимом возрастании вызванной секреции АХ под влиянием агониста ДФПП и ее угнетении при действии антагонистов тубокурарина и веркурония при высокой частоте стимуляции двигательного нерва [20]. Аутоингибирование происходит при активации АХР агонистом цитизином, а блокирование рецепторов тубокурарином и гексаметонием повышает секрецию при низкой частоте стимуляции. Оба эффекта не зависят от активности кальмодулиновой системы, но устраняются блокатором кальциевых каналов верапамилом [19], поскольку на нервных окончаниях зарегистрированы верапамил-чувствительные кальциевые токи [60]. Тем не менее, события, последующие за активацией верапамил-чувствительных кальциевых каналов и приводящие к снижению уровня секреции, еще не выявлены.
Обобщая имеющиеся экспериментальные данные и гипотезы о механизмах влияния активаторов и ингибиторов никотиновых АХР на секрецию АХ из двигательных нервных окончаний, можно сказать, что существуют два разнонаправленных изменения уровня вызванного освобождения медиатора. Облегчающее действие или аутофасилитация, вероятнее всего, опосредована никотиновыми рецепторами, содержащими α3β2 субъединицы, действующими как кратковременный пресинаптический усилитель для увеличения коэффициента надежности синаптической передачи, если требуется увеличение силы мышечных сокращений [20, 26, 61]. Возможно, использование α-конотоксина LvIA (пептид из яда плотоядного морского брюхоногого моллюска Conus lividus), который на сегодняшний день считают наиболее селективным ингибитором никотиновых α3β2 АХР, позволит получить прямые доказательства участия этих рецепторов в аутофасилитации [62]. Аутоингибирование, по-видимому, связанное с активацией рецепторов с α7 субъединицей, необходимо для предотвращения избыточной траты нейромедиатора из пула везикул, готового к немедленному освобождению.
Однако, принимая во внимание ряд исследований, появившихся в последнее пятилетие, необходимо сказать, что могут быть альтернативные механизмы холинергической регуляции секреции АХ в нервно-мышечном синапсе.
На Шванновской клетке иммуногистохимическим методом показано наличие нейрональных АХР, содержащих α7 субъединицу, которые могут контролировать распространение АХ в области синапса [43]. Активация этих рецепторов избытком АХ в условиях ингибирования ацетихолин- и бутирилхолинэстеразы в синаптической зоне приводит к освобождению глиотрансмиттера (предположительно гамма-аминомасляной кислоты), который оказывает ингибирующее действие на освобождение АХ. Возможные варианты накопления эндогенного АХ – это снижение активности холинэстераз при патологических состояниях (снижение экспрессии самих ферментов, при применении лекарственных ингибиторов). Вместе с тем, получены данные о том, что существуют эндогенные ингибиторы ацетихолинэстеразы, например, оксид азота, интенсивное освобождение которого в синаптическую щель при ритмической стимуляции приводит к снижению активности фермента [63]. С другой стороны, активация никотиновых рецепторов на Шванновской клетке приводит к увеличению кальциевого транзиента и, весьма вероятно, к выходу предполагаемого глиотрансмиттера даже без предварительного ингибирования холинэстеразы [43]. Недавно было показано, что индуцированная ритмической стимуляцией активация α7 АХР на Шванновской клетке управляет спилловером АХ в нервно-мышечном синапсе при высокочастотном режиме работы синапса за счет выделения Шванновской клеткой другого активного соединения – аденозина [64], пресинаптическое действие которого приводит к уменьшение секреции АХ из нервных терминалей [65]. Таким образом, Шванновскую клетку нельзя исключить из вероятных участников процессов, модулирующих синаптическую передачу в определенных условиях (интенсивный выброс медиатора, недостаточность холинэстераз).
Интерес вызывает возникшая относительно недавно гипотеза о пресинаптической гомеостатической пластичности в нервно-мышечном синапсе, которая подразумевает изменение интенсивности секреторного процесса в ответ на снижение постсинаптической активности. Это означает, что блокирование постсинаптических АХР токсинами или миорелаксантами вызывает повышение количества высвобождаемых синаптических везикул, т.е. квантового состава [66]. Механизм, лежащий в основе этой пластичности, неизвестен. Было показано, что токсины, селективно блокирующие никотиновые рецепторы мышечного волокна, содержащие α1 субъединицу, вызывали повышение квантового состава. Влияние антагониста на синапсы мышей, нокаутированных по α7 субъединице АХР, приводило к увеличению квантового состава, аналогичному в синапсах животных дикого типа. Эти данные показали, что блокирование пресинаптических нейрональных рецепторов не вносит вклад в облегчение секреторного процесса. Авторы считают, что помимо инициирования мышечного сокращения, ионотропные никотиновые АХР мышечного волокна могут служить сигнальными молекулами, которые участвуют в синаптической пластичности. По-видимому, доказательства такой неканонической регуляции процесса секреции АХ в нервно-мышечном синапсе ожидают нас в ближайшем будущем.
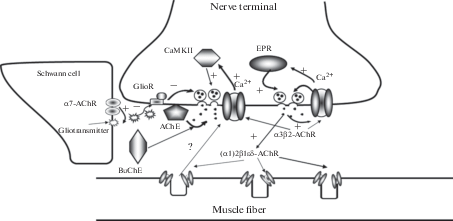
На основании анализа рассмотренных данных можно предложить гипотетическую схему реализации действия эндогенного АХ (а также, вероятно, и экзогенных активаторов АХР) на процесс вызванной секреции квантов АХ из двигательных нервных окончаний (рис. 1). Учитывая наблюдения о том, что активация АХР приводит к эффектам разной направленности и результативности, трудно представить, что они реализуются системой с одним “входным” звеном. Мы предполагаем, что облегчающий секрецию эффект может быть опосредован активацией пресинаптических рецепторов нейронального типа, содержащих α3β2 субъединицы. АХ, освободившийся в ответ на нервный стимул, активирует эти рецепторы, что приводит к входу ионов кальция в аксоплазму. Повышенная внутриклеточная концентрация кальция может стимулировать кальций-индуцированное освобождение этих ионов из эндоплазматического ретикулума или активировать кальмодулинкиназу. Эти процессы могут облегчать активацию кальций-зависимых белков экзоцитоза и повышать выброс медиатора. Аутоингибирование секреторного процесса можно связать с нейрональными рецепторами с α7 субъединицей, обнаруженными на Шванновских клетках. Повышение частоты стимуляции, вызывающее возрастание концентрации АХ, особенно при недостаточной активности холинэстераз, позволяет молекулам АХ достичь мембраны Шванновской клетки и активировать α7 АХР. Это приводит к освобождению глиотрансмиттера (предположительно, гамма-аминомасляной кислоты или аденозина), который, взаимодействуя с собственными рецепторами, уменьшает интенсивность секреции АХ. Согласно гипотезе последних лет о пресинаптической гомеостатической пластичности, из постсинаптической мембраны мышечного волокна при активации АХР мышечного типа, содержащих субъединицы (α1)2β1εδ, может освобождаться некий, пока неустановленный фактор, способный оказывать влияние на секрецию медиатора из нервных окончаний. Таким образом, по-видимому, именно наличие нескольких “входных” звеньев и многоступенчатость последующих событий является причиной неоднозначности эффектов при действии холинергических соединений на вызванную секрецию квантов АХ в нервно-мышечном синапсе.
Рис. 1.
Схема процессов в синаптическом контакте нервно-мышечного препарата. α3β2-AChR – нейрональный никотиновый рецептор на нервном окончании; (α1)2β1εδ-AChR – мышечный никотиновый рецептор на постсинаптической мембране; α7-AChR – нейрональный никотиновый рецептор на Шванновской клетке; EPR – эндоплазматический ретикулум; AChE – ацетилхолинэстераза; BuChE – бутирилхолинэстераза; GlioR – глиотрансмиттер; CaMKII – кальций-кальмодулин-зависимая киназа II. Знак + указывает на стимулирующее действие; знак – указывает на ингибирующее действие, знак ? указывает на неизвестный фактор, выделяющийся из мышечного волокна, участвующий в реализации пресинаптической гомеостатической пластичности.
Независимо от того, посредством каких механизмов и при участии каких сигнальных систем агонисты и антагонисты модулируют секрецию АХ из двигательных нервных окончаний, важность и физиологическое значение этого влияния трудно переоценить. Многие нервно-мышечные патологии, связанные с синаптическим дефектом и приводящие к развитию мышечной слабости, требуют восстановления функциональной синаптической передачи. Необходимость выявления наиболее эффективных соединений, способных реализовать это требование, будет стимулировать исследователей к изучению процессов, регулирующих пресинаптические функции.
Список литературы
Kamenskaya M.A., Magazanik L.G., Kotova E.R., Samybaldina N.K., Miroshnikov A.I., Apsalon U.R. (1979) Effect of presynaptic neurotoxins from bee and cobra venom on spontaneous mediator secretion from mouse motor-nerve ending. Bull. Exp. Biol. Med. 87(5): 402–405. https://doi.org/10.1007/BF00806665
Magazanik L.G., Nikol’ski E.E. (1979) Pre- and postsynaptic action of a cholinomimetic (subecholine) under a voltage clamp of the postsynaptic membrane. Dokl. Akad. Nauk. SSSR. 249(6): 1488–1491.
Magazanik L.G., Potapjeva N.N., Fedorova I.M. (1998) Modulation of transmitter release from motor nerve endings by muscarinic and adenosine receptors. J. Neurochem. 71: S4.
Nikolsky E.E., Vyskocil F., Bukharaeva E.A., Samigullin D., Magazanik L.G. (2004) Cholinergic regulation of the evoked quantal release at frog neuromuscular junction. J. Physiol. 560: 77–88. https://doi.org/10.1113/jphysiol.2004.065805.4
Bukharaeva E.A., Samigullin D., Nikolsky E.E., Magazanik L.G. (2007) Modulation of the kinetics of evoked quantal release at mouse neuromuscular junctions by calcium and strontium. J. Neurochem. 100(4): 939–949. https://doi.org/10.1111/j.1471-4159.2006.04282.x
Santafé M.M., Salon I., Garcia N., Lanuza M.A., Uchitel O.D., Tomàs J. (2003) Modulation of ACh release by presynaptic muscarinic autoreceptors in the neuromuscular junction of the newborn and adult rat. Eur. J. Neurosci. 17(1): 119–127. https://doi.org/10.1046/j.1460-9568.2003.02428.x
Khaziev E.F., Fatikhov N.F., Samigullin D.V., Barrett G.L., Bukharaeva E.A., Nikolsky E.E. (2012) Decreased entry of calcium into motor nerve endings upon activation of presynaptic cholinergic receptors. Dokl. Biol. Sci. 446: 283–285. https://doi.org/10.1134/S0012496612050080
Connor E.A., Dunaevsky A., Griffiths D.J., Hardwick J.C., Parsons R.L. (1997) Transmitter release differs at snake twitch and tonic endplates during potassium-induced nerve terminal depolarization. J. Neurophysiol. 77(2): 749–760. https://doi.org/10.1152/jn.1997.77.2.749
Foldes F.F., Chaudhry I.A., Kinjo M., Nagashima H. (1989) Inhibition of mobilization of acetylcholine: the weak link in neuromuscular transmission during partial neuromuscular block with d-tubocurarine. Anesthesiology. 71: 218–223.
Somogyi G.T., Vizi E.S., Chaudhry I.A., Nagashima H., Duncalf D., Foldes F.F., Goldiner P.L. (1987) Modulation of stimulation-evoked release of newly formed acetylcholine from mouse hemidiaphragm preparation. Naunyn Schmiedebergs Arch. Pharmacol. 336(1): 11–15. https://doi.org/10.1007/BF00177744
Katz B., Miledi R. (1979) Estimates of quantal content during 'chemical potentiation' of transmitter release Proc. R. Soc. Lond. B. Biol. Sci. 205(1160): 369–378. https://doi.org/10.1098/rspb.1979.0070
Li R.A., Ennis I.L., Vélez P., Tomaselli G.F., Marbán E. (2000) Novel structural determinants of mu-conotoxin (GIIIB) block in rat skeletal muscle (mu1) Na+channels. J. Biol. Chem. 275(36): 27551–27558. https://doi.org/10.1074/jbc.M909719199
Wessler I., Halank M., Rasbach J., Kitbinger H. (1986) Presynaptic nicotine receptors mediating a positive feed-back on transmitter release from the rat phrenic nerve. Naunyn-Schmiedebergs Arch. Pharmacol. 334: 365–372. https://doi.org/10.1007/BF00569371
Wessler I., Apel C., Garmsen M., Klein A. (1992) Effects of nicotine receptor agonists on acetylcholine release from the isolated motor nerve, small intestine and trachea of rats and guinea-pigs. Clin. Investig. 70(3–4): 182–189. https://doi.org/10.1007/BF00184649
Wessler I., Scheuer B., Kilbinger H. (1987) [3H]acetylcholine release from the phrenic nerve is increased or decreased by activation or desensitization of nicotine receptors. Eur. J. Pharmacol. 135(1): 85–87. https://doi.org/10.1016/0014-2999(87)90760-6
Giniatullin R.A., Magazanik L.G. (1998) Does desensitisation of acetylcholine receptors play a physiological role in the neuromuscular synapse? Ross. Fiziol. Zh. Im. I. M Sechenova. 84(1–2): 3–14.
Wilson D.F. (1982) Influence of presynaptic receptors on neuromuscular transmission in rat. Am. J. Physiol. 242(5): 366–372. https://doi.org/10.1152/ajpcell.1982.242.5.C366
Prior C., Dempster J., Marshall I.G. (1993) Electrophysiological analysis of transmission at the skeletal neuromuscular junction. J. Pharmacol. Toxicol. Met. 30: 1–17. https://doi.org/10.1016/1056-8719(93)90002-V
Prior C., Singh S. (2000) Factors influencing the low-frequency associated nicotinic ACh autoreceptor-mediated depression of ACh release from rat motor nerve terminals. Br. J. Pharmacol. 129(6): 1067–1074. https://doi.org/10.1038/sj.bjp.0707442
Singh S., Prior C. (1998) Prejunctional effects of the nicotinic ACh receptor agonist dimethylphenylpiperazinium at the rat neuromuscular junction. J. Physiol. 511: 451–560. https://doi.org/10.1111/j.1469-7793.1998.451bh.x
Balezina O.P., Fedorin V.V., Gaidukov A.E. (2006) Effect of nicotine on neuromuscular transmission in mouse motor synapses. Bull. Exp. Biol. Med. 142(1): 17–21. https://doi.org/10.1007/s10517-006-0280-32006
Gaydukov A.E., Bogacheva P.O., Tarasova E.O., Balezina O.P. (2014) The mechanism of choline-mediated inhibition of acetylcholine release in mouse motor synapses. Acta Naturae. 6(4): 110–115. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4273098/pdf/AN20758251-23-110.pdf
Apel C., Rícný J., Wagner C., Wessler I. (1995) alpha-Bungarotoxin, kappa-bungarotoxin, alpha-cobratoxin and erabutoxin-b do not affect [3H]acetylcholine release from the rat isolated left hemidiaphragm. Naunyn Schmiedebergs Arch. Pharmacol. 352(6): 646–652. https://doi.org/10.1007/BF00171324
Vizi E.S., Chaudhry I.A., Goldiner P.L., Ohta Y., Nagashima H., Foldes F.F. (1991) The pre- and postjunctional components of the neuromuscular effect of antibiotics. J. Anesth. 5(1): 1–9. https://doi.org/10.1007/s0054010050001
Vizi E.S., Somogyi G.T., Nagashima H., Duncalf D., Chaudhry I.A., Kobayashi O., Goldiner P.L., Foldes F.F. (1987) Tubocurarine and pancuronium inhibit evoked release of acetylcholine from the mouse hemidiaphragm preparation. Br. J. Anaesth. 59(2): 226–231. https://doi.org/10.1093/bja/59.2.226
Faria M., Oliveira L., Timoteo M.A., Lobo M.G., Correia-De-Sa P. (2003) Blockade of neuronal facilitatory nicotinic receptors containing alpha 3 beta 2 subunits contribute to tetanic fade in the rat isolated diaphragm. Synapse. 49: 77–88 https://doi.org/10.1002/syn.10211
Vizi E.S., Somogyi G.T. (1989) Prejunctional modulation of acetylcholine release from the skeletal neuromuscular junction: link between positive (nicotinic)-and negative (muscarinic)-feedback modulation. Br. J. Pharmacol. 97(1): 65–70. https://doi.org/10.1111/j.1476-5381.1989.tb11924.x
Wessler I. (1992) Acetylcholine at motor nerves: storage, release, and presynaptic modulation by autoreceptors and adrenoceptors. Int. Rev. Neurobiol. 34: 283–384. https://doi.org/10.1016/s0074-7742(08)60100-2
Kimura I., Okazaki M., Uwano T., Kobayashi S., Kimura M. (1991) Succinylcholine-induced acceleration and suppression of electrically evoked acetylcholine release from mouse phrenic nerve-hemidiaphragm muscle preparation. Jpn. J. Pharmacol. 57(3): 397–403. https://doi.org/10.1254/jjp.57.397
Tian L., Prior C., Dempster J., Marshall I.G. (1994) Nicotinic antagonist-produced frequency-dependent changes in acetylcholine release from rat motor nerve terminals. J. Physiol. 476(3): 517–529. https://doi.org/10.1113/jphysiol.1994.sp020151
Ferry C.B., Kelly S.S. (1988) The nature of the presynaptic effects of (+)-tubocurarine at the mouse neuromuscular junction. J. Physiol. 403: 425–437. https://doi.org/10.1113/jphysiol.1988.sp017257
Wilson D.F. (1982) Influence of presynaptic receptors on neuromuscular transmission in rat. Am. J. Physiol. 242: 366–373. https://doi.org/10.1152/ajpcell.1982.242.5.C366
Wilson D.F., Thomsen R.H. (1991) Nicotinic receptors on the rat phrenic nerve: evidence for negative feedback. Neurosci. Lett. 132(2): 163–166. https://doi.org/10.1016/0304-3940(91)90292-2
Tian L., Prior C., Dempster J., Marshall I.G. (1997) Hexamethonium- and methyllycaconitine-induced changes in acetylcholine release from rat motor nerve terminals. Br. J. Pharmacol. 122(6):1025–1034. https://doi.org/10.1038/sj.bjp.0701481
Wilson D.F., Thomsen R.H. (1992) Effects of hexamethonium on transmitter release from the rat phrenic nerve. Neurosci. Lett. 143(1–2): 79–82. https://doi.org/10.1016/0304-3940(92)90237-2
Wilson D.F., West A.E., Lin Y. (1995) Inhibitory action of nicotinic antagonists on transmitter release at the neuromuscular junction of the rat. Neurosci. Lett. 186(1): 29–32. https://doi.org/10.1016/0304-3940(95)11274-Z
Domet M.A., Webb C.E., Wilson D.F. (1995) Impact of alpha-bungarotoxin on transmitter release at the neuromuscular junction of the rat. Neurosci. Lett. 199(1): 49–52. https://doi.org/10.1016/0304-3940(95)12013-t
Prior C., Tian L., Dempster J., Marshall I.G. (1995) Prejunctional actions of muscle relaxants: Synaptic vesicles and transmitter mobilization as sites of action. Gen. Pharmacol. 26: 659–666. https://doi.org/10.1016/0306-3623(94)00246-j
Bowman W.C., Prior C., Marshall I.G. (1990) Presynaptic Receptors in the Neuromuscular Junction. Ann. N. Y. Acad. Sci. 604: 69–81. https://doi.org/10.1111/j.1749-6632.1990.tb31983.x
Bowman W.C., Marshall L.G., Gibb A.G., Harbome A.J. (1988) Feedback control of transmitter release at the neuromuscular junction. Trends Pharmacol. Sci. 9: 16–20.
Jones W., Salpeter M. (1983) Absence of [125I] alpha-bungarotoxin binding to motor nerve terminals of frog, lizard and mouse muscle. J. Neurosci. 3(2): 326–331. https://doi.org/10.1523/JNEUROSCI.03-02-00326
Tsuneki H., Kimura I., Dezaki K., Kimura M., Sala C., Fumagalli G. (1995) Immunohistochemical localization of neuronal nicotinic receptor subtypes at the pre- and postjunctional sites in mouse diaphragm muscle. Neurosci. Lett. 196: 13–16. https://doi.org/10.1016/0304-3940(95)11824-G
Petrov K.A., Girard E., Nikitashina A.D., Colasante C., Bernard V., Nurullin L., Leroy J., Samigullin D., Colak O., Nikolsky E., Plaud B., Krejci E. (2014) Schwann cells sense and control acetylcholine spillover at the neuromuscular junction by α7 nicotinic receptors and butyrylcholinesterase. J. Neurosci. 34(36): 11870–11883. https://doi.org/10.1523/JNEUROSCI.0329-14.2014
Fagerlund M.J., Eriksson L.I. (2009) Current concepts in neuromuscular transmission. Br. J. Anaesth. 103(1): 108–114. https://doi.org/10.1093/bja/aep150
Jonsson M., Gurley D., Dabrowski M., Larsson O., Johnson E.C., Eriksson L.I. (2006) Distinct pharmacologic properties of neuromuscular blocking agents on human neuronal nicotinic acetylcholine receptors: a possible explanation for the train-of-four fade. Anesthesiology. 105(3): 521–533. https://doi.org/10.1097/00000542-200609000-00016
Wessler I. (1989) Control of transmitter release from the motor nerve by presynaptic nicotinic and muscarinic autoreceptors. Trends Pharmacol. Sci. 10(3): 110–114. https://doi.org/10.1016/0165-6147(89)90208-3
Vizi E.S., Kiss J., Elenkov I.J. (1991) Presynaptic modulation of cholinergic and noradrenergic neurotransmission: interaction between them. NIPS. 6: 119–123.
Miledi R., Molenaar P.C, Polak RL (1978) Alpha-Bungarotoxin enhances transmitter “released” at the neuromuscular junction. Nature. 272(5654): 641–643. https://doi.org/10.1038/272641a0
Kabbani N., Nichols R.A. (2018) Beyond the Channel: Metabotropic Signaling by Nicotinic Receptors. Trends Pharmacol. Sci. 39(4): 354–366. https://doi.org/10.1016/j.tips.2018.01.002
Tsuneki H., Klink R., Léna C., Korn H., Changeux J.P. (2000) Calcium mobilization elicited by two types of nicotinic acetylcholine receptors in mouse substantia nigra pars compacta. Eur. J. Neurosci. 12(7): 2475–2485. https://doi.org/10.1046/j.1460-9568.2000.00138.x
Djemil S., Chen X., Zhang Z., Lee I., Rauf M., Pak D., Dzakpasu R. (2020) Activation of nicotinic acetylcholine receptors induces potentiation and synchronization within in vitro hippocampal networks. J. Neurochem. 153(4): 468–484. https://doi.org/10.1111/jnc.14938
Mukhamedyarov M., Kochunova J., Yusupova E. (2010) The contribution of calcium/calmodulin-dependent protein-kinase II (CaMKII) to short-term plasticity at the neuro-muscular junction. Brain Res. Bull. 81(6): 613–616. https://doi.org/10.1016/j.brainresbull.2009.12.010
Kulak J.M., McIntosh J.M., Yoshikami D., Olivera B.M. (2001) Nicotine-evoked transmitter release from synaptosomes: functional association of specific presynaptic acetylcholine receptors and voltage-gated calcium channels. J. Neurochem. 77(6): 1581–1589. https://doi.org/10.1046/j.1471-4159.2001.00357.x
Bowman W.C. (1989) Presynaptic nicotinic autoreceptors. Trends Pharmacol. Sci. 10(4): 136–137. https://doi.org/10.1016/0165-6147(89)90162-4
Tarasova E.O., Gaydukov A.E., Balezina O.P. (2015) Methods of activation and the role of calcium/calmodulin-dependent protein kinase II in the regulation of acetylcholine secretion in the motor synapses of mice. Neurochem. J. 9(2): 101–107. https://doi.org/10.1134/S1819712415020099
Gaydukov A.E., Balezina O.P. (2017) CaMKII Is Involved in the Choline-Induced Downregulation of Acetylcholine Release in Mouse Motor Synapses. Acta Naturae. 9(4): 110–113.
Gaydukov A.E., Bogacheva P.O., Balezina O.P. (2019) The Participation of Presynaptic Alpha7 Nicotinic Acetylcholine Receptors in the Inhibition of Acetylcholine Release during Long-Term Activity of Mouse Motor Synapses. Neurochem. J. 13(1): 20–27. https://doi.org/10.1134/S1819712419010082
Shen J.X., Yakel J.L. (2009) Nicotinic acetylcholine receptor-mediated calcium signaling in the nervous system. Acta Pharmacol. Sin. 30(6):673–680. https://doi.org/10.1038/aps.2009.64
King J.R., Ullah A., Bak E., Jafri M.S., Kabbani N. (2018) Ionotropic and metabotropic mechanisms of allosteric modulation of α7 nicotinic receptor intracellular calcium. Mol. Pharmacol. 93: 601–61. https://doi.org/10.1124/mol.117.111401
Penner R., Dreyer F. (1986) Two different presynaptic calcium currents in mouse motor nerve terminals. Pfugers Arch. 406: 190–197. https://doi.org/10.1007/BF00586682
Wood S.J., Slater C.R. (2001) Safety factor at the neuromuscular junction. Prog. Neurobiol. 64(4): 393–429. https://doi.org/10.1016/s0301-0082(00)00055-1
Zhangsun D., Zhu X., Wu Y., Hu Y., Kaas Q., Craik D.J., McIntosh J.M., Luo S. (2015). Key residues in the nicotinic acetylcholine receptor β2 subunit contribute to α-conotoxin LvIA binding. J. Biol. Chem. 290(15): 9855–9862. https://doi.org/10.1074/jbc.M114.632646
Petrov K.A., Malomouzh A.I., Kovyazina I.V., Krejci E., Nikitashina A.D., Proskurina S.E, Zobov V.V., Nikolsky E.E. (2013) Regulation of acetylcholinesterase activity by nitric oxide in rat neuromuscular junction via N-methyl-D-aspartate receptor activation. Eur. J. Neurosci. 37(2): 181–189. https://doi.org/10.1111/ejn.12029
Noronha-Matos J.B., Oliveira L., Peixoto A., Almeida L., Castellão-Santana M.L., Ambiel C.R., Alves-do Prado W., Correia-de-Sá P. (2020) Nicotinic α7 receptor-induced adenosine release from perisynaptic Schwann cells controls acetylcholine spillover from motor endplates. J. Neurochem. 154(3): 263–283. https://doi.org/10.1111/jnc.14975
Correia-de-Sá P., Sebastião A.M., Ribeiro J.A. (1991) Inhibitory and excitatory effects of adenosine receptor agonists on evoked transmitter release from phrenic nerve ending of the rat. Br. J. Pharmacol. 103: 1614–1620. https://doi.org/10.1111/j.1476-5381.1991.tb09836.x
Wang X., McIntosh J.M., Rich M.M. (2018) Muscle Nicotinic Acetylcholine Receptors May Mediate Trans-Synaptic Signaling at the Mouse Neuromuscular Junction. J. Neurosci. 38(7): 1725–1736. https://doi.org/10.1523/JNEUROSCI.1789-17.2018
Дополнительные материалы отсутствуют.
Инструменты
Российский физиологический журнал им. И.М. Сеченова