

Российский физиологический журнал им. И.М. Сеченова, 2021, T. 107, № 4-5, стр. 474-491
Роль нейро-кардиального соединения в симпатической регуляции сердца
Ю. Г. Одношивкина 1, А. М. Петров 1, 2, *
1 Казанский государственный медицинский университет
Казань, Россия
2 Казанский институт биохимии и биофизики Казанского научного центра РАН
Казань, Россия
* E-mail: fysio@rambler.ru
Поступила в редакцию 19.01.2021
После доработки 02.02.2021
Принята к публикации 03.02.2021
Аннотация
Один из важных механизмов сердечной регуляции реализуется через иннервацию кардиомиоцитов нейронами симпатической нервной системы. Симпатические аксоны ветвятся и формируют на своем протяжении расширения (варикозы), содержащие синаптические везикулы с основным нейромедиатором (норадреналином) и ко-нейромедиаторами. Варикозы тесно контактируют с кардиомиоцитами, в результате могут формироваться нейро-кардиальные соединения, имеющие синапс-подобную организацию – специализированные пре- и постсинаптические регионы, разделенные узкой щелью. Эти синаптические образования подвержены пластичности и освобождение нейромедиатора из пресинаптических варикозов плотно регулируется, в том числе, со стороны ауторецепторов. Нейро-кардиальная передача имеет быстрые хронотропный и инотропный эффекты, а также управляет трофическими процессами, определяющими размеры кардиомиоцитов и архитектуру сердечной стенки. Разные подтипы постсинаптических адренорецепторов вовлечены в эти кратковременные и долговременные эффекты нейро-кардиальных взаимодействий. Изменения в адренергической нейропередаче в сердце часто сопровождают многие распространенные патологии (сердечная недостаточность, аритмии, гипертония), внося вклад в их развитие. В представленном обзоре мы систематизировали и обобщили экспериментальные данные, свидетельствующие о существовании в сердце синаптической передачи, которая может иметь решающее значение в коммуникации между мозгом и сердцем.
ВВЕДЕНИЕ
Главный путь регуляции мозгом деятельности сердца реализуется через симпатическую и парасимпатическую нервную систему. За счет этой коммуникации контролируется ритм сердца, артериальное давление и трофические процессы. В представленном обзоре мы сосредоточились на симпатической ветви нейро-кардиальных взаимодействий. Рабочий миокард густо иннервируется симпатическими нейронами, изменение активности которых происходит при многих патологических состояниях. В частности, хроническая гиперактивность симпатических нейронов часто наблюдается при аритмиях, гипертензии, сердечной недостаточности и гипертрофии, а острая гиперактивация симпатической нервной системы увеличивает риск возникновения аритмий и внезапной смерти [1, 2]. Учитывая широкую распространенность данных состояний, бета-блокаторы, угнетающие эффекты симпатической нервной системы на сердце, являются одними из самых часто используемых препаратов. С другой стороны, снижение эффективности действия симпатической нервной системы на сердце (за счет уменьшения плотности симпатической иннервации, содержания симпатического нейромедиатора – норадреналина (НА)) может наблюдаться с возрастом [3, 4]. Более того, при болезни Паркинсона общим немоторным симптомом является сердечная дизавтономия, которая проявляется на ранней стадии заболевания и сопряжена с уменьшением симпатической иннервации сердца, а также захвата НА в синаптические везикулы симпатических нервных окончаний [5, 6]. Снижение функциональной активности симпатических нервных окончаний может сопровождаться усилением предсердных аритмий [7]. Низкая экспрессия фермента дофамин-бета-гидроксилазы, приводящая к снижению синтеза НА, симпатической иннервация сердца и захвата НА в синаптические везикулы, встречается при семейных формах дизавтономии, которые вызывают нарушения ритма и сократимости сердца, а также увеличивают риск внезапной смерти [8]. Все это указывает на важное значение коммуникации между симпатическими нервами и кардиомиоцитами в обеспечении жизнедеятельности организма.
СИМПАТИЧЕСКАЯ ИННЕРВАЦИЯ СЕРДЦА И ПРЕСИНАПТИЧЕСКИЕ ВАРИКОЗЫ
Шейно-грудной симпатический звездчатый ганглий, расположенный рядом с T1–T4 сегментами, преимущественно иннервирует сердце. Выходящие из ганглия симпатические аксоны проникают в сердце через эпикард, где ветвятся и проходят через миокард, располагаясь близко к сосудам [9]. Отростки симпатических аксонов имеют характерную морфологию с повторяющимися расширениями (варикозами), содержащими заполненные нейромедиатором синаптические везикулы (размером 30–60 нм). Длина отдельного варикоза ~1–2 мкм и ширина ~0.6 мкм, варикозы обычно сгруппированы, и дистанция между ними составляет несколько микрон, разделенных тонким (0.1–0.2 мкм) отрезком аксона [10, 11]. Почти все кардиомиоциты имеют контакты с несколькими симпатическими варикозами [12, 13]. Судя по иммуногистохимическим данным, симпатические варикозы содержат типичный пресинаптический белковый аппарат. В частности, в них локализуются белки, необходимые для экзо- и эндоцитоза синаптических везикул, например, синтаксин 1, синапсин 1, SNAP-25, синаптотагмин-1, SV-2 [14–17]. Пресинаптический SNARE белок синтаксин формирует скопления (0.2 мкм2) в мембране варикоза, рядом с которыми распределены синаптические везикулы, это указывает на формирования специализированных сайтов освобождения нейромедиатора – активных зон [10, 16]. Количество докированных везикул в отдельном варикозе может составлять от 40 до 120 [10]. Экзоцитоз этих синаптических везикул запускается за счет входа Са2+ через потенциал-зависимые Са2+-каналы N-типа в ответ на потенциал действия. Дополнительно Са2+-вызванное освобождение Са2+ из эндоплазматического ретикулума через рианодиновые рецепторы 2 типа вносит вклад в Са2+-транзиент в симпатическом варикозе [18].
Варикозы симпатической нервной системы содержат синаптические везикулы, заполненные основным нейромедиатором – НА. После освобождения концентрация НА в примыкающем к симпатическую варикозу пространстве быстро уменьшается, что зависит от обратного захвата нейромедиатора Na+-зависимым транспортером SLC6A2 в нервные окончания [26]. Мутации в гене SLC6A2 ассоциируется с сердечной недостаточностью и тахикардией [27]. Мы предполагаем, что по аналогии с нервно-мышечными синапсами в нейро-кардиальных адренергических синапсах, вероятно, существует спонтанная и неквантовая секреция, уровень которой может влиять на установление базального ритма сердца и сократимости. Наличие секреции НА в покое согласуется с наблюдениями о том, что низкие дозы β-блокаторов могут снижать в покое ритм сердца, но не способны устранить тахикардию, вызванную освобождением НА при стимуляции кардиальных симпатических аксонов [15]. Это коррелирует с клиническими данными о том, что пациенты, получающие β-блокаторы, сохраняют частично хронотропный рефлекс, опосредуемый сильной активацией симпатической нервной системы. Следует отметить, что существование неквантовой секреции предполагается для холинергических нервных окончаний в сердце [19] и симпатических варикозов, иннервирующих гладкую мускулатуру [20].
Возможными дополнительными комедиаторами в синаптических везикулах могут быть гистамин и β-никотинамидадениндинуклеотид (β-НАД) [21–23]. Помимо синаптических везикул, в варикозах обнаруживаются гетерогенные по размерам электронно-плотные гранулы (обычно больше чем синаптические везикулы, до 130 нм в диаметре), которые расположены вдали от активной зоны [11]. Эти гранулы заполнены пептидами, в основном – нейропептидом Y, также могут содержать галанин и мозговой нейротрофический фактор. Освобождение пептид-содержащих гранул обычно происходит при высокочастотной активности симпатических нейронов. Пептиды связываются с высокой аффинностью с рецепторами и медленно расщепляются во внеклеточной среде, поэтому они способны диффузно передавать сигнал на соседние клетки или даже действовать системно. Нейропептид Y может ингибировать освобождение ацетилхолина (АХ) и НА через пресинаптические Y2-рецепторы и оказывать синергичный с НА положительный инотропный эффект, действуя через Y1-рецерторы кардиомиоцитов желудочков [1, 24–27]. Однако нейропептид Y может вызывать отрицательный инотропный эффект в препаратах предсердий и желудочков [28, 29], а также по-разному влиять на сократимость миокарда в зависимости от стадии постнатального онтогенеза [30]. Следовательно, конечный эффект нейропептида Y может варьировать в зависимости от условий, в том числе, от возраста [31, 32]. Экспрессия Y1- и Y2-рецепторов в сердце крыс увеличивалась после рождения, тогда как Y5-рецепторов, наоборот, снижалась [33]. При этом стимуляция Y5-рецепторов кардиомиоцитов, вероятно, имеет главным образом длительные эффекты, способствуя развитию гипертрофии сердца в ответ на хроническую гиперактивацию симпатической нервной системы [34, 35]. Мозговой нейротрофический фактор через TrkB-рецепторы тонически увеличивает сократимость кардиомиоцитов и скорость их расслабления [36].
В синаптических везикулах и электронно-плотных гранулах содержится также АТФ. После экзоцитоза в синаптической щели АТФ расщепляется до аденозина, способного ограничивать стимулирующие эффекты НА и нейропептида Y на сердце [37]. Сам АТФ через активацию метаботропных (P2Y1, P2Y2, P2Y4, P2Y6, P2Y11) и ионотропных (P2X, всех подтипов) пуринорецепторов кардиомиоцитов регулирует многие аспекты деятельности сердца, при этом часто проявляя стимулирующие эффекты [37, 38]. Более того, активация пуринорецепторов может усиливать позитивное инотропное действие стимуляции β-АР [39].
НЕЙРО-КАРДИАЛЬНЫЕ СОЕДИНЕНИЯ
До недавнего времени полагалось, что сердечные симпатические нервы выделяют НА в интерстиций миокарда, где НА диффундирует, проникая в межклеточные пространства и капилляры. Однако обнаружение с помощью электронной микроскопии относительно узкой синаптической щели (меньше 100 нм, и объемом меньше 0.1 мкм3) между симпатическими варикозами и кардиомиоцитами, а также оценка локальной концентрации НА в синаптической щели (около 100 нM) указывают на формирование нейро-кардиального контакта (рис. 1) по аналогии с классическим нервно-мышечным соединением [13, 15]. При этом размер щели варьирует от менее чем 20 до 100 нм [1], а с рабочими кардиомиоцитами обычно контактируют несколько варикозов от одного или нескольких нейронов [40], что напоминает полинейрональную иннервацию некоторых мышечных волокон [41]. Теоретически, формирование таких тесных нейро-кардиальных контактов позволяет обойти следующие ограничения эффективности “диффузного” освобождения НА: (1) суммарно, рабочие кардиомиоциты имеют на несколько порядков больший объем, чем иннервирующие миокард симпатические варикозы; (2) интенсивный коронарный кровоток способен быстро вымывать НА из интерстиция миокарда; (3) обилие разрушающих катехоловые амины ферментов в сердце, в частности, моноаминоксидаз, активность которых сопровождается продукцией активных форм кислорода [13, 42, 43]. Следовательно, точечная коммуникация через нейро-кардиальные соединения, вероятно, позволяет избежать избыточного выделения НА, что может быть токсично для кардиомиоцитов, а также требовать высокую активность ферментов, ответственных за синтез НА в симпатических нейронах. При существовании нейро-кардиальных соединений (рис. 1) только в условиях высокочастотной активности симпатического нейрона, когда способность транспортера повторно захватывать НА превышена, возможна значительная диффузия НА за пределы синаптической щели (“перелив”) и его глобальное действие на кардиомиоциты, в том числе, неиннервированные (в норме если они есть, то они имеют минорное значение [40]).
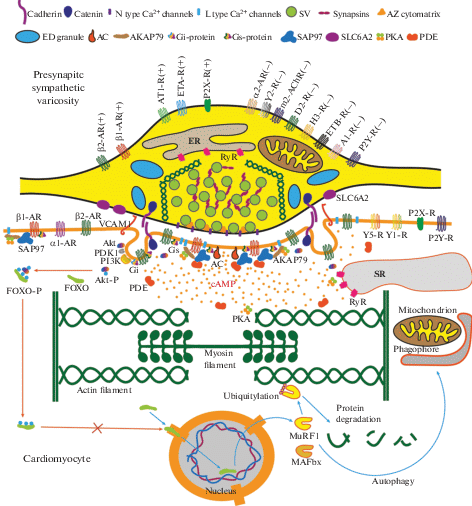
Рис. 1.
Схема нейро-кардиального соединения. Показан симпатический варикоз, контактирующий с кардиомиоцитом. Внутри варикоза синаптические везикулы (SVs) формируют скопления, часть SVs прикреплена к пресинаптической мембране в специализированной области – активной зоне (AZ), где сконцентрированы белки экзоцитоза и потенциал-зависимые Са2+-каналы N-типа. Дополнительно выход Са2+ через рианодиновые рецепторы (RyRs) из эндоплазматического ретикулума (ER) может усиливать освобождение нейромедиатора. Освобождение норадреналина из SVs может регулироваться со стороны многих пресинаптических рецепторов: активация β1-адренорецепторов (β1-АR), β2-АR, ангиотензиновых AT1-рецепторов (AT1-R), эндотелиновых ETA-рецепторов (ETA-R), пуриновых P2X-рецепторов (P2X-R) – усиливает (+) секрецию норадреналина, тогда как стимуляция α2-АR, Y2-рецепторов к нейропептиду Y (Y2-R), мускариновых m2-холинорецепторов (m2-АChR), дофаминовых D2-рецепторов (D2-R), гистаминовых H3-рецепторов (H3-R), эндотелиновых ETB-рецепторов (ETВ-R), пуриновых P2Y-рецепторов (P2Y-R), аденозиновых A1-рецепторов (A1-R) наоборот, ослабляет (–) секрецию норадреналина. После экзоцитоза норадреналин захватывается обратно в нервное окончание транспортером SLC6A2. В варикозах также присутствуют электронно-плотные гранулы (ED-granules), которые освобождают нейропептиды (в основном нейропептид Y) в ответ на высокочастотную активность. Молекулы адгезии (cadherins, catenin, VCAM1) обеспечивают сцепление пре- и постсинаптических мембран. На постсинаптической мембране кардиомиоцита локализуются в основном β1-ARs, также на периферии в кавеолах (липидных рафтах) экспрессируются β2-ARs. α1-ARs, вероятно, расположены преимущественно в экстрасинаптических областях кардиомиоцитов. Также рецепторы к нейропептиду Y (Y1-R, Y5-R) и ATФ (P2Y-R, P2Х-R) могут локализоваться на плазматической мембране в непосредственной близости к синаптическому региону. Каркасные белки (SAP97, AKAP79) формируют сайты для кластеризации β1-AR, Gs-белка, аденилатциклазы (AC), протеинкиназы А (PKA), фосфодиэстеразы (PDE), потенциал-зависимого Са2+-канала L типа, на их основе создаются сигнальные комплексы. При активации постсинаптических β-AR образуются локальные пулы цАМФ (cAMP), в зону действия которых попадают специфические комплексы PKA и белка-мишени (L-типа Са2+_канал, RyR саркоплазматического ретикулума (SR), тропонин и др.). Фосфорилирование последних вызывает реализацию быстрых эффектов активации симпатических нейронов. Тоническая стимуляция постсинаптических β2-AR запускает цепочку событий (в которых принимают участие Gi-белок, фосфоинозитид-3-киназа (PI3K), киназа PDK1), ведущую к фосфорилирование киназы Akt. Затем Akt фосфорилирует транскрипционный фактор FOXO, в итоге FOXO теряет способность проникать в ядро и усиливать экспрессию убиквинтинлигаз (MuRF1, MAFbx). Снижение интенсивности убиквинтилирования замедляет распад миокардиальных белков и интенсивность аутофагии, способствуя увеличению размеров иннервируемых кардиомиоцитов. Это медленный трофический эффект нейро-кардиальной передачи.
Основной механизм действия НА в сердце связан со стимуляцией β1-адренорецепторов (АР). После взаимодействия с агонистом β1-АР быстро (десятки миллисекунд) активирует Gs-белок, который увеличивает ферментативную активность аденилатциклазы, превращающую АТФ в цАМФ. В результате, в течение нескольких секунд после активации β1-АР уровень цАМФ увеличивается в объеме цитоплазмы (компартменте), непосредственно прилегающем к плазматической мембране, где локализуются стимулированные β1-АР. Активация одновременно многих β1-АР, распределенных по всей поверхности мембраны, ведет к глобальному повышению цАМФ в кардиомиоците. Подробная цепочка событий детально изложена в многочисленных обзорах (например, [44, 45]), и здесь мы остановимся только на некоторых моментах, которые особенно важны для понимания нейро-кардиальных взаимодействий.
Функциональные доказательства формирования нейро-кардиальных соединений были получены в экспериментах с ко-культурами (кардиомиоциты и симпатические нейроны). В этих экспериментах с помощью генетического сенсора оценивалась локальная концентрация вторичного посредника цАМФ в кардиомиоцитах. Было показано, что в ответ на освобождение НА из симпатических варикозов увеличение цАМФ наблюдалось только в иннервируемых (постсинаптических) кардиомиоцитах. Причем, в ближних к варикозу регионах кардиомиоцита локальный уровень цАМФ возрастал быстрее и сильнее, чем в дистальных регионах того же кардиомиоцита. Локальная концентрация НА в нейро-кардиальной щели была оценена в ~100 нМ, и экзогенная аппликация эквивалентной концентрации НА вызывала в десять раз более амплитудный ответ (рост цАМФ), вероятно, за счет активации экстрасинаптических рецепторов [15]. Эксперименты in vivo с оптогенетической активацией иннервирующих сердце симпатических нейронов [46] показали, что 10-миллисекундная стимуляция светом, вызывающая освобождение НА, достаточна для зависимого от β-АР ускорения сердечного ритма с задержкой только в ~170 мс [15]. Это указывает скорее на синаптическую передачу, нежели на действие НА, диффундирующего из варикозов через интерстициальную жидкость к кардиомиоцитам, хотя это время на порядок больше, чем синаптическая задержка в нервно-мышечных синапсах [47]. Однако стоит учитывать, что в кардиомиоцитах постсинаптический ответ медленный и связан с активаций, главным образом, метаботропных рецепторов, которые модулируют ионные токи.
ПОСТСИНАПТИЧЕСКАЯ СПЕЦИАЛИЗАЦИЯ НЕЙРО-КАРДИАЛЬНОГО СОЕДИНЕНИЯ
Относительно специализации постсинаптического региона в нейро-кардиальном соединении известно мало. В процессе развития формирование контакта между пресинаптическим варикозом и кардиомиоцитом требует взаимодействия интегрина α4β1 на пресинаптической стороне и молекулы адгезии VCAM-1 на кардиомиоцитах [48]. В ко-культуре в формировании нейро-кардиального соединения принимают участие кадгерины и β-катенины, формирующие адгезивные комплексы, стабилизирующие синаптический контакт [14]. Схожий механизм работает при образовании и стабилизации нервно-мышечных синапсов [49, 50]. Постсинаптические мембраны должны заякоривать рецепторные и сигнальные молекулы, обеспечивающие ответ на освобождение нейромедиатора. Различные каркасные (рапсин, MACF1) и цитоскелетные белки, наряду с липидными микродоменами (рафтами) участвуют в заякоривании постсинаптических сигнальных комплексов в нервно-мышечных синапсах [51, 54]. В нейро-кардиальном соединении обнаруженное формирование локального пула цАМФ в околомембранном пространстве, прилежащем к постсинаптической мембране [15], подразумевает компартментализацию сигнализации через путь β-АР – аденилатциклаза – цАМФ – протеинкиназа А, который активируется НА. При этом барьерами на пути диффузии цАМФ в кардиомиоците могут быть как внутриклеточные органеллы, мембранные инвагинации, так и заякоренные на цитоскелете фосфодиэстеразы, гидролизующие цАМФ [40, 55]. В ко-культуре, в месте контакта симпатического варикоза с мембраной кардиомиоцита, формируются специализированные зоны, где концентрируются каркасные белки SAP97 и AKAP79/150 вместе с β1- и β2-АР, а также молекулами адгезии (рис. 1). Рецепторная композиция таких постсинаптических регионов подвержена зависимой от активности пластичности, и кратковременная стимуляция симпатических нервных окончаний вызывает интернализацию избирательно β2-АР, в результате в постсинаптической зоне остаются преимущественно β1-АР [14]. В предсердиях β2-АР также могут концентрироваться в регионах кардиомиоцита, непосредственно окружающих пресинаптический варикоз, где могут ко-локализоваться с белком липидных рафтов – кавеолином 3 и сигнальными молекулами [56, 57]. Причем нарушение целостности липидных рафтов кардиомиоцитов за счет удаления или окисления их основного компонента – холестерина существенно подавляет вызванное активацией β2-АР увеличение Са2+-транзиента и сократимости [58, 59]. Следует отметить, что часто ассоциированные с липидными рафтами P2X1-рецепторы также могут формировать кластеры поблизости от симпатического варикоза в предсердиях и желудочках [60]. Возможно, обогащенные холестерином микродомены принимают участие наряду с каркасными белками в структурной и (или) функциональной организации постсинаптического региона в кардиомиоцитах, как это предполагается в нервно-мышечных синапсах [54, 61, 62].
Следует отметить, что кардиомиоциты контактируют с несколькими варикозами, которые могут принадлежать разным симпатическим нейронам [40]. Это потенциально создает возможность для формирования разных локальных пулов цАМФ и соответственно фосфорилирования разных белков-мишеней в одном кардиомиоците при активации разных симпатических нейронов. С другой стороны, формирование нескольких микропулов цАМФ в кардиомиоците, взамен глобального повышения цАМФ, может быть более энергетически эффективно, поскольку подвергаются фосфорилированию протеинкиназой А только специфичные белки. Локализация АР (особенно β2-подтипа) в отдельных регионах мембраны создает условиях для компартментализации сигнализации. Более того, в кардиомиоцитах β2-АР способны контролировать сигнализацию через основные β1-АР [59, 63, 64]. В частности, сильная стимуляция β2-АР может усиливать рекрутирование цАМФ-гидролизующей фосфодиэстеразы 4 к β1-АР, в результате активация последних ведет только к локальному повышению цАМФ и меньшему увеличению Са2+-транзинета. Подобная ситуация проявляется при стимуляции кардиомиоцитов желудочков одновременно неселективным β-агонистом (изопротеренолом, НА) в присутствии селективного агониста β2-АР (сальбутонола, зинтерола) [63]. Однако нейрональный метаболит холестерина 24-гидроксихолестерин (в нано- и микромолярных концентрациях) способен ослаблять вызванные изопротеренолом и зависимые, в основном, от β1-АР увеличение силы сокращений и Са2+-транзиента за счет опосредованной β2-АР активации фосфодиэстеразы 4 в предсердиях [64]. Более того, окисление мембранного холестерина способствует зависимому от β2-АР увеличению продукции активных форм кислорода, что значительно угнетает стимулирующие эффекты активации β1-АР (на сократимость, Са2+ транзиент и продукцию NO) в предсердиях мышей [59].
РЕГУЛЯЦИЯ ОСВОБОЖДЕНИЯ НЕЙРОМЕДИАТОРА В НЕЙРО-КАРДИАЛЬНЫХ СОЕДИНЕНИЯХ
Регуляция синаптической передачи основывается на контроле пресинаптических процессов, обеспечивающих освобождение нейромедиатора, и постсинаптических процессов рецепции молекул нейромедиатора. В отношении специфики регуляции постсинаптических рецепторов в нейро-кардиальных соединениях известно крайне мало, поэтому мы сосредоточимся на пресинаптических феноменах.
В любом синапсе функционируют петли обратной связи, направленные на оптимизацию секреции нейромедиатора, нейро-кардиальное соединение не исключение. НА, действуя через ауторецепторы (β1-, β2- и α2-АР) на нервных окончаниях, регулирует свое собственное освобождение в сердце (рис. 1). Активация пресинаптических β1/β2-АР и ангиотензиновых AT1-рецепторов может увеличивать освобождение НА в симпатических варикозах, тогда как α2-АР, мускариновых m2 ацетилхолиновых, дофаминовых D2- и гистаминовых H3-рецепторов имеет обратный эффект [2, 21, 65, 66]. Активация эндотелиновых рецепторов ETA и ETB на симпатических варикозах в желудочках может усиливать и угнетать освобождение НА соответственно; при этом стимулирующий эффект ETA-рецепторов преобладает при экзогенной аппликации эндотелина-1 [67]. Освобождаемый вместе с НА и пептидами АТФ может иметь быстрые эффекты, реализуемые через ионотропные P2X- (вероятно, P2X2- и P2X3-) рецепторы в сердце [37]. В частности, АТФ способен усиливать освобождение НА [68, 69]. Однако АТФ и его производное аденозин способны также тормозить освобождение НА в предсердиях, действуя через пресинаптические P2Y- и A1-аденозиновые рецепторы соответственно [70]. Отдельные субтипы P2Y, обеспечивающие контроль освобождения НА в сердце, остаются неизвестными. Другой ко-медиатор в симпатических варикозах – β-НАД может действовать через пуринорецепторы, как АТФ и также тормозить освобождение парасимпатического нейромедиатора – АХ [22].
При патологических условиях пресинаптические механизмы регуляции освобождения НА могут сильно модифицироваться, внося вклад в развитие заболевания. В ко-культурах, полученных из клеток гипертензивных животных, было показано, что усиленный прирост цАМФ в кардиомиоцитах при гипертензии связан преимущественно с облегчением освобождения НА из симпатических варикозов, а не с большей чувствительностью кардиомиоцитов к НА [71]. Подобное облегчение освобождения НА может определяться гиперактивацией пресинаптических ауторецепторов (β1-АР и особенно β2-АР), усиливающих освобождение НА; также вклад может вносить снижение обратного захвата НА в пресинаптические нервные окончания и увеличение экспрессии потенциал-зависимых Ca2+-каналов N-типа, обеспечивающих основной приток Са2+ в варикоз [2, 72, 73]. Усиление активации пресинаптических ангиотензиновых AT1- рецепторов способно увеличивать освобождение НА при гипертонии, а их делеция в симпатических нейронах снижает гипертензию, вызванную ангиотензином II [74]. Кроме того, AT1-рецепторы могут формировать гетеродимеры с пресинаптическими ингибиторными α2-АР, в результате последние теряют способность угнетать освобождение НА; более того, теперь при активации α2-АР запускается атипическая сигнализация, увеличивающая секрецию НА [75]. В том же ключе, формирование комплекса AT1-рецепторов со стимулирующими пресинаптическими β2-АР усиливает стабильность β2-АР на плазматической мембране и вызванное активацией β2-АР увеличение освобождения НА [76]. Повышенный адреналин в кровотоке тоже может значительно усиливать освобождение НА из симпатических варикозов в сердце, действуя через пресинаптические β-АР. Причем, в отличие от нормы, когда симпатические нейроны звездчатого ганглия выделят преимущественно НА, в условиях предгипертонии симпатические нейроны могут освобождать наряду с НА существенные порции адреналина из синаптических везикул. Это происходит за счет повышения активности в варикозах фермента фенилэтаноламин-N-метилтрансферазы (PNMT), превращающего НА в адреналин [2, 77]. При ишемии миокарда АТФ, способный разнонаправленно регулировать освобождение НА, преимущественно усиливает его освобождение, действуя через P2X-рецепторы [78]. Это может определяться изменением соотношения подтипов пуринорецепторов, в частности, повышением экспрессии P2X4-рецепторов [79]. В постишемический период, наоборот, часто наблюдается торможение экзоцитоза НА в сердце, что в значительной степени определяется действием повышенного уровня аденозина на A1-рецепторы [80]. Примечательно, что после инфаркта миокарда симпатические нервы могут подвергаться “трансдифференциации” и начинать освобождать наряду с НА и АХ из синаптических везикул [81]. Освобождение АХ может выступать в роли компенсаторного механизма, ограничивающего проаритмогенное действие НА [82]. Подобная холинергическая трансдифференциация симпатических нервов наблюдается и у пациентов с хронической сердечной недостаточностью [83]. Эти данные коррелируют с представлениями, что один синапс может использовать два основных нейромедиатора, упакованных в одни и те же или разные популяции синаптических везикул. Подобный двойственный фенотип может ярко проявляться в ходе развития или повреждений [84].
В целом, пресинаптические изменения, опосредующие увеличение освобождения НА, могут вносить вклад в развитие сердечных патологий (например, при сердечной недостаточности и гипертонии), хотя и угнетение освобождения НА также может иметь негативные последствия. Так, снижение способности накапливать и освобождать норадреналин из варикозов у B6CBAF1 мышей сопровождается уменьшением симпатического компонента контроля ритма сердца и усилением проаритмической активности (фибрилляции предсердий), в том числе, в ответ на экзогенное введение НА [7].
ТРОФИЧЕСКИЕ ЭФФЕКТЫ НЕЙРО-КАРДИАЛЬНОЙ КОММУНИКАЦИИ
Общепринято, что нейро-кардиальные взаимодействия имеют важные кратковременные последствия (контролируют хронотропию и инотропию). Однако меньшее внимание уделяется долговременным эффектам симпатической иннервации, которые определяют трофические процессы и ремоделирование миокарда.
В течение эмбрионального развития, выделяемые сердцем нейротрофические факторы контролируют установление симпатической иннервации и выживаемость симпатических нейронов; доступность этих факторов (в основном, нейронального фактора роста – NGF) определяет распределение и созревание симпатических отростков, формирующих контакты с кардиомиоцитами [85]. У мышей с делецией NGF или его рецептора TrkA наблюдается полная потеря постганглионарных симпатических нейронов [86, 87]. В процессе развития кардиомиоцитами выделяются ограниченные количества NGF, и его хватает, чтобы поддержать выживание только части симпатических нейронов [88]. Симпатические нейроны, полученные от человеческих плюрипотентных стволовых клеток, образовывали при ко-культивировании стабильные контакты с кардиальными мышечными клетками, но не со скелетными миотубами. Формирование этих контактов усиливало созревание нейронов [89]. Таким образом, в ходе развития миоциты сердца в значительной степени определяют размер пула симпатических нейронов, иннервирующих сердце, а также размер “сердечной двигательной единицы”.
Существуют и обратные влияния симпатических нейронов на трофические процессы в кардиомиоцитах. Было показано, что раннее установление симпатической иннервации важно для перехода кардиомиоцитов из фазы пролиферативного роста в фазу гипертрофического роста [90]. Также была обнаружена положительная корреляция между симпатической иннервацией и размером кардиомиоцитов. Во-первых, в ко-культуре кардиомиоциты, обладающие нейро-кардиальными контактами (иннервированные), имели существенно больший размер, чем неиннервированные кардиомиоциты. Во-вторых, in vivo в сердце кардиомиоциты, контактирующие с большим числом пресинаптических варикозов, имеют больший размер, чем те кардиомиоциты, что образуют соединения с меньшим числом симпатических варикозов [40, 91]. Хотя основные АР в кардиомиоцитах желудочков β1-АР и, затем, по уровню экспрессии следуют α1B-АР [92], трофический эффект симпатической иннервации на размер кардиомиоцитов зависит от активации β2-АР (рис. 1). Фармакологическое ингибирование β2-АР, как и абляция симпатических нейронов вызывает атрофическое ремоделирование кардиомиоцитов, сопровождающееся усиленной деградацией белков (за счет активации мышце-специфичных E3-убиквинтинлигаз MAFbx и MuRF1) с последующей активацией аутофаго-лизосомной системы [40, 91]. При этом контроль экспрессии убиквинтинлигаз осуществляется через путь Akt-FOXO. В частности, нейрокардиальная коммуникация приводит к зависимой от β2-АР активации Akt-киназы, которая фосфорилирует транскрипционный фактор FOXO, в результате последний перестает усиливать экспрессию данных лигаз, что препятствует избыточному протеолизу [13]. β2-АР активируют Akt опосредованно, что требует участия фосфоинозитол-3-киназы и Gi-белка и(или) бета-аррестина [45]. В частности, опосредуемое фосфоинозитид-3-киназой превращение фосфатидилинозитол-4,5-бифосфатов цитоплазматического слоя мембраны в фосфатидилинозитол-3,4,5-трифосфаты создает в мембране площадку (микродомен), необходимую для активации Akt посредством ее фосфорилирования зависимой от фосфоинозитидов киназой PDK1 [93]. Более того, способность β2-АР активировать Akt зависит от холестерина и липидных рафтов [57, 58], что дополнительно указывает на компартментализацию этой сигнализации в определенном регионе кардиомиоцита. В то же время трофический эффект нейро-кардиальных взаимодействий вряд ли связан с влиянием синаптической активности на FOXO-опосредуемую транскрипцию в особом “cубсинаптическом” ядре, поскольку в кардиомиоцитах обычно имеется 2–3 ядра, расположенных на отдалении от клеточной мембраны. В отличие от этого, в нервно-мышечном синапсе специфические субсинаптические ядра прилежат к постсинаптической мембране, обеспечивая зависимую от синаптической активности экспрессию специфичных генов, продукты которых необходимы для построения синаптического аппарата [94].
Следует отметить, что нейро-кардиальные взаимодействия, сопряженные с β2-АР могут быть вовлечены в ремоделирование сердца при патологиях, сопряженных с изменением экспрессии АР. Так, при сердечной недостаточности различного генеза доля β2-АР может увеличиваться, а β1-АР снижаться [59, 95]. Однако напрямую этот вопрос не исследован. Интересно, что β2-АР в изобилии экспрессируются на сердечных фибробластах и эндотелиальных клетках, однако симпатические нервные окончания не образуют стабильных контактов с этими клетками [13]. Следовательно, симпатические аксоны должны иметь механизмы селективного распознавания кардиомиоцитов и дальнейшего формирования точечного контакта. Даже после повреждения сердца симпатические нервы избирательно прорастают по направлению к кардиомиоцитам. Эта реиннервация важна для регенерации неонатального сердца. Так, симпатическая денервация сердца полностью подавляет регенерацию сердца у неонатальных мышей после удаления верхушки левого желудочка [96].
Кардиомиоциты в сердечной стенке, соединяясь щелевыми контактами, образуют слои, которые накладываются друг на друга. В слоях кардиомиоциты отличаются размером, ориентацией и паттерном экспрессии ионных каналов. Такая организация необходима для обеспечения механических свойств – развития оптимальной силы сокращения при сохранении прочности. Учитывая, что формирование синаптического контакта между симпатическим варикозом и кардиомиоцитом способствует гипертрофии последнего [91], установление нейро-кардиальных контактов может дирижировать формированием архитектуры стенки сердца. Более плотная симпатическая иннервация обнаруживается в эпикардиальных регионах (наружные слои миокарда) по сравнению с субэндокардиальными участками. Это показано как у грызунов, так и у людей [40]. Подобное распределение симпатических волокон положительно коррелирует с размером кардиомиоцитов в этих регионах сердечной стенки, а удаление симпатической иннервации, так же, как и системное хроническое применение агониста бета-АР, подавляет проявление трансмуральной гетерогенности размера кардиомиоцитов [40]. Безусловно, помимо неравномерности симпатической иннервации, нагрузка также влияет на размер кардиомиоцита, поскольку разные слои испытывают отличное активное и пассивное натяжение [97]. Потенциально гетерогенность электрокардиографических характеристик кардиомиоцитов частично может быть связана с неравномерностью симпатической иннервации. Так, гетерогенность симпатической иннервации влияет на динамику локальной реполяризации [98], а аритмии часто сопровождаются альтерациями симпатической иннервации и захвата меченых аналогов катехоловых аминов в отдельных регионах сердца [99].
Следует отметить, что вероятно не только НА, выделяемый из симпатических нервных окончаний, но и комедиаторы и нейропептиды играют роль в трофическом контроле [31, 32]. В частности, нейропептид Y может увеличивать количество белка в культивированных кардиомиоцитах желудочков, уменьшая расщепление белков и усиливая их синтез [100]. Более того, нейропептид Y способен усиливать гипертрофическое действие НА на кардиомиоциты желудочков [101]. Эти данные клеточных исследований согласуются со способностью нейропептида Y усиливать патологические изменения кардиомиоцитов в ряде моделей сердечной гипертрофии [34, 102].
ЗАКЛЮЧЕНИЕ
Сердце – самая сложноорганизованная мышца, которая обеспечивает создание давления для кровообращения. Регуляция деятельности сердца со стороны симпатической нервной системы является ключевой для адаптации кровообращения к потребностям организма. Данные последних лет указывают на осуществление прецизионной коммуникации симпатических нейронов с миоцитами сердца через нейро-кардиальные соединения – подобные синапсам образования. Такой тип взаимодействий обеспечивает возможность тонкой регуляции функционирования отдельных кардиомиоцитов и их групп, и в то же время позволяет избежать освобождения избытка нейромедиатора и нейромодуляторов из пресинаптических варикозов. Коммуникация через нейро-кардиальные соединения вызывает быстрые (хроно- и инотропные) и долговременные (трофические) сердечные эффекты, которые преимущественно реализуются за счет разных подтипов постсинаптических АР. Нейро-кардиальная передача подвержена пластичности и регулируется на пресинаптическом уровне за счет ауторецепторов и рецепторов, чувствительных к основным регуляторами кровообращения (ангиотензину II, эндотелинам, ацетилхолину). При многих заболеваниях (сердечная недостаточность, аритмии, гипертония) происходят изменения нейро-кардиальных связей, которые могут вносить значительный вклад в распространение патологий. Разработка фармакологических препаратов, нацеленных на коррекцию нейро-кардиальной передачи, может быть перспективным направлением в терапии широкого спектра сердечно-сосудистых нарушений. Однако перед этим предстоит предпринять большие усилия для понимания фундаментальных принципов организации фокусной коммуникации между симпатической нервной системой и сердцем.
Список литературы
Fedele L., Brand T. (2020) The Intrinsic Cardiac Nervous System and Its Role in Cardiac Pacemaking and Conduction. J. Cardiovasc. Dev. Dis. 7: 54. https://doi.org/10.3390/jcdd7040054
Bardsley E.N., Paterson D.J. (2020) Neurocardiac regulation: from cardiac mechanisms to novel therapeutic approaches. J. Physiol. 598: 2957–2976. https://doi.org/10.1113/JP276962
Francis Stuart S.D., Wang L., Woodard W.R., Ng G.A., Habecker B.A. Ripplinger C.M. (2018) Age-related changes in cardiac electrophysiology and calcium handling in response to sympathetic nerve stimulation. J. Physiol. 596: 3977–3991. https://doi.org/10.1113/JP276396
McLean M.R., Goldberg P.B., Roberts J. (1983) An ultrastructural study of the effects of age on sympathetic innervation and atrial tissue in the rat. J. Mol. Cell Cardiol. 15: 75–92. https://doi.org/10.1016/0022-2828(83)90284-5
Lamotte G., Holmes C., Wu T., Goldstein D.S. (2019) Long-term trends in myocardial sympathetic innervation and function in synucleinopathies. Parkinsonism Relat. Disord. 67: 27–33. https://doi.org/10.1016/j.parkreldis.2019.09.014
Safandeev V.V., Kolacheva A.A., Ugrumov M.V. (2019) Estimation of Metabolism of Catecholamines in Peripheral Organs As an Indicator of Their Desympathization under the Influence of Neurotoxins. Dokl. Biochem. Biophys. 486: 171–174. https://doi.org/10.1134/S1607672919030037
Kuzmin V.S., Potekhina V.M., Odnoshivkina Y.G., Chelombitko M.A. Fedorov A.V., Averina O.A. et al. (2020) Proarrhythmic atrial ectopy associated with heart sympathetic innervation dysfunctions is specific for murine B6CBAF1 hybrid strain. Life Sci. 266: 118887. https://doi.org/10.1016/j.lfs.2020.118887
Goldstein D.S., Eldadah B., Sharabi Y., Axelrod F.B. (2008) Cardiac sympathetic hypo-innervation in familial dysautonomia. Clin. Auton. Res. 18: 115–119. https://doi.org/10.1007/s10286-008-0464-1
Kimura K., Ieda M., Fukuda K. (2012) Development, maturation, and transdifferentiation of cardiac sympathetic nerves. Circ. Res. 110: 325–336. https://doi.org/10.1161/CIRCRESAHA.111.257253
Bennett M.R., Cheung A., Brain K.L. (1998) Sympathetic neuromuscular transmission at a varicosity in a syncytium. Microsc. Res. Tech. 42: 433–450. https://doi.org/10.1002/(SICI)1097-0029(19980915)42:6<433::AID-JEMT6>3.0.CO;2-N
Goyal R.K., Chaudhury A. (2013) Structure activity relationship of synaptic and junctional neurotransmission. Auton. Neurosci. 176: 11–31. https://doi.org/10.1016/j.autneu.2013.02.012
Freeman K., Tao W., Sun H., Soonpaa M.H., Rubart M. (2014) In situ three-dimensional reconstruction of mouse heart sympathetic innervation by two-photon excitation fluorescence imaging. J. Neurosci. Methods. 221: 48–61. https://doi.org/10.1016/j.jneumeth.2013.09.005
Zaglia T., Mongillo M. (2017) Cardiac sympathetic innervation, from a different point of (re)view. J. Physiol. 595: 3919–3930.https://doi.org/10.1113/JP273120
Shcherbakova O.G., Hurt C.M., Xiang Y., Dell’Acqua M.L., Zhang Q. Tsien R.W., Kobika B.K. (2007) Organization of beta-adrenoceptor signaling compartments by sympathetic innervation of cardiac myocytes. J. Cell Biol. 176: 521–533. https://doi.org/10.1083/jcb.200604167
Prando V., Da Broi F., Franzoso M., Plazzo A.P., Pianca N., Francolini M., Basso C., Kay M.W., Zaglia T., Mongillo M. (2018) Dynamics of neuroeffector coupling at cardiac sympathetic synapses. J. Physiol. 596: 2055–2075. https://doi.org/10.1113/JP275693
Brain K.L., Cottee L.J., Bennett M.R. (1997) Varicosities of single sympathetic nerve terminals possess syntaxin zones and different synaptotagmin N-terminus labelling following stimulation. J. Neurocytol. 26: 491–500. https://doi.org/10.1023/a:1018533524643
Sung U., Apparsundaram S., Galli A., Kahlig K.M., Savchenko V., Schroeter S., Quick M.W., Blakely R.D. (2003) A regulated interaction of syntaxin 1A with the antidepressant-sensitive norepinephrine transporter establishes catecholamine clearance capacity. J. Neurosci. 23: 1697–709. https://doi.org/10.1523/JNEUROSCI.23-05-01697.2003
Li D., Paterson D.J. (2019) Pre-synaptic sympathetic calcium channels, cyclic nucleotide-coupled phosphodiesterases and cardiac excitability. Semin. Cell Dev. Biol. 94: 20–27. https://doi.org/10.1016/j.semcdb.2019.01.010
Abramochkin D.V., Nurullin L.F., Borodinova A.A., Tarasova N.V., Sukhova G.S., Nikolsky E.E., Rosenshtraukh L.V. (2010) Non-quantal release of acetylcholine from parasympathetic nerve terminals in the right atrium of rats. Exp. Physiol. 95: 265–273. https://doi.org/10.1113/expphysiol.2009.050302
Bennett M.R., Farnell L., Gibson W.G., Lin Y.Q., Blair D.H. (2001) Quantal and non-quantal current and potential fields around individual sympathetic varicosities on release of ATP. Biophys. J. 80: 1311–1328. https://doi.org/10.1016/S0006-3495(01)76105-X
Li M., Hu J., Chen Z., Meng J., Wang H., Ma X., Luo X. (2006) Evidence for histamine as a neurotransmitter in the cardiac sympathetic nervous system. Am. J. Physiol. Heart Circ. Physiol. 291: H45–H51.https://doi.org/10.1152/ajpheart.00939.2005
Pustovit K.B., Potekhina V.M., Ivanova A.D., Petrov A.M., Abramochkin D.V., Kuzmin V.S. (2019) Extracellular ATP and beta-NAD alter electrical properties and cholinergic effects in the rat heart in age-specific manner. Purinergic Signal. 15: 107–117. https://doi.org/10.1007/s11302-019-09645-6
Smyth L.M., Bobalova J., Mendoza M.G., Lew C., Mutafova-Yambolieva V.N. (2004) Release of beta-nicotinamide adenine dinucleotide upon stimulation of postganglionic nerve terminals in blood vessels and urinary bladder. J. Biol. Chem. 279: 48893–48903. https://doi.org/10.1074/jbc.M407266200
Herring N., Lokale M.N., Danson E.J., Heaton D.A., Paterson D.J. (2008) Neuropeptide Y reduces acetylcholine release and vagal bradycardia via a Y2 receptor-mediated, protein kinase C-dependent pathway. J. Mol. Cell Cardiol. 44: 477–485. https://doi.org/10.1016/j.yjmcc.2007.10.001
Herring N., Cranley J., Lokale M.N., Li D., Shanks J., Alston E.N.,et al. (2012) The cardiac sympathetic co-transmitter galanin reduces acetylcholine release and vagal bradycardia: implications for neural control of cardiac excitability. J. Mol. Cell Cardiol. 52: 667–676. https://doi.org/10.1016/j.yjmcc.2011.11.016
Heredia Mdel P., Delgado C., Pereira L., Perrier R., Richard S., Vassort G. et al. (2005) Neuropeptide Y rapidly enhances [Ca2+]i transients and Ca2+ sparks in adult rat ventricular myocytes through Y1 receptor and PLC activation. J. Mol. Cell Cardiol. 38: 205–212. https://doi.org/10.1016/j.yjmcc.2004.11.001
Rump L.C., Riess M., Schwertfeger E., Michel M.C., Bohmann C., Schollmeyer P. (1997) Prejunctional neuropeptide Y receptors in human kidney and atrium. J. Cardiovasc. Pharmacol. 29: 656–661.https://doi.org/10.1097/00005344-199705000-00014
Oki Y., Teraoka H., Kitazawa T. (2017) Neuropeptide Y (NPY) inhibits spontaneous contraction of the mouse atrium by possible activation of the NPY1 receptor. Auton. Autacoid. Pharmacol. 37: 23–28. https://doi.org/10.1111/aap.12055
Piper H.M., Millar B.C., McDermott B.J. (1989) The negative inotropic effect of neuropeptide Y on the ventricular cardiomyocyte. Naunyn. Schmiedebergs Arch. Pharmacol. 340: 333–337. https://doi.org/10.1007/BF00168519
Zverev A.A., Anikina T.A., Maslyukov P.M., Zefirov T.L. (2014) Role of neuropeptide Y in myocardial contractility of rats during early postnatal ontogeny. Bull. Exp. Biol. Med. 157: 421–423. https://doi.org/10.1007/s10517-014-2581-2
Widiapradja A., Chunduri P., Levick S.P. (2017) The role of neuropeptides in adverse myocardial remodeling and heart failure. Cell Mol. Life. Sci. 74: 2019–2038. https://doi.org/10.1007/s00018-017-2452-x
Protas L., Qu J., Robinson R.B. (2003) Neuropeptide y: neurotransmitter or trophic factor in the heart? News. Physiol. Sci. 18: 181–185. https://doi.org/10.1152/nips.01437.2003
Masliukov P.M., Moiseev K., Emanuilov A.I., Anikina T.A., Zverev A.A., Nozdrachev A.D. (2016) Development of neuropeptide Y-mediated heart innervation in rats. Neuropeptides. 55: 47–54. https://doi.org/10.1016/j.npep.2015.10.007
Bell D., Allen A.R., Kelso E.J., Balasubramaniam A., McDermott B.J. (2002) Induction of hypertrophic responsiveness of cardiomyocytes to neuropeptide Y in response to pressure overload. J. Pharmacol. Exp. Ther. 303: 581–591. https://doi.org/10.1124/jpet.102.038448
Pellieux C., Sauthier T., Domenighetti A., Marsh D.J., Palmiter R.D., Brunner H.R. et al. (2000) Neuropeptide Y (NPY) potentiates phenylephrine-induced mitogen-activated protein kinase activation in primary cardiomyocytes via NPY Y5 receptors. Proc. Natl. Acad. Sci. USA. 97: 1595–1600. https://doi.org/10.1073/pnas.030533197
Feng N., Huke S., Zhu G., Tocchetti C.G., Shi S., Aiba T., Kaludercic N., Hoover D.B., Beck S.E., Mankowski J.L., Tomaselli G.F., Bers D.M., Kass D.A., Paolocci N. (2015) Constitutive BDN-F/TrkB signaling is required for normal cardiac contraction and relaxation. Proc. Natl. Acad. Sci. USA. 112: 1880–1885. https://doi.org/10.1073/pnas.1417949112
Burnstock G. (2017) Purinergic Signaling in the Cardiovascular System. Circ. Res. 120: 207–228. https://doi.org/10.1161/CIRCRESAHA.116.309726
Mei Q., Liang B.T. (2001) P2 purinergic receptor activation enhances cardiac contractility in isolated rat and mouse hearts. Am. J. Physiol. Heart. Circ. Physiol. 281: H334–H341. https://doi.org/10.1152/ajpheart.2001.281.1.H334
Anikina T.A., Zverev A.A., Sitdikov F.G., Anisimova I.N. (2013) Interaction of adrenergic and purinergic receptors in the regulation of rat myocardial contractility in postnatal ontogeny. Russ. J. Dev. Biol. 44: 296–301. https://doi.org/10.1134/S1062360413060027
Pianca N., Di Bona A., Lazzeri E., Costantini I., Franzoso M., Prando V. et al. (2019) Cardiac sympathetic innervation network shapes the myocardium by locally controlling cardiomyocyte size through the cellular proteolytic machinery. J. Physiol. 597(14): 3639–3656. https://doi.org/10.1113/JP276200
Vyskocil F., Magazanik L.G. (1977) Dual end-plate potentials at the single neuromuscular junction of the adult frog. Pflugers Arch. 368: 271–273. https://doi.org/10.1007/BF00585207
Thaemert J.C. (1966) Ultrastructure of cardiac muscle and nerve contiguities. J. Cell. Biol. 29: 156–162. https://doi.org/10.1083/jcb.29.1.156
Kaludercic N., Takimoto E., Nagayama T., Feng N., Lai E.W., Bedja D., Chen K., Gabrielson K.L., Blakely R.D., Shin J.C., Pacak K., Kass D.A., Lisa F.D., Paolocci N. (2010) Monoamine oxidase A-mediated enhanced catabolism of norepinephrine contributes to adverse remodeling and pump failure in hearts with pressure overload. Circ. Res. 106: 193–202. https://doi.org/10.1161/CIRCRESAHA.109.198366
Bhogal N.K., Hasan A., Gorelik J. (2018) The Development of Compartmentation of cAMP Signaling in Cardiomyocytes: The Role of T-Tubules and Caveolae Microdomains. J. Cardiovasc. Dev. Dis. 5: 25.https://doi.org/10.3390/jcdd5020025
Wang J., Gareri C., Rockman H.A. (2018) G-Protein-Coupled Receptors in Heart Disease. Circ. Res. 123: 716–735. https://doi.org/10.1161/CIRCRESAHA.118.311403
Wengrowski A.M., Wang X., Tapa S., Posnack N.G., Mendelowitz D., Kay M.W. (2015) Optogenetic release of norepinephrine from cardiac sympathetic neurons alters mechanical and electrical function. Cardiovasc. Res. 105: 143–150. https://doi.org/10.1093/cvr/cvu258
Tsentsevitsky A.N., Zakyrjanova G.F., Petrov A.M. (2020) Cadmium desynchronizes neurotransmitter release in the neuromuscular junction: Key role of ROS. Free Radic Biol. Med. 155: 19–28. https://doi.org/10.1016/j.freeradbiomed.2020.05.017
Wingerd K.L., Goodman N.L., Tresser J.W., Smail M.M., Leu S.T., Rohan S.J., Pring J.L., Jacksan D.Y., Clegg D.O. (2002) Alpha 4 integrins and vascular cell adhesion molecule-1 play a role in sympathetic innervation of the heart. J. Neurosci. 22: 10772–10780. https://doi.org/10.1523/JNEUROSCI.22-24-10772.2002
Wu H., Lu Y., Barik A., Joseph A., Taketo M.M. Xiong W.C. (2012) beta-Catenin gain of function in muscles impairs neuromuscular junction formation. Development. 139: 2392–2404. https://doi.org/10.1242/dev.080705
Cifuentes-Diaz C., Nicolet M., Goudou D., Rieger F., Mege R.M. (1994) N-cadherin expression in developing, adult and denervated chicken neuromuscular system: accumulations at both the neuromuscular junction and the node of Ranvier. Development. 120: 1–11.
Krivoi I.I., Petrov A.M. (2019) Cholesterol and the Safety Factor for Neuromuscular Transmission. Int. J. Mol. Sci. 20: 1046.https://doi.org/10.3390/ijms20051046
Chen P.J., Martinez-Pena Y.V.I., Aittaleb M., Akaaboune M. (2016) AChRs Are Essential for the Targeting of Rapsyn to the Postsynaptic Membrane of NMJs in Living Mice. J. Neurosci. 36: 5680–5685. https://doi.org/10.1523/JNEUROSCI.4580-15.2016
Oury J., Liu Y., Topf A., Todorovic S., Hoedt E., Preethish-Kumar V. et al. (2019) MACF1 links Rapsyn to microtubule- and actin-binding proteins to maintain neuromuscular synapses. J. Cell Biol. 218: 1686–1705. https://doi.org/10.1083/jcb.201810023
Marchand S., Devillers-Thiery A., Pons S., Changeux J.P., Cartaud J. (2002) Rapsyn escorts the nicotinic acetylcholine receptor along the exocytic pathway via association with lipid rafts. J. Neurosci. 22: 8891–8901. https://doi.org/10.1523/JNEUROSCI.22-20-08891.2002
Petrov A.M., Zefirov A.L. (2013) Cholesterol and lipid rafts in the biological membranes. Role in the release, reception and ion channel functions. Usp. Fiziol. Nauk. 44: 17–38.
Odnoshivkina U.G., Sytchev V.I., Nurullin L.F., Giniatullin A.R., Zefirov A.L., Petrov A.M. (2015) β2-adrenoceptor agonist-evoked reactive oxygen species generation in mouse atria: implication in delayed inotropic effect. Eur. J. Pharmacol. 765: 140–153. https://doi.org/10.1016/j.ejphar.2015.08.020
Sytchev V.I., Odnoshivkina Y.G., Ursan R.V., Petrov A.M. (2017) Oxysterol, 5α-cholestan-3-one, modulates a contractile response to β2-adrenoceptor stimulation in the mouse atria: Involvement of NO signaling. Life Sci. 188: 131–140. https://doi.org/10.1016/j.lfs.2017.09.006
Odnoshivkina Y.G., Sytchev V.I., Petrov A.M. (2017) Cholesterol regulates contractility and inotropic response to β2-adrenoceptor agonist in the mouse atria: Involvement of G i -protein–Akt–NO-pathway. J. Mol. Cell Cardiol. 107: 27–40. https://doi.org/10.1016/j.yjmcc.2016.05.001
Ursan R., Odnoshivkina U.G., Petrov A.M. (2019) Membrane cholesterol oxidation downregulates atrial beta-adrenergic responses in ROS-dependent manner. Cell Signal. 67: 109503. https://doi.org/10.1016/j.cellsig.2019.109503
Hansen M.A., Bennett M.R., Barden J.A. (1999) Distribution of purinergic P2X receptors in the rat heart. J. Auton. Nerv. Syst. 78: 1–9. https://doi.org/10.1016/s0165-1838(99)00046-6
Petrov A.M., Kravtsova V.V., Matchkov V.V., Vasiliev A.N., Zefirov A.L., Chibalin A.V. et al. (2017) Membrane lipid rafts are disturbed in the response of rat skeletal muscle to short-term disuse. Am. J. Physiol. Cell Physiol. 312: C627–C637. https://doi.org/10.1152/ajpcell.00365.2016
Kravtsova V.V., Petrov A.M., Vasil’ev A.N., Zefirov A.L., Krivoi I.I. (2015) Role of cholesterol in the maintenance of endplate electrogenesis in rat diaphragm. Bull. Exp. Biol. Med. 158(3): 298–300. https://doi.org/10.1007/s10517-015-2745-8
Yang H.Q., Wang L.P., Gong Y.Y., Fan X.X., Zhu S.Y., Wang X.T. (2019) beta2-Adrenergic Stimulation Compartmentalizes beta1 Signaling Into Nanoscale Local Domains by Targeting the C-Terminus of beta1-Adrenoceptors. Circ. Res. 124: 1350–1359. https://doi.org/10.1161/CIRCRESAHA.118.314322
Odnoshivkina U.G., Sytchev V.I., Starostin O., Petrov A.M. (2019) Brain cholesterol metabolite 24-hydroxycholesterol modulates inotropic responses to beta-adrenoceptor stimulation: The role of NO and phosphodiesterase. Life Sci. 220: 117–126. https://doi.org/10.1016/j.lfs.2019.01.054
Abadie C., Foucart S., Page P., Nadeau R. (1996) Modulation of noradrenaline release from isolated human atrial appendages. J. Auton. Nerv. Syst. 61: 269–276. https://doi.org/10.1016/s0165-1838(96)00093-8
Rump L.C., Riera-Knorrenschild G., Schwertfeger E., Bohmann C., Spillner G., Schollmeyer P. (1995) Dopaminergic and alpha-adrenergic control of neurotransmission in human right atrium. J. Cardiovasc. Pharmacol. 26: 462–470. https://doi.org/10.1097/00005344-199509000-00017
Isaka M., Kudo A., Imamura M., Kawakami H., Yasuda K. (2007) Endothelin receptors, localized in sympathetic nerve terminals of the heart, modulate norepinephrine release and reperfusion arrhythmias. Basic Res. Cardiol. 102: 154–162. https://doi.org/10.1007/s00395-006-0623-2
Sperlagh B., Erdelyi F., Szabo G., Vizi E.S. (2000) Local regulation of [(3)H]-noradrenaline release from the isolated guinea-pig right atrium by P(2X)-receptors located on axon terminals. Br. J. Pharmacol. 131: 1775–1783. https://doi.org/10.1038/sj.bjp.0703757
Machida T., Heerdt P.M., Reid A.C., Schafer U., Silver R.B. Broekman M.J. (2005) Ectonucleoside triphosphate diphosphohydrolase 1/CD39, localized in neurons of human and porcine heart, modulates ATP-induced norepinephrine exocytosis. J. Pharmacol. Exp. Ther. 313: 570–577. https://doi.org/10.1124/jpet.104.081240
von Kugelgen I., Stoffel D., Starke K. (1995) P2-purinoceptor-mediated inhibition of noradrenaline release in rat atria. Br. J. Pharmacol. 115: 247–254. https://doi.org/10.1111/j.1476-5381.1995.tb15870.x
Larsen H.E., Lefkimmiatis K., Paterson D.J. (2016) Sympathetic neurons are a powerful driver of myocyte function in cardiovascular disease. Sci. Rep. 6: 38898. https://doi.org/10.1038/srep38898
Larsen H.E., Bardsley E.N., Lefkimmiatis K., Paterson D.J. (2016) Dysregulation of Neuronal Ca2+ Channel Linked to Heightened Sympathetic Phenotype in Prohypertensive States. J. Neurosci. 36: 8562–8573. https://doi.org/10.1523/JNEUROSCI.1059-16.2016
Shanks J., Mane S., Ryan R., Paterson D.J. (2013) Ganglion-specific impairment of the norepinephrine transporter in the hypertensive rat. Hypertension. 61: 187–193. https://doi.org/10.1161/HYPERTENSIONAHA.112.202184
Jancovski N., Bassi J.K., Carter D.A., Choong Y.T., Connelly A., Nguyen T.P. (2013) Stimulation of angiotensin type 1A receptors on catecholaminergic cells contributes to angiotensin-dependent hypertension. Hypertension. 62: 866–871. https://doi.org/10.1161/HYPERTENSIONAHA.113.01474
Bellot M., Galandrin S., Boularan C., Matthies H.J., Despas F., Denis C. (2015) Dual agonist occupancy of AT1-R-alpha2C-AR heterodimers results in atypical Gs-PKA signaling. Nat. Chem. Biol. 11: 271–279. https://doi.org/10.1038/nchembio.1766
Toth A.D., Gyombolai P., Szalai B., Varnai P., Turu G., Hunyady L. (2017) Angiotensin type 1A receptor regulates beta-arrestin binding of the beta2-adrenergic receptor via heterodimerization. Mol. Cell Endocrinol. 442: 113–124. https://doi.org/10.1016/j.mce.2016.11.027
Bardsley E.N., Davis H., Buckler K.J., Paterson D.J. (2018) Neurotransmitter Switching Coupled to beta-Adrenergic Signaling in Sympathetic Neurons in Prehypertensive States. Hypertension. 71: 1226–1238. https://doi.org/10.1161/HYPERTENSIONAHA.118.10844
Sperlagh B., Heinrich A., Csolle C. (2007) P2 receptor-mediated modulation of neurotransmitter release-an update. Purinergic Signal. 3: 269–284. https://doi.org/10.1007/s11302-007-9080-0
Braganca B., Nogueira-Marques S., Ferreirinha F., Fontes-Sousa A.P., Correia-de-Sa P. (2019) The Ionotropic P2X4 Receptor has Unique Properties in the Heart by Mediating the Negative Chronotropic Effect of ATP While Increasing the Ventricular Inotropy. Front. Pharmacol. 10: 1103. https://doi.org/10.3389/fphar.2019.01103
Burgdorf C., Richardt D., Kurz T., Seyfarth M., Jain D., Katus H.A. (2001) Adenosine inhibits norepinephrine release in the postischemic rat heart: the mechanism of neuronal stunning. Cardiovasc. Res. 49: 713–720. https://doi.org/10.1016/s0008-6363(00)00309-6
Olivas A., Gardner R.T., Wang L., Ripplinger C.M., Woodward W.R., Habecker B.A. (2016) Myocardial Infarction Causes Transient Cholinergic Transdifferentiation of Cardiac Sympathetic Nerves via gp130. J. Neurosci. 36(2): 479–488. https://doi.org/10.1523/JNEUROSCI.3556-15.2016
Wang L., Olivas A., Francis Stuart S.D., Tapa S., Blake M.R., Woodward W.R (2020) Cardiac sympathetic nerve transdifferentiation reduces action potential heterogeneity after myocardial infarction. Am. J. Physiol. Heart. Circ. Physiol. 318: H558–H565. https://doi.org/10.1152/ajpheart.00412.2019
Kanazawa H., Ieda M., Kimura K., Arai T., Kawaguchi-Manabe H., Matsuhashi (2010) Heart failure causes cholinergic transdifferentiation of cardiac sympathetic nerves via gp130-signaling cytokines in rodents. J. Clin. Invest. 120: 408–421. https://doi.org/10.1172/JCI39778
Vaaga C.E., Borisovska M., Westbrook G.L. (2016) Dual-transmitter neurons: functional implications of co-release and co-transmission. Curr. Opin. Neurobiol. 29: 25–32. https://doi.org/10.1016/j.conb.2014.04.010
Franzoso M., Zaglia T., Mongillo M. (2016) Putting together the clues of the everlasting neuro-cardiac liaison. Biochim. Biophys. Acta. 1863: 1904–1915. https://doi.org/10.1016/j.bbamcr.2016.01.009
Crowley C., Spencer S.D., Nishimura M.C., Chen K.S., Pitts-Meek S., Armanini M.P., Ling L.M., McMahon S.B., Shelton D.L, Levinson A.D. (1994) Mice lacking nerve growth factor display perinatal loss of sensory and sympathetic neurons yet develop basal forebrain cholinergic neurons. Cell. 76: 1001–1011. https://doi.org/10.1016/0092-8674(94)90378-6
Smeyne R.J., Klein R., Schnapp A., Long L.K., Bryant S., Lewin A., Lira S.A. Barbacid M. (1994) Severe sensory and sympathetic neuropathies in mice carrying a disrupted Trk/NGF receptor gene. Nature. 368(6468): 246–249. https://doi.org/10.1038/368246a0
Mok S.A., Lund K., Campenot R.B. (2009) A retrograde apoptotic signal originating in NGF-deprived distal axons of rat sympathetic neurons in compartmented cultures. Cell Res. 19: 546–560. https://doi.org/10.1038/cr.2009.11
Oh Y., Cho G.S., Li Z., Hong I., Zhu R., Kim M.J. (2016) Functional Coupling with Cardiac Muscle Promotes Maturation of hPSC-Derived Sympathetic Neurons. Cell Stem. Cell 19: 95–106. https://doi.org/10.1016/j.stem.2016.05.002
Kreipke R.E., Birren S.J. (2015) Innervating sympathetic neurons regulate heart size and the timing of cardiomyocyte cell cycle withdrawal. J. Physiol. 593: 5057–5073. https://doi.org/10.1113/JP270917
Zaglia T., Milan G., Franzoso M., Bertaggia E., Pianca N., Piasentini E., Voltarelli V.A., Chiavegato D., Brum P.C., Glass D.J., Schaffino S., Sandri M., Mongillo M. (2013) Cardiac sympathetic neurons provide trophic signal to the heart via beta2-adrenoceptor-dependent regulation of proteolysis. Cardiovasc. Res. 97: 240–250. https://doi.org/10.1093/cvr/cvs320
Myagmar B.E., Flynn J.M., Cowley P.M., Swigart P.M., Montgomery M.D., Thai K., Nair D., Gupta R., Deng Dx, Hosoda C., Melov S., Baker A.J., Simpson P.C. (2017) Adrenergic Receptors in Individual Ventricular Myocytes: The Beta-1 and Alpha-1B Are in All Cells, the Alpha-1A Is in a Subpopulation, and the Beta-2 and Beta-3 Are Mostly Absent. Circ. Res. 120: 1103–1115. https://doi.org/10.1161/CIRCRESAHA.117.310520
Gao X., Lowry P.R., Zhou X., Depry C., Wei Z., Wong G.W. Zhang Y. (2011) PI3K/Akt signaling requires spatial compartmentalization in plasma membrane microdomains. Proc. Natl. Acad. Sci. USA. 108: 14509–14514. https://doi.org/10.1073/pnas.1019386108
Belotti E., Schaeffer L. (2020) Regulation of Gene expression at the neuromuscular Junction. Neurosci. Lett. 735: 135163 https://doi.org/10.1016/j.neulet.2020.135163
Wang J., Gareri C., Rockman H.A. (2018) G-Protein–Coupled Receptors in Heart Disease. Circul. Res. 123: 716–735. https://doi.org/10.1161/circresaha.118.311403
White I.A., Gordon J., Balkan W., Hare J.M. (2015) Sympathetic Reinnervation Is Required for Mammalian Cardiac Regeneration. Circ. Res. 117(12): 990–994 https://doi.org/10.1161/CIRCRESAHA.115.307465
Hoffman J.I. (1987) Transmural myocardial perfusion. Prog. Cardiovasc. Dis. 29: 429–464. https://doi.org/10.1016/0033-0620(87)90016-8
Yoshioka K., Gao D.W., Chin M., Stillson C., Penades E., Lesh M. et al. (2000) Heterogeneous sympathetic innervation influences local myocardial repolarization in normally perfused rabbit hearts. Circulation. 101: 1060–1066. https://doi.org/10.1161/01.cir.101.9.1060
Momose M., Tyndale-Hines L., Bengel F.M., Schwaiger M. (2001) How heterogeneous is the cardiac autonomic innervation? Basic Res. Cardiol 96: 539–546. https://doi.org/10.1007/s003950170004
Millar B.C., Schluter K.D., Zhou X.J., McDermott B.J., Piper H.M. (1994) Neuropeptide Y stimulates hypertrophy of adult ventricular cardiomyocytes. Am. J. Physiol. 266: C1271–1277. https://doi.org/10.1152/ajpcell.1994.266.5.C1271
Kanevskij M., Taimor G., Schafer M., Piper H.M., Schluter K.D. (2002) Neuropeptide Y modifies the hypertrophic response of adult ventricular cardiomyocytes to norepinephrine. Cardiovasc. Res. 53: 879–887. https://doi.org/10.1016/s0008-6363(01)00517-x
Wang J., Hao D., Zeng L., Zhang Q., Huang W. (2021) Neuropeptide Y mediates cardiac hypertrophy through microRNA-216b/FoxO4 signaling pathway. Int. J. Med. Sci. 18: 18–28. https://doi.org/10.7150/ijms.51133
Дополнительные материалы отсутствуют.
Инструменты
Российский физиологический журнал им. И.М. Сеченова